[level-membership-for-pediatrics-category]
Chapter 589 Neurocutaneous Syndromes
589.1 Neurofibromatosis
Clinical Manifestations and Diagnosis
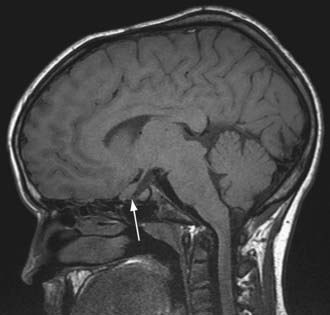
NF-1 is the most prevalent type, with an incidence of 1/3,000, and is diagnosed when any 2 of the following 7 features are present: (1) Six or more café-au-lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals. Café-au-lait spots are the hallmark of neurofibromatosis and are present in almost 100% of patients. They are present at birth but increase in size, number, and pigmentation, especially during the first few yrs of life (Fig. 589-1). The spots are scattered over the body surface, with predilection for the trunk and extremities but sparing the face. (2) Axillary or inguinal freckling consisting of multiple hyperpigmented areas 2-3 mm in diameter. Skinfold freckling usually appears between 3 and 5 yr of age. The frequency of axillary and inguinal freckling has been reported to be greater than 80% by 6 yr of age. (3) Two or more iris Lisch nodules. Lisch nodules are hamartomas located within the iris and are best identified by a slit-lamp examination (Fig. 589-2). They are present in >74% of patients with NF-1 but are not a component of NF-2. The prevalence of Lisch nodules increases with age, from only 5% of children <3 yr of age, to 42% among children 3-4 yr of age, and virtually 100% of adults ≥21 yr of age. (4) Two or more neurofibromas or 1 plexiform neurofibroma. Neurofibromas typically involve the skin, but they may be situated along peripheral nerves and blood vessels and within viscera including the gastrointestinal tract. These lesions appear characteristically during adolescence or pregnancy, suggesting a hormonal influence. They are usually small, rubbery lesions with a slight purplish discoloration of the overlying skin. Plexiform neurofibromas are usually evident at birth and result from diffuse thickening of nerve trunks that are frequently located in the orbital or temporal region of the face. The skin overlying a plexiform neurofibroma may be hyperpigmented to a greater degree than a café-au-lait spot. Plexiform neurofibromas may produce overgrowth of an extremity and a deformity of the corresponding bone. (5) A distinctive osseous lesion such as sphenoid dysplasia (which may cause pulsating exophthalmos) or cortical thinning of long bones (e.g., of the tibia) with or without pseudoarthrosis. (6) Optic gliomas are present in approximately 15% of patients with NF-1 and represent mostly low-grade astrocytomas. They are the main CNS tumor with a marked increased frequency in NF-1. Because of their growth, it is recommended that all children age 10 yr or younger with NF-1 undergo annual ophthalmologic examinations. When they progress, visual symptoms are produced because the tumors enlarge and put pressure on the optic nerves and chiasm resulting in impaired visual acuity and visual fields. Extension into the hypothalamus can lead to endocrine deficiencies or failure to thrive. The MRI findings of an optic glioma include diffuse thickening, localized enlargement, or a distinct focal mass originating from the optic nerve or chiasm (Fig. 589-3). (7) A first-degree relative with NF-1 whose diagnosis was based on the aforementioned criteria.
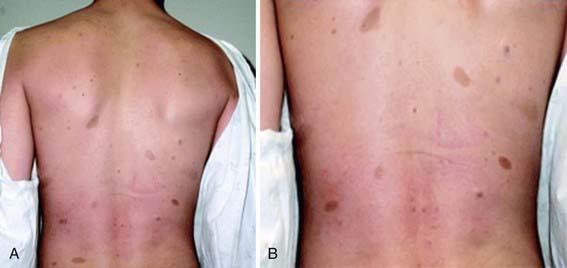
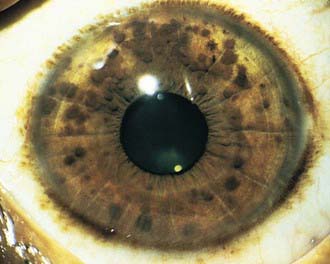
Figure 589-2 Neurofibromatosis 1. Pigmented hamartomas of the iris (Lisch nodules).
(From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 4, St Louis, 2002, Mosby, p 507.)
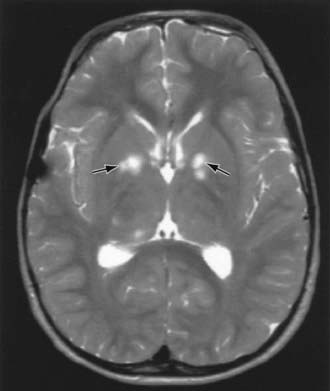
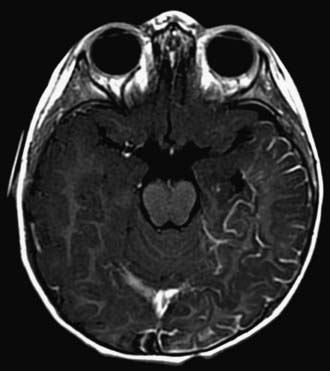
Children with NF-1 are susceptible to neurologic complications. MRI studies of selected children have shown abnormal hyperintense T2 weighted signals in the optic tracts, brainstem, globus pallidus, thalamus, internal capsule, and cerebellum (Fig. 589-4). These signals, “unidentified bright objects (UBOs),” tend to disappear with age; most have disappeared by 30 yr of age. It is unclear what the UBOs represent pathologically, and there is disagreement as to the presence and number of UBOs and their relationship to learning disabilities, attention deficit disorders, behavioral and psychosocial problems, and abnormalities of speech among affected children. Therefore, imaging studies such as brain MRIs should be reserved only for patients with clinical symptoms.
One of the most common complications is learning disability affecting approximately 30% of NF-1 children. Seizures are observed in approximately 8% of NF-1 patients. The cerebral vessels may develop aneurysms, or stenosis resulting in moyamoya disease (Chapter 594.1). Neurologic sequelae of these vascular abnormalities include transient cerebrovascular ischemic attacks, hemiparesis, and cognitive defects. Precocious puberty may become evident in the presence or absence of lesions of the optic chiasm and hypothalamus. Malignant neoplasms are also a significant problem in patients with NF-1, affecting approximately 3% of patients. A neurofibroma occasionally differentiates into a malignant peripheral nerve sheath tumor (MPNST). The incidence of pheochromocytoma, rhabdomyosarcoma, leukemia, and Wilms tumor is higher than in the general population. Scoliosis is a common complication found in about 10% of the patients. Patients with NF-1 are at risk for hypertension, which may result from renal vascular stenosis or a pheochromocytoma.
NF-2 is a rarer condition, with an incidence of 1/25,000, and may be diagnosed when 1 of the following 4 features is present: (1) bilateral vestibular schwannomas; (2) a parent, sibling, or child with NF-2 and either unilateral vestibular schwannoma or any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, or posterior subcapsular lenticular opacities; (3) unilateral vestibular schwannoma and any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, or posterior subcapsular lenticular opacities; (4) multiple meningiomas (2 or more) and unilateral vestibular schwannoma or any 2 of the following: schwannoma, glioma, neurofibroma, or cataract. Symptoms of tinnitus, hearing loss, facial weakness, headache, or unsteadiness may appear during childhood, although signs of a cerebellopontine angle mass are more commonly present in the 2nd and 3rd decades of life. Although café-au-lait spots and skin neurofibromas are classic findings in NF-1, they are much less common in NF-2. Posterior subcapsular lens opacities are identified in about 50% of patients with NF-2. The NF2 gene (also known as merlin or schwannomin) is located on chromosome 22q1.11. The frequency of lesions associated with NF-2 is noted in Table 589-1.
Table 589-1 FREQUENCY OF LESIONS ASSOCIATED WITH NEUROFIBROMATOSIS TYPE 2
| FREQUENCY OF ASSOCIATION WITH NF2 | |
|---|---|
| NEUROLOGIC LESIONS | |
| Bilateral vestibular schwannomas | 90-95% |
| Other cranial nerve schwannomas | 24-51% |
| Intracranial meningiomas | 45-58% |
| Spinal tumors | 63-90% |
| Extramedullary | 55-90% |
| Intramedullary | 18-53% |
| Peripheral neuropathy | Up to 66% |
| OPHTHALMOLOGIC LESIONS | |
| Cataracts | 60-81% |
| Epiretinal membranes | 12-40% |
| Retinal hamartomas | 6-22% |
| CUTANEOUS LESIONS | |
| Skin tumors | 59-68% |
| Skin plaques | 41-48% |
| Subcutaneous tumors | 43-48% |
| Intradermal tumors | Rare |
From Asthagiri AR, Parry DM, Butman JA, et al: Neurofibromatosis type 2, Lancet 373:1974–1984, 2009, Table 1.)
American Academy of Pediatrics Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics. 1995;96:368-372.
Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet. 2009;373:1974-1984.
Baser ME, R Evans DG, Gutmann DH. Neurofibromatosis 2. Curr Opin Neurol. 2003;16(1):27-33.
Cnossen MH, de Goede-Bolder A, van den Broek KM, et al. A prospective 10 year follow up study of patients with neurofibromatosis type 1. Arch Dis Child. 1998;78:408-412.
DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics Mar. 2000;105:608-614.
Evans DGR. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16.
Hersh JH, Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics. 2008;121:633-642.
Hofman KJ, Harris EL, Bryan RN, et al. Neurofibromatosis type 1: the cognitive phenotype. J Pediatr. 1994;124:S1-S8.
Krab LC, Aarsen FK, de Goede-Bolder A, et al. Impact of neurofibromatosis type 1 on school performance. J Child Neurol. 2008;23:1002-1010.
Messiaen L, Yao S, Brems H, et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndromes. JAMA. 2009;302:2111-2118.
Plotkin SR, Stemmer-Rachamimov AO, Barker FGII, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361:358-367.
Rizzo JF, Lessell S. Cerebrovascular abnormalities in neurofibromatosis type 1. Neurology. 1994;44:1000-1002.
Williams VC, Lucas J, Babcock MA, et al. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124-133.
589.2 Tuberous Sclerosis
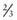 of the cases. Molecular genetic studies have identified 2 foci for TSC: the TSC1 gene is located on chromosome 9q34, and the TSC2 gene is on chromosome 16p13. The TSC1 gene encodes a protein called hamartin. The TSC2 gene encodes the protein tuberin. Within a cell, these 2 proteins bind to one another and work together. That is why a mutation in the TSC1 gene and a mutation in the TSC2 gene result in a similar disease in people. The loss of either tuberin or hamartin results in the formation of numerous benign tumors (hamartomas). Thus, the TSC1 and TSC2 genes are tumor suppressor genes. Tuberin and hamartin are involved in a key pathway in the cell that regulates protein synthesis and cell size. One of the ways cells regulate their growth is by controlling the rate of protein synthesis. A protein called mTOR was identified as 1 of the master regulators of cell growth. mTOR, in turn, is controlled by rheb, a small cytoplasmic GTPase. When rheb is activated, the protein synthesis machinery is turned on, most likely via mTOR, and the cell grows in size.
of the cases. Molecular genetic studies have identified 2 foci for TSC: the TSC1 gene is located on chromosome 9q34, and the TSC2 gene is on chromosome 16p13. The TSC1 gene encodes a protein called hamartin. The TSC2 gene encodes the protein tuberin. Within a cell, these 2 proteins bind to one another and work together. That is why a mutation in the TSC1 gene and a mutation in the TSC2 gene result in a similar disease in people. The loss of either tuberin or hamartin results in the formation of numerous benign tumors (hamartomas). Thus, the TSC1 and TSC2 genes are tumor suppressor genes. Tuberin and hamartin are involved in a key pathway in the cell that regulates protein synthesis and cell size. One of the ways cells regulate their growth is by controlling the rate of protein synthesis. A protein called mTOR was identified as 1 of the master regulators of cell growth. mTOR, in turn, is controlled by rheb, a small cytoplasmic GTPase. When rheb is activated, the protein synthesis machinery is turned on, most likely via mTOR, and the cell grows in size.Clinical Manifestations and Diagnosis
Definite TSC is diagnosed when at least 2 major or 1 major plus 2 minor features are present see Tables 589-2 and 589-3 for major and minor features, respectively).
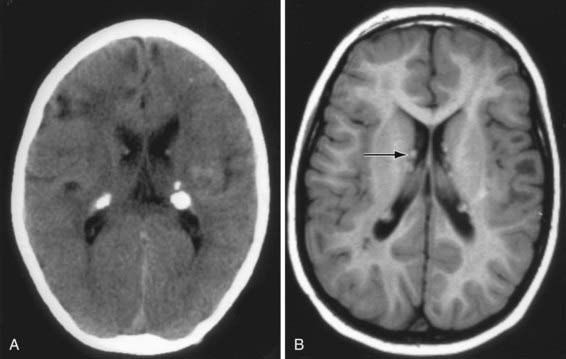
The hallmark of TSC is the involvement of the CNS (Fig. 589-5). Retinal lesions consist of 2 types: hamartomas (elevated mulberry lesions or plaquelike lesions) and white depigmented patches (similar to the hypopigmented skin lesions) (Fig. 589-6). The characteristic brain lesion is a cortical tuber (see Fig. 589-5). Brain MRI is the best way of identifying cortical tubers. Based on fetal MRI studies, we know that cortical tubers are formed while in utero. Subependymal nodules are lesions found along the wall of the lateral ventricles where they undergo calcification and project into the ventricular cavity, producing a candle-dripping appearance. These lesions do not cause any problems; in 5-10% of cases, these benign lesions can grow into subependymal giant cell astrocytomas (SEGAs). These tumors can grow and block the circulation of cerebrospinal fluid (CSF) around the brain and cause hydrocephalus, which requires immediate neurosurgical intervention.
The most common neurologic manifestations of TSC consist of epilepsy, cognitive impairment, and autism spectrum disorders. TSC may present during infancy with infantile spasms and a hypsarrhythmic electroencephalogram (EEG) pattern. It is important to remember that you can have hypsarrhythmia without infantile spasms and infantile spasms without hypsarrhythmia especially in TSC patients. The seizures may be difficult to control and, at a later age, they may develop into myoclonic epilepsy (Chapter 586). In Europe and Canada, infantile spasms associated with TSC are treated with vigabatrin (rather than ACTH) with good results. In the USA, ACTH had been the drug of choice until vigabatrin was approved by the U.S. Food and Drug Administration (FDA) for use in infantile spasms. Many patients with TSC have normal intelligence and few, if any, neurologic abnormalities.
Skin Lesions
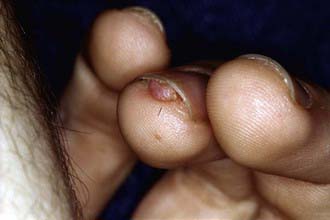
More than 90% of cases show the typical hypomelanotic macules that have been likened to an ash leaf on the trunk and extremities. Visualization of the hypomelanotic macule is enhanced by the use of a Wood ultraviolet lamp (Chapter 645). To count as a major feature, at least 3 hypomelanotic macules must be present (see Fig. 589-6). Facial angiofibromas develop between 4 and 6 yr of age; they appear as tiny red nodules over the nose and cheeks and are sometimes confused with acne (see Fig. 589-6). Later, they enlarge, coalesce, and assume a fleshy appearance. A shagreen patch is also characteristic of TSC and consists of a roughened, raised lesion with an orange-peel consistency located primarily in the lumbosacral region. During adolescence or later, small fibromas or nodules of skin may form around fingernails or toenails in 15-20% of the TSC patients (Fig. 589-7).
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345-1356.
Curatolo P, Bombardieri R, Jazwiak S. Tuberous sclerosis. Lancet. 2008;372:657-668.
Datta AN, Hahn CD, Sahin M. Clinical presentation and diagnosis of tuberous sclerosis complex in infancy. J Child Neurol. 2008;23:268-273.
Ewalt DH, Diamond N, Rees C, et al. Long-term outcome of transcatheter embolization of renal angiomyolipomas due to tuberous sclerosis complex. J Urol. Nov 2005;174(5):1764-1766.
O’Callaghan FJK, Martyn CN, Renowden S, et al. Subependymal nodules, giant cell astrocytomas and the tuberous sclerosis complex: a population-based study. Arch Dis Child. 2008;93:751-754.
Osborne JP, Merrifield J, O’Callaghan FJK. Tuberous sclerosis—what’s new? Arch Dis Child. 2008;93:728-731.
Roach ES, Miller VS. Neurocutaneous disorders. Cambridge, United Kingdom: Cambridge University Press; 2004.
Tworetzky W, McElhinney DB, Margossian R, et al. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003;92(4):487-489.
589.3 Sturge-Weber Syndrome
Diagnosis
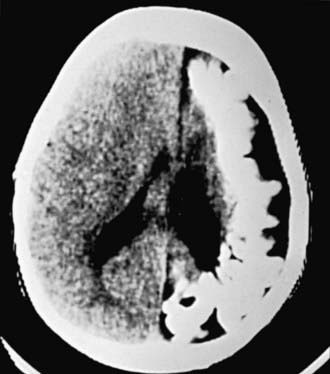
MRI with contrast is the imaging modality of choice for demonstrating the leptomeningeal angioma in SWS (Figure 589-8). White matter abnormalities are common and are thought to be a result of chronic hypoxia. Often, atrophy is noted ipsilateral to the leptomeningeal angiomatosis. Calcifications can be seen best with a head CT (Fig. 589-9). Ophthalmologic evaluation for glaucoma is also necessary. Based on the involvement of the brain and the face, there are 3 types according to the Roach Scale:
Comi AM. Advances in Sturge-Weber syndrome. Curr Opin Neurol. 2006;19(2):124-128.
Cordisco MR. An update on lasers in children. Curr Opin Pediatr. 2009;21(4):499-504.
Kossoff EH, Buck C, Freeman JM. Outcomes of 32 hemispherectomies for Sturge-Weber syndrome worldwide. Neurology. 2002;59:1735-1738.
Tan OT, Sherwood K, Gilchrest BA. Treatment of children with port-wine stains using the flashlamp-pulsed tunable dye laser. N Engl J Med. 1989;320:416-421.
589.4 Von Hippel–Lindau Disease
Gläsker S. Central nervous system manifestations in VHL: genetics, pathology and clinical phenotypic features. Fam Cancer. 2005;4(1):37-42.
Kaelin WGJr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8(11):865-873.
Latif F, Tory K, Gmarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317-1320.
Maher ER, Kaelin WGJr. von Hippel-Lindau disease. Medicine. 1997;76:381-391.
589.7 Incontinentia Pigmenti
Clinical Manifestations and Diagnosis
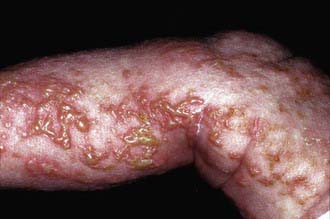
This disease has 4 phases, not all of which may occur in a given patient. The 1st phase is evident at birth or in the 1st few weeks of life and consists of erythematous linear streaks and plaques of vesicles (Fig. 589-10) that are most pronounced on the limbs and circumferentially on the trunk. The lesions may be confused with those of herpes simplex, bullous impetigo, or mastocytosis, but the linear configuration is unique. Histopathologically, epidermal edema and eosinophil-filled intraepidermal vesicles are present. Eosinophils also infiltrate the adjacent epidermis and dermis. Blood eosinophilia as high as 65% of the white blood cell count is common. The 1st stage generally resolves by 4 mo of age, but mild, short-lived recurrences of blisters may develop during febrile illnesses. In the 2nd phase, as blisters on the distal limbs resolve, they become dry and hyperkeratotic, forming verrucous plaques. The verrucous plaques rarely affect the trunk or face and generally involute within 6 mo. Epidermal hyperplasia, hyperkeratosis, and papillomatosis are characteristic. The 3rd or pigmentary stage is the hallmark of incontinentia pigmenti. It generally develops over weeks to months and may overlap the earlier phases, be evident at birth, or, more commonly, begin to appear in the 1st few weeks of life. Hyperpigmentation is more often apparent on the trunk than the limbs and is distributed in macular whorls, reticulated patches, flecks, and linear streaks that follow Blaschko lines. The axillae and groin are invariably affected. The sites of involvement are not necessarily those of the preceding vesicular and warty lesions. The pigmented lesions, once present, persist throughout childhood. They generally begin to fade by early adolescence and often disappear by age 16 yr. Occasionally, the pigmentation remains permanently, particularly in the groin. The lesion, histopathologically, shows vacuolar degeneration of the epidermal basal cells and melanin in melanophages of the upper dermis as a result of incontinence of pigment. In the 4th stage, hairless, anhidrotic, hypopigmented patches or streaks occur as a late manifestation of incontinentia pigmenti; they may develop, however, before the hyperpigmentation of stage 3 has resolved. The lesions develop mainly on the flexor aspect of the lower legs and less often on the arms and trunk.
 of affected children. Ocular anomalies, such as neovascularization, microphthalmos, strabismus, optic nerve atrophy, cataracts, and retrolenticular masses, occur in >30% of children. Nonetheless, >90% of patients have normal vision. Less common abnormalities include dystrophy of nails (ridging, pitting) and skeletal defects.
of affected children. Ocular anomalies, such as neovascularization, microphthalmos, strabismus, optic nerve atrophy, cataracts, and retrolenticular masses, occur in >30% of children. Nonetheless, >90% of patients have normal vision. Less common abnormalities include dystrophy of nails (ridging, pitting) and skeletal defects.Bruckner AL. Incontinentia pigmenti: a window to the role of NF-kappaB function. Semin Cutan Med Surg. 2004;23:116-124.
Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature. 2000;405(6785):466-472.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 589 Neurocutaneous Syndromes
589.1 Neurofibromatosis
Clinical Manifestations and Diagnosis
NF-1 is the most prevalent type, with an incidence of 1/3,000, and is diagnosed when any 2 of the following 7 features are present: (1) Six or more café-au-lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals. Café-au-lait spots are the hallmark of neurofibromatosis and are present in almost 100% of patients. They are present at birth but increase in size, number, and pigmentation, especially during the first few yrs of life (Fig. 589-1). The spots are scattered over the body surface, with predilection for the trunk and extremities but sparing the face. (2) Axillary or inguinal freckling consisting of multiple hyperpigmented areas 2-3 mm in diameter. Skinfold freckling usually appears between 3 and 5 yr of age. The frequency of axillary and inguinal freckling has been reported to be greater than 80% by 6 yr of age. (3) Two or more iris Lisch nodules. Lisch nodules are hamartomas located within the iris and are best identified by a slit-lamp examination (Fig. 589-2). They are present in >74% of patients with NF-1 but are not a component of NF-2. The prevalence of Lisch nodules increases with age, from only 5% of children <3 yr of age, to 42% among children 3-4 yr of age, and virtually 100% of adults ≥21 yr of age. (4) Two or more neurofibromas or 1 plexiform neurofibroma. Neurofibromas typically involve the skin, but they may be situated along peripheral nerves and blood vessels and within viscera including the gastrointestinal tract. These lesions appear characteristically during adolescence or pregnancy, suggesting a hormonal influence. They are usually small, rubbery lesions with a slight purplish discoloration of the overlying skin. Plexiform neurofibromas are usually evident at birth and result from diffuse thickening of nerve trunks that are frequently located in the orbital or temporal region of the face. The skin overlying a plexiform neurofibroma may be hyperpigmented to a greater degree than a café-au-lait spot. Plexiform neurofibromas may produce overgrowth of an extremity and a deformity of the corresponding bone. (5) A distinctive osseous lesion such as sphenoid dysplasia (which may cause pulsating exophthalmos) or cortical thinning of long bones (e.g., of the tibia) with or without pseudoarthrosis. (6) Optic gliomas are present in approximately 15% of patients with NF-1 and represent mostly low-grade astrocytomas. They are the main CNS tumor with a marked increased frequency in NF-1. Because of their growth, it is recommended that all children age 10 yr or younger with NF-1 undergo annual ophthalmologic examinations. When they progress, visual symptoms are produced because the tumors enlarge and put pressure on the optic nerves and chiasm resulting in impaired visual acuity and visual fields. Extension into the hypothalamus can lead to endocrine deficiencies or failure to thrive. The MRI findings of an optic glioma include diffuse thickening, localized enlargement, or a distinct focal mass originating from the optic nerve or chiasm (Fig. 589-3). (7) A first-degree relative with NF-1 whose diagnosis was based on the aforementioned criteria.
Figure 589-2 Neurofibromatosis 1. Pigmented hamartomas of the iris (Lisch nodules).
(From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 4, St Louis, 2002, Mosby, p 507.)
Children with NF-1 are susceptible to neurologic complications. MRI studies of selected children have shown abnormal hyperintense T2 weighted signals in the optic tracts, brainstem, globus pallidus, thalamus, internal capsule, and cerebellum (Fig. 589-4). These signals, “unidentified bright objects (UBOs),” tend to disappear with age; most have disappeared by 30 yr of age. It is unclear what the UBOs represent pathologically, and there is disagreement as to the presence and number of UBOs and their relationship to learning disabilities, attention deficit disorders, behavioral and psychosocial problems, and abnormalities of speech among affected children. Therefore, imaging studies such as brain MRIs should be reserved only for patients with clinical symptoms.
One of the most common complications is learning disability affecting approximately 30% of NF-1 children. Seizures are observed in approximately 8% of NF-1 patients. The cerebral vessels may develop aneurysms, or stenosis resulting in moyamoya disease (Chapter 594.1). Neurologic sequelae of these vascular abnormalities include transient cerebrovascular ischemic attacks, hemiparesis, and cognitive defects. Precocious puberty may become evident in the presence or absence of lesions of the optic chiasm and hypothalamus. Malignant neoplasms are also a significant problem in patients with NF-1, affecting approximately 3% of patients. A neurofibroma occasionally differentiates into a malignant peripheral nerve sheath tumor (MPNST). The incidence of pheochromocytoma, rhabdomyosarcoma, leukemia, and Wilms tumor is higher than in the general population. Scoliosis is a common complication found in about 10% of the patients. Patients with NF-1 are at risk for hypertension, which may result from renal vascular stenosis or a pheochromocytoma.
NF-2 is a rarer condition, with an incidence of 1/25,000, and may be diagnosed when 1 of the following 4 features is present: (1) bilateral vestibular schwannomas; (2) a parent, sibling, or child with NF-2 and either unilateral vestibular schwannoma or any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, or posterior subcapsular lenticular opacities; (3) unilateral vestibular schwannoma and any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, or posterior subcapsular lenticular opacities; (4) multiple meningiomas (2 or more) and unilateral vestibular schwannoma or any 2 of the following: schwannoma, glioma, neurofibroma, or cataract. Symptoms of tinnitus, hearing loss, facial weakness, headache, or unsteadiness may appear during childhood, although signs of a cerebellopontine angle mass are more commonly present in the 2nd and 3rd decades of life. Although café-au-lait spots and skin neurofibromas are classic findings in NF-1, they are much less common in NF-2. Posterior subcapsular lens opacities are identified in about 50% of patients with NF-2. The NF2 gene (also known as merlin or schwannomin) is located on chromosome 22q1.11. The frequency of lesions associated with NF-2 is noted in Table 589-1.
Table 589-1 FREQUENCY OF LESIONS ASSOCIATED WITH NEUROFIBROMATOSIS TYPE 2
| FREQUENCY OF ASSOCIATION WITH NF2 | |
|---|---|
| NEUROLOGIC LESIONS | |
| Bilateral vestibular schwannomas | 90-95% |
| Other cranial nerve schwannomas | 24-51% |
| Intracranial meningiomas | 45-58% |
| Spinal tumors | 63-90% |
| Extramedullary | 55-90% |
| Intramedullary | 18-53% |
| Peripheral neuropathy | Up to 66% |
| OPHTHALMOLOGIC LESIONS | |
| Cataracts | 60-81% |
| Epiretinal membranes | 12-40% |
| Retinal hamartomas | 6-22% |
| CUTANEOUS LESIONS | |
| Skin tumors | 59-68% |
| Skin plaques | 41-48% |
| Subcutaneous tumors | 43-48% |
| Intradermal tumors | Rare |
From Asthagiri AR, Parry DM, Butman JA, et al: Neurofibromatosis type 2, Lancet 373:1974–1984, 2009, Table 1.)
American Academy of Pediatrics Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics. 1995;96:368-372.
Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet. 2009;373:1974-1984.
Baser ME, R Evans DG, Gutmann DH. Neurofibromatosis 2. Curr Opin Neurol. 2003;16(1):27-33.
Cnossen MH, de Goede-Bolder A, van den Broek KM, et al. A prospective 10 year follow up study of patients with neurofibromatosis type 1. Arch Dis Child. 1998;78:408-412.
DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics Mar. 2000;105:608-614.
Evans DGR. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16.
Hersh JH, Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics. 2008;121:633-642.
Hofman KJ, Harris EL, Bryan RN, et al. Neurofibromatosis type 1: the cognitive phenotype. J Pediatr. 1994;124:S1-S8.
Krab LC, Aarsen FK, de Goede-Bolder A, et al. Impact of neurofibromatosis type 1 on school performance. J Child Neurol. 2008;23:1002-1010.
Messiaen L, Yao S, Brems H, et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndromes. JAMA. 2009;302:2111-2118.
Plotkin SR, Stemmer-Rachamimov AO, Barker FGII, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361:358-367.
Rizzo JF, Lessell S. Cerebrovascular abnormalities in neurofibromatosis type 1. Neurology. 1994;44:1000-1002.
[/not-level-membership-for-pediatrics-category]
