56 Neuroblastoma
Epidemiology, Screening, Etiology, and Genetics
Neuroblastoma is the most common extracranial solid tumor in children and the most common malignancy of infants. Approximately 650 new cases are diagnosed annually in the United States, with an incidence of 0.9 per 100,000.1 The tumor accounts for 8% to 10% of all childhood cancers and is slightly more common in boys than in girls, with a male-to-female ratio of 1.1 : 1. Review of more than 3000 patients registered on studies at Pediatric Oncology Group (POG) institutions from 1990 to 2000 and at Children’s Cancer Group (CCG) institutions from 1991 to 1995 show the median age at diagnosis is 17.3 months; 40.1% are younger than 1 year of age, 89.4% are younger than 5 years of age, and 97.8% are younger than 10 years of age.2
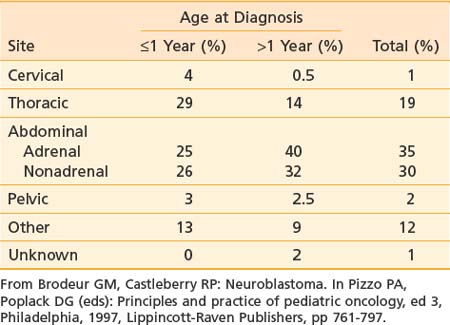
Neuroblastoma arises from primitive adrenergic neuroblasts of neural crest tissue. This tissue forms columns that are the precursors of spinal ganglia, dorsal spinal nerve roots, and chromaffin cells of the adrenal medulla. Thus, the majority of neuroblastoma cases occur in an anatomic distribution consistent with the location of neural crest tissue. The location of tumors at the time of diagnosis varies with age (Table 56-1). The adrenal gland is the most common location for the development of neuroblastoma, accounting for 35% of cases (Figure 56-1). Paraspinal ganglia in the low thoracic and abdominal chains constitute 30% of all cases, the posterior mediastinum accounts for 19%, and pelvic and cervical ganglia account for 2% or fewer.3 It has been reported that microscopic neuroblastic nodules, resembling neuroblastoma in situ, were found frequently at autopsy in infants younger than 3 months of age who died of unrelated causes.4 Initially, this was believed to mean the incidence of neuroblastoma was much higher during early infancy, but these tumors regressed spontaneously and were never clinically detected. However, studies have demonstrated that these nodules occur uniformly in all fetuses, peak between 17 and 20 weeks of gestation, and gradually regress by the time of birth or shortly thereafter.5 Thus, the development and subsequent regression of neuroblastoma nodules most likely represents a normal embryologic event, and the development of clinically detectable neuroblastoma is a consequence of a disruption of this process.
Screening Studies
The most extensive data regarding neuroblastoma screening come from Japan, Germany, and a combined Canadian and U.S. study. Neuroblastoma screening has been in place in Japan since 1981. During a 12-year screening period, the incidence rate in infants younger than 1 year of age rose from 28 to 260 per million, but there was no reduction in the incidence rate in children older than 1 year of age.6 The Japanese have also noted a reduction in overall mortality since the inception of mass screening.7 However, this reduced mortality may reflect the improving treatments that have arisen during the same time and not be a true reflection of the benefits of screening. In Canada, they have also seen an increase in the incidence of early stage disease in children less than 1 year of age.8 There has been no reduction in the incidence of advanced stage disease in children older than 1 year of age or in the mortality from neuroblastoma based on population screening at 3 weeks and 6 months of age in Canada.9 Screening at 1 year of age in Germany also did not reduce the incidence of advanced-stage disease in children or the mortality of children diagnosed with neuroblastoma.10 In addition, the psychological affect of false-positive tests and the potential risk incurred by interventions performed on clinically insignificant tumors has not been fully quantified, but could be significant. Many of the tumors detected by mass screening have now been observed, and more than one-third spontaneously regress.11 Thus the benefits of mass screening seem limited and do not appear to warrant the costs.
Genetics
The cause of neuroblastoma is unknown in most cases. No prenatal or postnatal exposure to drugs, chemicals, or radiation has unequivocally demonstrated an increased incidence of neuroblastoma.12,13 Genetics, however, play an extremely important role. A case control study in France (ESCALE) showed a positive association between congenital malformation and neuroblastoma, particularly in infants less than one year of age.14 Familial neuroblastoma is also well described and presents at an earlier median age (9 months versus 22 months for sporadic cases). At least 20% of patients with familial neuroblastoma have bilateral or multifocal disease.15 Genetic studies of hereditary disease have been impeded by the rarity of the condition and the small size of pedigrees caused by the lethality of neuroblastoma in early childhood. A family history of neuroblastoma is obtained in only approximately 1% of patients. Studies of such families suggest locus heterogeneity; no commonly mutated gene has been identified, and the penetrance appears to be low.16 Genetic loci in different families include germline mutations in PHOX2B, seen in patients with associated abnormalities such as congenital hypoventilation syndrome or Hirschsprung disease, and abnormalities of the short arm of 16.17–20 Neuroblastoma has also been seen occasionally in patients with constitutional chromosomal rearrangements, including deletions overlapping putative tumor suppressor loci at chromosome bands 1p36 and 11q14-23.21–23 Thus neuroblastoma predisposition is genetically heterogeneous, and initiation of tumorigenesis may require multiple alterations. A recent study using whole-genome single-nucleotide polymorphism analysis revealed a common genetic variation at chromosome band 6p22 that is associated with susceptibility to neuroblastoma, particularly with high-risk disease.24 Very recently, a number of studies have appeared showing that germline mutations or amplifications in the ALK gene are found in approximately 10% of sporadic neuroblastoma and germline mutations in the majority of heritable neuroblastoma.25
Deletion of the distal short arm of chromosome 1 (1p) was the first described genetic mutation in neuroblastoma tumors and is the most consistently reported abnormality, occurring in 70% to 80% of the karyotyped near-diploid tumors.26 This most likely represents a deletion (and loss of heterozygosity [LOH]) of a tumor suppressor gene and is found more commonly in advanced disease. Homogeneous staining regions (HSRs) and double-minute chromatin bodies (dmins) are a manifestation of gene amplification. Both of these are derived from the distal short arm of chromosome 2 (2p), which contains the proto-oncogene MYCN.27
MYCN amplification occurs in roughly 25% of primary neuroblastoma cases, 5% to 10% in patients with low stages of disease and stage IV-S, and 30% to 40% in patients with advanced disease.28 The MYCN proto-oncogene is derived from the c-myc viral oncogene and, as mentioned previously, is located on 2p, thus correlating with the cytogenetic abnormalities of HSRs and dmins. If it is going to occur, MYCN amplification is almost always present by the time of diagnosis. This suggests that it is an intrinsic property of a subset of aggressive tumors with a poor prognosis.27 The development of an overexpressing TH-MYCN transgenic mouse that spontanously develops neuroblastoma is further evidence of a role of this oncogene in tumorigenesis.29 Molecular studies have shown a strong correlation between 1p LOH and MYCN, suggesting these two genetic events may be related.26
Chromosomal ploidy is another marker of prognosis that is particularly useful in infants younger than 18 months of age. Near-diploid or pseudodiploid tumors have near-normal nuclear DNA content but often have structural chromosomal abnormalities, including MYCN amplification. Hyperdiploid or near-triploid tumors typically lack MYCN amplification and 1p deletion and therefore have an excellent prognosis.30,31
Telomerase activity has been correlated with MYCN amplification and poor outcomes while increased expression of high-affinity nerve growth factor receptor (gp140TRK-A) has been associated with favorable outcomes. Conversely, the lack of expression of gp140TRK-A has been associated with MYCN amplification and poor survival.27
Pathologic Conditions
Neuroblastoma is a classic “small round blue cell” tumor. Others include Ewing sarcoma, non-Hodgkin lymphoma, primitive neuroectodermal tumors, and rhabdomyosarcoma. Histologic subtypes, including neuroblastoma, ganglioneuroblastoma, and ganglioneuroma, represent different points along the maturation pathway in order of increasing differentiation. The typical neuroblastoma is composed of small, uniform cells containing dense, hyperchromatic nuclei and scant cytoplasm. The presence of neuritic processes (neuropil) is pathognomonic. Homer-Wright pseudorosettes, neuroblasts surrounding areas of eosinophilic neuropil, are seen in 15% to 50% of cases. Ganglioneuromas are composed primarily of mature ganglion cells, neuropil, and Schwann cells, and behave in a benign fashion. Ganglioneuroblastomas have histopathologic characteristics of both neuroblastomas and ganglioneuromas. Because histopathologic features may vary within any single tumor, multiple sections must be examined.32,33 Distinguishing neuroblastomas from other small round blue cell tumors requires special techniques including immunohistochemistry and electron microscopy. Neuroblastoma stains with monoclonal antibodies that recognize neuron-specific enolase, synaptophysin, and neurofilament. Electron microscopy reveals neurosecretory granules that contain catecholamines, microfilaments, and parallel arrays of microtubules within the neuropil.34
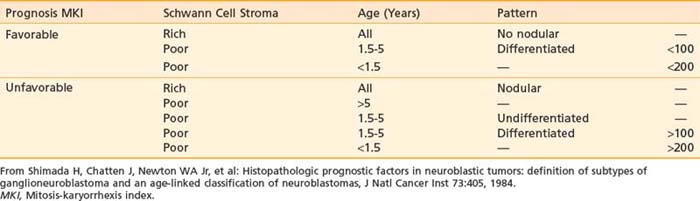
Although many different classification systems have been used to help define the prognosis for neuroblastoma, the International Neuroblastoma Pathology Commmittee (INPC) defined and tested a modification of the Shimada system, now widely accepted and validated.32 It is divided into favorable and unfavorable prognostic groups based on histologic category, age, amount of Schwann cell stroma, degree of differentiation; and the mitosis-karyorrhexis index. Each of these features are also independently prognostic.35 In addition, nodular versus diffuse histologic pattern is noting macro nodules, which tend to be associated with a poor prognosis compared with intermixed nodules. Characteristics of the INPC system are listed in Table 56-2.
Clinical Presentation
As mentioned previously, primary tumor location varies with age (see Table 56-1). Extent of disease is also age-dependent; the majority of children younger than 1 year of age have localized disease at the time of diagnosis, whereas the majority of children older than 1 year of age have disseminated disease (Table 56-3). Signs and symptoms of neuroblastoma depend on location of the primary tumor but can also reflect lymph node status and metastatic spread. Bone marrow, bone, liver, and skin are the most common sites for hematogenous spread. Rarely, disease spreads to the lungs and brain. Table 56-4 lists the associated signs and symptoms for each anatomic area of disease. Because of the type of catecholamines secreted from most neuroblastomas, hypertension, flushing, and tachycardia are uncommon symptoms. Other rare paraneoplastic symptoms are secretory diarrhea, resulting from secretion of vasoactive intestinal peptide,36 and opsoclonus-myoclonus ataxia syndrome, most likely resulting from antineuronal antibodies that cross-react with cerebellar tissue.37
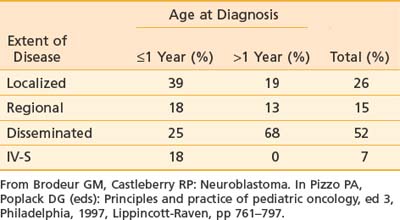
Table 56-4 Signs and Symptoms of Disease Based on Anatomic Location of Primary Tumor and Metastases
| Primary Site | Signs and Symptoms |
|---|---|
| Abdomen |
Diagnostic and Staging Studies
In terms of regional and distant disease, CT and MRI can assess nodal disease, hepatic metastases, and intraspinal extension. CT and MRI are also useful in determining whether metastases to the skull, orbit, mandible, or brain are present. Conventional radiographs may be used to assess painful bone metastases, but the standard and most sensitive agent for detection of osteomedullary involvement is 123I-metaiodobenzylguanidine (MIBG) scan, which concentrates as well in primary tumors and soft tissue metastases. MIBG is a guanethidine derivative and an analogue of norepinephrine, and therefore specifically taken up and stored in tumors of sympathetic origin, which express the norepinephrine transporter.38 Because of the high specificity and sensitivity in neuroblastoma, 123I-MIBG has superseded the use of technetium bone scans for the detection of skeletal metastases in the majority of children with neuroblastoma tumors, which take up the chemical in greater than 90% of cases, and has been recommended by the last international consensus conference as a standard element of staging and response evaluation.39,40 A 99mTc bone scan is indicated for detection of bone metastases in the occasional patient whose tumor does not take up MIBG. 18F-deoxyglucose positron emission tomography is also a useful complementary imaging modality that reflects the metabolic activity of primary and metastatic lesions, but its exact role in staging and response is still under investigation.41,42 Metastatic lesions are most often located in the periorbital region, in the metaphyses of long bones, and in the axial skeleton. Complete staging includes two bilateral posterior iliac crest bone marrow aspirates and biopsies. A single positive result is sufficient for the documentation of bone marrow involvement.39
Excess catecholamines are produced in nearly 90% of cases. Therefore, part of the neuroblastoma workup includes measuring urine catecholamines and their metabolites, specifically norepinephrine, vanillylmandelic acid, 3-methoxy-4-hydroxylphenylglycol, or homovanillic acid. Urine or serum dopamine may be measured. Urinary catecholamines are often given as ratios to urinary creatinine.43
According to the international criteria for diagnosing, staging, and assessing response to treatment, the diagnosis of neuroblastoma is established under one of the following circumstances: (1) An unequivocal pathologic diagnosis is made from tumor tissue by light microscopy (with or without immunohistology, electron microscopy, increased urine or serum catecholamines or metabolites), or (2) Bone marrow aspirate or trephine biopsy contains unequivocal tumor cells and urine or serum catecholamines or metabolites are increased.39
Staging and Prognostic Factors
Several different staging systems have been used in neuroblastoma. The Evans and D’Angio classification, historically used by the CCG classification, is the original system. It is based on the extent of the primary tumor and the presence or absence of distant disease. It includes stages I, II, III, IV, and IV-S.44 The POG used an alternative staging system that included initial resectability of the primary and lymph node status, although the importance of lymph node involvement in neuroblastoma is unclear. It includes stages A, B, C, D, and D-S.45,46 The differences between these two systems can be substantial, depending on the stage. Therefore, a group of investigators met at two international conferences to arrive at a consensus regarding neuroblastoma staging. Criteria for diagnosing, staging, and assessing response to treatment was set forth, and the International Neuroblastoma Staging System (INSS) is now generally accepted as the standard staging system (Table 56-5). To interpret the literature, it is important to recognize the similarities between the INSS and the other two major staging systems. Stage 1 is similar to stages I and A. Stage 4 is essentially the same as stages IV and D, and stage 4S is the same as IV-S and D-S. The greatest disagreement concerns the middle stages (II and III; B and C). Therefore, the INSS divided stage 2 into 2A (incompletely excised tumor) and 2B (ipsilateral lymph node involvement) to evaluate whether patients with 2B disease behave more like stage 2A patients or stage 3 patients. In addition to revising staging, the INSS delineated standard definitions of response to treatment (Table 56-6). These revisions and standard definitions were designed to make interpreting the literature and making management decisions uniform.39
* The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column.
† Marrow involvement in stage 4S should be minimal (i.e., <10% of total nucleated cells identified as malignant on bone marrow biopsy or on marrow aspirate). More extensive marrow involvement is considered to be stage 4. The metaiodobenzylguanidine scan (if performed) should be negative in the marrow.
From Brodeur GM, Seeger RC, Barrett A, et al: International criteria for diagnosis, staging and response to treatment in patients with neuroblastoma, J Clin Oncol 6:1874–1881, 1988; and Brodeur GM, Pritchard J, Berthold F, et al: Revisions in the international criteria neuroblastoma diagnosis, staging and response to treatment, J Clin Oncol 11:1466–1477, 1993.
Table 56-6 INSS Classification of Responses to Therapy
| Response | Metastatic Sites/Markers | Primary Tumor |
|---|---|---|
| Complete response (CR) | No tumor | No tumor; catecholamines normal |
| Very good partial response (VGPR) | Decreased by 90%-99% | No tumor; catecholamines normal; residual 99Tc |
| Partial response (PR) | Decreased by >50% | All measurable sites decreased by >50% |
| Bones and bone marrow | Number of positive bone sites decreased by >50%; no more than one positive bone marrow site allowed* | |
| Mixed response (MR) | No new lesions | >50% reduction of any measurable lesion (primary or metastases) with <50% reduction in any other; <25% increase in any existing lesion |
| No response (NR) | No new lesions | <50% reduction but <25% increase in any existing lesion |
| Progressive disease (PD) | Any new lesion | Increase of any measurable lesion by >25%; previous negative marrow biopsy positive for tumor |
INSS, International Neuroblastoma Staging System.
* One positive marrow aspirate or biopsy allowed for PR if this response represents a decrease from the number of positive sites at diagnosis.
From Brodeur GM, Pritchard J, Berthold F et al: Revisions in the international criteria neuroblastoma diagnosis, staging and response to treatment, J Clin Oncol 11:1466–1477, 1993.
A more recent international consensus conference was held in 2005 to further refine the staging and risk classification for neuroblastoma. This International Neuroblastoma Risk Group (INRG) Task Force proposed a simpler staging, based on whether or not a tumor had image-defined risk factors for surgery (L1: no risk factors; L2: risk factors present),47 and whether or not there was metastatic disease.48 In addition, further refinements were made to the response criteria, to incorporate semiquantitative scoring for the MIBG scan49,50 and standardization of bone marrow disease using immunocytology and real time–polymerase chain reaction.51,52
Treatment for neuroblastoma is based on risk stratification. Therefore, it is essential to understand the clinical and biologic variables predictive of disease outcome. The most important clinical risk factors are disease stage and age at diagnosis. Infants younger than 18 months have a more favorable outcome compared with older children with the same disease stage, as shown in a retrospective analysis of 3666 patients (1986 to 2001) from patients in the Children’s Oncology Group (COG) with documented follow-up data. Using a cut-off of 460 days of age, the 4-year event-free survival (EFS) for those younger than 460 days was 82% versus 42% for those older than 460 days (P < 0.0001); using a cut-off of 573 days, the 4-year EFS was 74% and 38%, respectively (P < 0.0001). The 18-month (547-day) cut off was chosen for convenience, because the difference in EFS was slight over the range of 15 to 18 months. MYCN gene amplification and stage 4 were also more frequent in the older patients, but even after adjustment for these factors, age remained significant.53 Similarly, several biologic variables have prognostic significance. A new histopathologic classification, the INPC, based on the original Shimada system, is now the international standard. Like the Shimada system, this classifies patients into favorable and unfavorable groups based on age, amount of Schwann cell stroma, degree of differentiation, and mitotic-karyorrhectic index (see Table 56-2). Further analyses of an international database of 8800 neuroblastoma patients showed that each of the components of the INPC had independent prognostic significnace within a given age group.35 Serum markers including ferritin, neuron-specific enolase, lactate dehydrogenase, and GD2 each have prognostic value (increased levels associated with worse prognosis); however, they are not currently used in risk stratification because their multivariate importance is less significant than other factors. Unfavorable genetic prognostic markers that are most significant in multivariable analysis include MYCN gene amplification, diploidy, and11q LOH. Other genetic changes with prognostic value in univariate analysis include 1p LOH, TRKA expression, and 17q additions, though these are not currently used in risk stratification.35
Standard Therapeutic Approaches
Surgery
Surgery is used to establish the diagnosis, provide tissue for biologic studies, determine stage, establish vascular access for chemotherapy, and accomplish therapeutic excision of the primary tumor and involved regional lymph nodes. The operative approach depends on the initial risk evaluation, which in turn is a function of tumor location, mobility, relationship to major vessels and nerves, ability to control blood supply, presence of distant metastases, and overall prognosis. More recently, image-defined risk factors have been used to determine resectability.54 These risk factors are based in the main on vascular or neural encasement as determined by imaging studies. In patients with high-risk disease, initial surgery is directed toward diagnosis, staging, vascular access, and adequate tissue sampling for diagnostic studies. Biopsy of the liver at the time of initial surgery is proposed for patients with abdominal tumors without clinical evidence of metastatic disease.2
Nonadherent, intracavitary lymph nodes should be sampled, including lymph nodes superior and inferior to the primary tumor. Lymph nodes adherent to the primary tumor have little relevance in predicting outcome but should be removed if grossly involved. High-risk patients always receive neoadjuvant chemotherapy prior to primary site resection as this increases the chance of complete removal and reduces the risk of complications, especially nephrectomy.55 In high-risk patients, gross total resection of the primary tumor site and involved regional lymphatics seems to be associated with improved outcome, and especially improved local control (LC).56–57
Radiation Therapy
Neuroblastoma is a relatively radiosensitive tumor. Data collected on neuroblastoma cell line show a D0 of 1.04 Gy and an n of 1.36, which is much more radiosensitive than other mammalian tumors.58 However, despite its radiosensitivity in the laboratory, its clinical response to irradiation is variable, which may be attributed to its variation in genomic amplification of MYCN.59 Radiation therapy is indicated mainly for high-risk disease, including bulky, unresectable, residual, and metastatic disease, or for palliation of painful recurrent lesions. In rare cases, radiation may be used for intermediate- or low-risk neuroblastoma, including symptomatic spinal cord compression unresponsive to chemotherapy, or life-threatening respiratory distress or abdominal distention in an infant with 4S neuroblastoma resulting from hepatomegaly. Doses have ranged from 15 to 30 Gy in 1.5 to 4 Gy per fraction. The role of radiation therapy for patients with locoregional disease is not clearly defined. A study by Paulino and colleagues60 evaluated locoregional control in infants with neuroblastoma who received radiation therapy. They reported 53 children younger than 1 year old who were retrospectively staged according to the INSS guidelines; 8 had stage 1, 7 stage 2A, 6 stage 2B, 15 stage 3, 6 stage 4, and 11 stage 4S. The primary tumor was located in the adrenal gland in 26 (49%), abdomen and nonadrenal in 14 (26%), thorax in 9 (17%), neck in 2 (4%), and pelvis in 2 (4%). All patients, except 11 with stage 4S and 4 with stage 4, had resection of the primary tumor. Postoperative doses ranged from 15 to 25 Gy, whereas preoperative doses ranged from 12 to 31 Gy using a median fraction size of 1.5 Gy. Chemotherapy was employed in 22 of 53 patients (42%). The 5-year overall survival (OS), freedom from progression (FFP), and LC rates were 79%, 81%, and 88%, respectively. INSS stage was a prognostic factor for OS and FFP. There were no isolated locoregional relapses. The 15-year musculoskeletal toxicity rate was 47.3% for those receiving radiation and 3.3% for the group who did not receive radiation. The authors concluded that most LN+ infants achieve locoregional control without radiation and infants younger than 6 months of age who received radiation were the most susceptible to musculoskeletal abnormalities.60
Treatment Technique
As with all radiation treatment planning, every attempt should be made to minimize exposure to normal tissues. For high-risk patients who require radiation to the primary site, gross target volume (GTV) is defined as the tumor remaining prior to attempted surgical resection as defined by CT, MRI, or MIBG scans. Uninvolved draining lymph node regions, including next-echelon lymph node regions, are not covered because it is rare to have an isolated next-echelon nodal failure.61 Primary tumors are often large and frequently expand into a body cavity, displacing normal structures, without infiltrating them. The primary tumor shrinks after induction chemotherapy, and then, after surgical resection, the normal structures now occupy the space previously occupied by the tumor mass. These normal structures do not need to be included in the GTV. Clinical target volume (CTV) typically includes a 1 to 1.5 cm margin on the GTV, and planning target volume (PTV) depends on patient immobilization but is approximately 0.5 cm on the CTV. CT-based treatment planning is recommended for treatment of the primary site. In many cases, anterior and posterior beams are best suited for sparing normal tissue, but in select cases 3-D conformal or intensity-modulated radiation therapy (IMRT) may be beneficial. Caution is advised with respect to IMRT in the thoracic region where there are significant heterogeneities and where tumor movement is difficult to track. Dose constraints to the contralateral kidney, the lung, and the liver are extremely important, and in girls, dose to the ovaries should be considered. Because the primary tumors are near midline, it is important to include an entire vertebral body in the PTV to prevent scoliosis.
Neuroblastoma frequently metastasizes to the skull, often diffusely involving the calvarium and skull base. Although radiating the sites of metastatic disease may improve LC, irradiating the brain in young patients is undesirable. Memorial Sloan-Kettering Cancer Center recently published their results with regard to technique, outcome, and toxicities with brain-sparing whole-skull radiotherapy.62
Dose
Historically, radiation was given to all patients except those with Stage I tumors. Doses less than 20 Gy were found to be acceptable for LC, although today the majority of those patients would not receive radiation therapy.63 In the most recently completed COG study (A3973)64 dose to both the primary site and metastatic sites was 21.6 Gy in 12 daily fractions (1.8 Gy qd). Another acceptable regimen is 21 Gy in 14 twice-daily fractions (1.5 Gy bid). Because of the high rates of local recurrence, particularly following an incomplete resection, in the ongoing COG study (ANBL0532),65 patients undergoing incomplete resections will receive the standard dose (21.6 Gy) to the postinduction chemotherapy, preoperative tumor volume followed by a boost of 14.4 Gy to the gross residual volume for a total dose of 36 Gy.
For infants with stage 4S disease with respiratory distress secondary to hepatomegaly, radiation therapy may be indicated.66 Effective doses are 2 to 6 Gy in single or multiple fractions and the entire liver need not be included in the treatment portal. This scheme may be repeated if necessary (liver may be slow to respond) as long as the kidney is shielded. Radiation (alone or with surgery) may also be used for spinal cord compression secondary to dumbbell extension of the primary tumor; however, studies have shown that chemotherapy alone is equally efficacious with fewer long-term side effects. Doses range from 7.5 Gy to 30 Gy.67–70 Total-body irradiation (TBI) can be used for patients undergoing transplant. Doses are 7.5 to 12 Gy in 3 to 6 fractions.71 Rare patients who develop parenchymal brain metastases, have a significant risk for the development of leptomeningeal disease. At Memorial Sloan-Kettering Cancer Center, a retrospective query identified 29 children with high-risk neuroblastoma treated since 1987 who received radiation therapy for central nervous system (CNS) relapse. All patients received appropriate initial therapies before demonstrating CNS recurrence. At the time of relapse, 16 patients received craniospinal irradiation (CSI) and 13 received focal radiation therapy that did not address the entire neuraxis. CSI was delivered to a median dose of 21.6 Gy, and 12 patients received a boost to a median dose of 25.4 Gy. The majority of those who underwent CSI also received intra-Ommaya radioimmunotherapy with 131I-8H9 or 131I-3F8. At a median follow-up of 21 months after CNS relapse, 12 patients in the CSI group are alive without evidence of CNS disease. All 13 patients in the non-CSI cohort died at a median of 8.8 months.72
Chemotherapy
Chemotherapy is the principal modality used in intermediate- and high-risk patients. Cyclophosphamide, platinum derivatives, doxorubicin, camptothecins and the epipodophyllotoxins have proven effective in several phase II studies and have thus become the backbone of induction therapy.73–77 Recent pilot studies in relapse patients show activity of temozolomide alone or with irinotecan, leading to an ongoing phase II study in the COG.78–80
Risk-Stratified Therapy
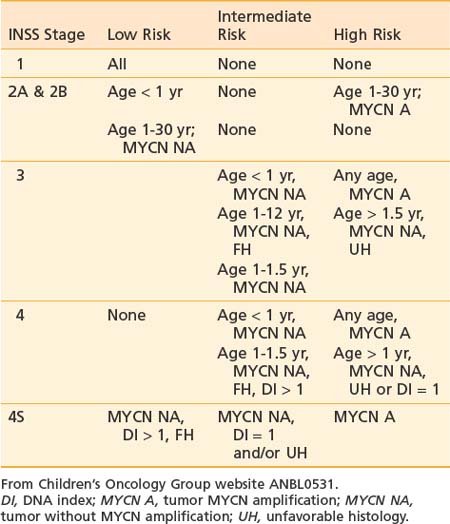
The COG Risk Stratification System (Table 56-7) uses patient age, INSS stage, DNA index (DI), INPC and MYCN amplification to assign patients to low-, intermediate-, and high-risk groups, which determine treatment. Further refinement has been added for intermediate-risk patients by intensifyng treatment in cases in which there is unbalanced loss of 11q.
Low-Risk Neuroblastoma
Children with localized neuroblastoma and favorable tumor biology may be cured with surgery alone, and, unlike other tumors, complete resection is unnecessary in these patients. Chemotherapy and radiation are reserved for progressive or recurrent disease or local disease-related symptoms such as spinal cord compression or respiratory distress. The CCG reported on the outcome of Evans stages I to II neuroblastoma treated with surgery as primary therapy. Patients were also assigned an INSS stage—All Evans stage I patients were INSS stage 1; nearly half of Evans stage II patients were also INSS stage 1, with the other half being INSS stage 2b; few were INSS stage 2a. EFS and OS for all stage I patients were 93% and 99% and for stage II patients were 81% and 98%. Additional therapy (radiation, surgery, or chemotherapy) was needed in only 10% of stage I patients and 20% of stage II patients.81 These results have been confirmed again in a large COG study, P9641.82 In this study, radiation was reserved for the rare situation in which patients with symptomatic spinal cord compression or hepatomegaly causing respiratory distress did not respond rapidly enough to chemotherapy. In addition, radiation therapy was given to patients with recurrent local-regional disease of unfavorable biology who received a partial response (PR) after treatment with courses I and II (eight cycles) of chemotherapy with or without subsequent operation. For children with disease other than stage 4S, the dose was 21 Gy in 14 daily fractions. For children with stage 4S disease, the dose was 4.5 Gy in three daily fractions. The target volume included the gross residual disease with a 2-cm margin. For patients who underwent an operative procedure prior to radiation therapy, the volume was the preoperative tumor, regardless of the extent of resection.
MYCN amplification is rare in localized neuroblastoma. The best method to manage low-risk patients who are MYCN-amplified remains controversial. In the aforementioned CCG study, seven patients had MYCN amplification. Four were INSS stage 1 and three were INSS stage 2b. Four of these patients relapsed 2 to 22 months from diagnosis and three died from progression of disease 8 to 25 months after relapse. For stage II patients, MYCN amplification, Shimada unfavorable, age older than 2 years, or positive lymph nodes predicted for a lower OS. However, because most patients were salvaged, multimodality therapy was recommended only for those with progressive disease.81 The POG reported that the presence of MYCN amplification in localized neuroblastoma did not necessarily portend an adverse outcome. Only 2% of stage A (three patients) and 7% of stage B (three patients) were MYCN amplified. Four of the six were without evidence of disease for 16 to 38 months or more. Two patients died of their disease; both had unfavorable histology (UH) and one had diploid DNA. The authors concluded that the prognostic significance of MYCN amplification may vary according to histologic features.83 A more recent study using the INRG database included 87 children with stage 1 or 2 neuroblastoma and tumor MYCN amplification. The 5-year EFS and OS were 53% and 72%, compared with 2573 stage 1 and 2 patients without MYCN amplification, whose 5-year EFS and OS were 90% and 98%, respectively.84
Patients with INSS stage 4S usually fall into the low-risk category with the exception of those patients with MYCN-amplified tumors, who are high-risk, or with either a UH or DI of 1, who are intermediate-risk. Although these patients have metastatic disease on presentation, spontaneous regression occurs frequently. The OS was 92% in a prospective CCG study of 80 patients, where cytotoxic therapy was only given to patients who were symptomatic.85 Other cooperative groups had similar results, with an OS of 85% to 88%, and worse outcome for the rare patients with tumors that had MYCN gene amplification, diploidy, and UH. Unfavorable clinical features were age less than 2 months, and patients who were symptomatic.30,86
Patients with 4S neuroblastoma who have unfavorable biologic factors have a poorer prognosis and are now considered intermediate- or high-risk. The CCG recently reported on outcome and prognostic factors in infants treated prospectively with supportive care only or, in symptomatic patients, with low-dose cytotoxic therapy (radiation, chemotherapy, or a combination of the two). The 5-year EFS and OS were 86% and 92%, respectively. Of these patients, 55% received supportive care only, and their 5-year OS was 100% compared with 81% survival in those patients who were symptomatic and required treatment. Infants younger than 2 months old at diagnosis with rapidly progressive abdominal disease accounted for five of the six deaths. Such infants may benefit from more intensive treatment.85,86
Although not yet standardized, recommended management for 4S disease begins with a diagnostic biopsy only, as resection of the primary tumor does not influence outcome.30,85,87 Patients with favorable biology are then observed for symptoms. Special attention is paid to infants younger than 2 months old. If respiratory compromise occurs, moderately intensive chemotherapy is used, with radiation therapy reserved for those who do not respond to chemotherapy. Lastly, patients with 4S disease with intraspinal involvement are managed with chemotherapy. Several retrospective studies and a French prospective study have shown that chemotherapy alone is a safe and effective therapy for intraspinal disease.67–70
Intermediate-Risk Neuroblastoma
Patients in this risk group have regionally advanced disease (stage 3 or stage 4 younger than 12-18 months of age) and are MYCN non-amplified. CCG-3881 was a prospective study of intermediate intensity chemotherapy for patients with Evans stage III disease and favorable biology at any age, or unfavorable biology and age less than 12 months; 92% of these patients fit the criteria for INSS stage 3, and 63% met the criteria for intermediate-risk disease. The patients were treated with chemotherapy including cyclophosphamide, doxorubicin, cisplatin, and etoposide followed by radiation for any gross residual disease following delayed surgery. The EFS rate at 4 years was 100% for patients with favorable biology regardless of age and 90% for those younger than 1 year of age with one unfavorable characteristic such as UH or MYCN amplification. The EFS for Evans stage III patients who were 1 year of age or older with unfavorable biology was 54%, despite more intensive therapy. In multivariate analysis, only age and MYCN status had independent prognostic value.88 These patients with the EFS of 54% and a portion of the patients with the EFS of 90% are thus appropriately now considered high-risk. Stage 4 patients less than 1 year of age were also treated in CCG-3881; before 1991, all stage 4 infants were treated in a similar fashion; after 1991, all infants whose tumors were MYCN-amplified were transferred to the more dose intensive high-risk protocol. The 3-year EFS and OS for all 134 infants were 63% and 71%, respectively. The MYCN-nonamplified infants (true intermediate risk) had 93% EFS, whereas the MYCN-amplified infants (high risk) had 10% EFS.89 These findings dramatically illustrate the prognostic significance of MYCN amplification status and support the need to stratify these infants into different risk groups.
The role of radiation therapy for intermediate-risk disease remains controversial; studies have provided conflicting results. The POG found the percent of patients achieving a complete response (CR), EFS, and OS were significantly higher in the group that received chemotherapy and radiation as compared to chemotherapy alone in patients with POG stage C neuroblastoma.90 However, the population was a mixture of intermediate- and high-risk patients, and the chemotherapy regimen was very modest in this study, consisting of only cyclophosphamide and doxorubicin. Conversely, a randomized controlled trial conducted by de Bernardi and colleagues demonstrated no difference in relapse rates among those patients who received radiation therapy for residual disease and those who did not.91
A recently completed phase III study of combination chemotherapy in 467 patients with intermediate-risk neuroblastoma showed that therapy could be markedly reduced from 9 months to 3 months in the majority of patients. All patients received carboplatin, cyclophosphamide, doxorubicin, and etoposide. Patients who did not achieve a CR after four cycles with favorable biology had surgery to remove or debulk residual disease. Patients with unfavorable biology or residual tumor after the first four cycles, received four additional cycles of chemotherapy and then undergo debulking surgery if they do not achieve a CR. The indications for radiation were (1) progressive clinical deterioration despite chemotherapy or surgery, or both; (2) stage 4s infants with respiratory compromise; (3) stage 3 or 4 patients with progressive neurologic decline from cord compression; (4) PR after eight cycles of chemotherapy and surgery in patients with unfavorable biology; and (5) PR after surgery for local recurrence greater than 3 months after completing protocol therapy. The radiation dose was 24 Gy in 1.5-Gy fractions; 4S patients with respiratory distress could receive 4.5 Gy to the liver in three fractions. The radiation volume included viable gross or microscopic residual disease determined by CT, MRI, or MIBG imaging with 2-cm margins.92
The current ongoing COG study (http://www.cancer.gov/clinicaltrials/COG-ANBL0531) further reduces cytotoxic therapy in a risk-based fashion, so that the most favorable patients in the intermediate group will receive only two cycles of chemotherapy. Toddlers age 12 to 18 months of age with stage 4 disease who have no tumor MYCN amplification or other unfavorable biologic features have now been shown to have an outcome similar to infant stage 4 who are less than 12 months, and therefore may be treated as intermediate risk.31,93
High-Risk Neuroblastoma
Survival rates for patients in this risk group have improved but remain unacceptably low despite aggressive treatment. The 4-year survival rate among the 507 stage 4 patients older than 1 year of age who were treated in the CCG studies from 1978 to 1985 was 9% compared with 30% among the 675 patients treated from 1991 to 1995.94 The improvement may be attributable to chemotherapy dose intensification or to the increasing use of high-dose myeloablative therapy with autologous or allogenic bone marrow transplantation. The CCG completed a randomized control trial that demonstrated intensive induction chemotherapy and radiation therapy followed by myeloablative chemotherapy, total body radiation, and purged autologous bone marrow transplantation (ABMT) is superior to conventional chemotherapy as consolidation therapy in children with high-risk neuroblastoma. The 3-year EFS rates were 34% and 22%, respectively. Furthermore, all patients who completed cytotoxic therapy without disease progression were then randomized to receive treatment with 13-cis-retinoic acid for 6 months or no further therapy. The 3-year EFS from time of second randomization for those patients who received 13-cis retinoic acid was 46%, as compared with 29% who received no further therapy.75 A long-term follow-up analysis continued to show that EFS for patients randomized to ABMT was significantly higher than those randomized to chemotherapy, with 5-year EFS of 30% versus 19% (P = 0.04). The 5-year EFS (42% versus 31% ) from time of second randomization was higher for cis-RA than for no further therapy, although not significant. OS at 5 years was significantly higher for each randomization by test of proportions (P < 0.01).95 Two other randomized trials in Europe compared myeloablative therapy with either no further treatment or with a low-dose oral maintenance chemotherapy, and both showed a significant advantage for the myeloablative chemotherapy.96,97 The most recently completed COG study (A3973), achieved similar EFS with less toxicity using a myeloablative chemotherapy regimen without total body radiation and using peripheral blood stem cells rather than bone marrow. This randomized trial showed the lack of advantage for EFS by purging the stem cells with immunomagnetic beads, compared with unpurged stem cells.98
Current treatment approaches include intensive induction chemotherapy, myeloablative consolidation chemotherapy with stem cell rescue, and targeted therapy for residual disease. The goal of induction therapy is to induce maximum reduction in tumor bulk. Most induction therapies currently employ combinations of four to six agents, including platinum compounds, cyclophosphamide or ifosfamide, anthracylines, vincristine, and epipodophyllotoxins. Some studies have shown that response rates are the greatest with high-dose intensities, specifically high doses of platinum compounds.99,100 The efficacy of induction therapy is typically assessed at the time of second-look surgery and is measured as a CR or a PR. Those who have a CR or very good PR (VGPR) have a better chance of cure, although some patients with a PR can be converted to a CR with high-dose myeloablative therapy.101 However, many of the recent attempts to improve response with dose-intensive induction have not improved the CR or VGPR rate using stringent restaging including 123I-MIBG scans to detect residual disease.102 The European Neuroblastoma Study Group recently performed a randomized control trial comparing two identical induction regimens that differ only in the rapidity of delivery. This directly tested the hypothesis that an increased dose intensity of induction chemotherapy improves response rate and OS twofold. This randomized trial of 262 patients with stage 4 neuroblastoma showed that increasing the dose intensity twofold resulted in improved 5-year EFS (18% versus 30%; P = 0.02), but not in OS.103 However, the overall lack of improvement in EFS in this group compared with other contemporary high-risk trials belies the apparent benefit.
The goal of subsequent therapy is to consolidate the response by eliminating any remaining tumor. This is usually accomplished with myeloablative chemotherapy with or without TBI and AHCT.2 Historically, autologous bone marrow was used as myeloablative rescue; however, using autologous peripheral blood stem cells is now a more common practice to reduce the risk of tumor cell contamination and to increase the rapidity of engraftment. EFS at 3 years ranges from 38% to 62% in the most recent POG and CCG trials.74,104–106 Investigations are underway to evaluate the efficacy of two myeloablative consolidations, known as “tandem transplants,” rather than one. George et al.107 reported the limited institution experience with tandem transplantation. Ninety seven high-risk patients were treated with five cycles of chemotherapy followed by two courses of myeloablative therapy and hematopoietic cell rescue. Progression-free survival at 3, 5, and 7 years for all patients was 55%, 47%, and 45% respectively. Although the majority (93%) underwent both courses of ablation, seven patients received only one course. The outcome appeared promising, so a large cooperative randomized study was undertaken by the COG to compare single versus double high-dose regimens, which opened in 2007.65
External-beam radiation therapy is generally used as consolidative treatment to initial sites of bulk disease, including the primary site and large or persistent metastatic deposits. Specific indications for radiation therapy vary for different protocols. Excellent LC rates have been achieved when delivering 21 Gy to the primary site and regional lymph nodes after resection.108–110 Bone metastases should be irradiated when they are persistent after chemotherapy or even after a CR if they were initially high volume (i.e., >3 cm). In the most recently completed COG study (A3973)64 and in the current, ongoing COG study (ANBL0532),65 radiation is given following myeloblative stem cell transplant to all areas of residual soft tissue disease. Volumes are defined by the CT immediately prior to surgical resection (the presurgical resection volume), regardless of extent and timing of the surgical resection or response to chemotherapy. If the primary tumor was grossly resected at diagnosis, the radiation therapy is given to the primary site based on the initial diagnostic tumor volume. In A3973, dose to both the primary site and metastatic sites was 21.6 Gy in 12 daily fractions. Because of the high rates of local recurrence, particularly following an incomplete resection, in ANBL0532 patients undergoing incomplete resections will receive the standard dose of 21.6 Gy to the postinduction chemotherapy, preoperative tumor volume followed by a boost of 14.4 Gy to the gross residual volume for a total dose of 36 Gy.
Targeted radiopharmaceuticals are also under investigation as an alternative means of delivering specific radiotherapy to metastatic tumor sites, either for refractory disease or as part of a myeloablative regimen. Targeted radioisotope therapy using anti-GD2 antibody or MIBG for delivery of radiation in the form of 131iodine has been tested extensively in clinical trials in relapsed neuroblastoma. Cheung et al.111 have reported on the use of 131I-3F8 (anti-GD2 antibody) for treatment of refractory neuroblastoma with documented responses. They have also treated newly diagnosed patients using 131I-3F8 in high doses followed by bone marrow rescue and further treatment with cold antibody after transplant.111
131I-MIBG, a norepinephrine analogue taken up by the norepinephrine transporter on neuroblastoma, has been widely shown in European and U.S. studies to elicit approximately a 30% to 40% response rate for refractory neuroblastoma.112–114 A phase 1 dose escalation trial of 131I-MIBG reported by Matthay et al.115 determined the maximal marrow nonablative dose (444 MBq/kg) and the maximal practical high dose with hematopoietic cell rescue (666 MBq/kg). In 30 patients, there was a 37% response rate and no significant toxicity other than hematologic. A follow-up phase II study of 164 patients who failed to achieve a partial or CR with prior therapy or who relapsed after high-dose therapy has since been completed.114 Use of 18 mCi/kg (666 MBq/kg) of 131I-MIBG supported by AHCT produced an overall response of 37% in this heavily pretreated cohort. A subsequqent study of two consecutive doses of 131I-MIBG therapy administered in rapid succession followed by HCT has recently been reported to show minimal toxicity and thus the ability to intensify 131I-MIBG dosing.95
The results in these relapsed patients argue strongly for the incorporation of 131I-MIBG therapy into regimens that include chemotherapeutic agents. Given the demonstration that haematopoiesis can be successfully restored after high-dose 131I-MIBG therapy, the combination of 131I-MIBG therapy with high-dose chemotherapy has been tested in several pilot studies, providing evidence of feasibility and engraftment with this approach.116 Subsequently, a phase I study incorporated escalating doses of 131I-MIBG followed by carboplatin, etoposide, and melphalan with hematopoietic stem cell infusions.117 The combination appeared feasible and effective at a dose of 12 mCi/kg of 131I-MIBG and chemotherapy doses minimally decreased from those feasible without the MIBG. Dose adjustments were required for patients with decreased renal function, associated with an increased risk of veno-occlusive disease. A phase II study is now underway in New Approaches to Neuroblastoma Therapy and a pilot study for newly diagnosed patients in the COG.
Several institutions have investigated the role of intraoperative radiation therapy (IORT) as an acceptable alternative to external-beam radiation as a way to deliver a high dose to the primary tumor site with minimal toxicity to normal structures.118–121 Gillis and colleagues from the University of California–San Francisco recently updated their data. Thirty-one patients with newly diagnosed high-risk neuroblastoma were treated with IORT as part of multimodality therapy. IORT to the primary site and associated lymph nodes achieved excellent LC at a median follow-up of 44 months. The 3-year estimate of the local recurrence rate was 15%, less than that of most previously published series. Only 1 of 22 patients who had undergone gross total resection developed recurrence at the primary tumor site. The 3-year estimate of LC, progression-free survival, and OS was 85%, 47%, and 60%, respectively. Side effects attributable to either the disease process or multimodality treatment were observed in seven patients who developed either hypertension or vascular stenosis. These late complications resulted in the death of two patients. The authors concluded that IORT at the time of primary resection achieved comparable control and survival rates while avoiding many side effects associated with external-beam radiotherapy in young children. Although complications were observed, additional analysis is needed to determine the relative contributions of the disease process and specific components of the multimodality treatment to these adverse events.122
After myeloablative therapy and local radiotherapy, microscopic residual disease frequently causes disease recurrence. The goal for using targeted therapy for residual disease is to eliminate any residual tumor cells that are chemoresistant. Although many patients are in a clinical and radiographic CR after consolidation, microscopic residual disease often remains, and thus many patients relapse. One agent that has proven effective is 13-cis-retinoic acid.75 Immunotherapy is being investigated as a means of targeting residual disease after induction and consolidation therapy in randomized trials in both the United States and Europe. Anti-GD2 antibodies, the murine monoclonal antibodies 3F8 and GD2a, and the human-mouse chimeric monoclonal antibody ch14.18 have shown the most promise.123,124
Recurrent Disease
Current approaches under investigation for treating recurrent or refractory neuroblastoma include novel cytotoxic agents, targeted delivery of radiopharmaceuticals (see previous discussion), retinoids, antiangiogenic agents, and other biologic agents’ genetic pathways. Investigation of novel cytotoxic agents has shown significant activity in phase I and II studies of the camptothecins, irinotecan125 and topotecan,126–128 and of temozolomide.80 Several studies have also been initiated or completed showing activity of these agents in combination therapy. Thus, topotecan with cyclophosphamide is now a recognized active combination in neuroblastoma that appears slightly superior to topotecan alone,129 and combinations of irinotecan and temozolomide have appeared promising in pilot studies and are currently under further investigation.76,130 Further enhancement of the temozolomide is being sought by adding O6BG, which may help to overcome resistance by inactivation of the DNA repair protein methylguanine-DNA methyltransferase.131 In addition to 13-cis-retinoic acid, new retinoids are being developed to treat recurrent or refractory disease. Fenretinide is one such compound that induces apoptosis even in cells resistant to 13-cis-retinoic acid.132,133 Phase I and II trials of oral fenretinide have been completed in the United States and Italy, and new studies of better absorbed formulations are underway.134–137 Lastly, as mentioned in the section on high-risk disease, immune-mediated therapy, including anti-GD2 antibodies, the murine monoclonal antibodies 3F8 and GD2a, and the human-mouse chimeric monoclonal antibody ch14.18 and a humanized version, Hu14.18, are currently under investigation for their role in recurrent or refractory disease.138–140 New immunocytokines with an anti-GD2 combined with IL2, which showed superior activity to naked antibody in preclinical studies, have completed phase I trials.141 Currently, agents targeting molecular pathways, such as Trk inhibitors, histone deacetylase inhibitors,142 antiangiogenic agents such as bevacizumab,143 and inhibitors of the IGF1R144 are under investigation either alone or as radiosensitizers, based on preclinical testing.
Sequelae of Radiation Therapy
As holds true for radiation in all pediatric malignancies, consideration for long-term complications is of vital importance. Long-term survivors of neuroblastoma often suffer from spinal deformities secondary to surgery and radiation. Among the most common are kyphosis and scoliosis.145 Factors that put the child at risk are (1) very young age, (2) orthovoltage irradiation, (3) asymmetric irradiation of the spine, (4) epidural spread of tumor, and (5) laminectomy (see earlier discussion). However, spinal deformities are also seen in patients who received megavoltage symmetric radiation. It is essential that these children are followed closely; early and proper bracing may help considerably.
The infant kidney is more sensitive to radiation than the adult or older child’s kidney. Abnormal creatine clearance has been reported in infants with stage IV-S disease who received 12.25 and 14 Gy. It has not been determined whether this is clinically significant.146
1 Gutierrez JC, Fischer AC, Sola JE, et al. Markedly improving survival of neuroblastoma: a 30-year analysis of 1,646 patients. Pediatr Surg Int. 2007;23:637.
2 Brodeur G, Maris J. Neuroblastoma. In: Pizzo P, Poplack D, editors. Principles and practice of pediatric oncology. ed 4. Philadelphia: Lippincott Williams & Wilkins Publishers; 2002:895.
3 Matthay KK, Haas-Kogan DH, Constine L. Neuroblastoma. In Pediatric Radiation oncology, ed 4, Philadelphia: Lippincott Williams & Wilkins; 2004:179-222.
4 Beckwith J, Perrin E. In situ neuroblastomas: A contribution to the natural history of neural crest tumors. Am J Pathol. 1963;43:1089.
5 Ikeda Y, Lister J, Bouton JM, et al. Congenital neuroblastoma, neuroblastoma in situ, and the normal fetal development of the adrenal. J Pediatr Surg. 1981;16:636.
6 Yamamoto K, Hayashi Y, Hanada R, et al. Mass screening and age-specific incidence of neuroblastoma in Saitama Prefecture, Japan. J Clin Oncol. 1995;13:2033.
7 Asami T, Otabe N, Watabayashi M, et al. Screening for neuroblastoma: A 9-year birth cohort-based study in Niigata, Japan. Acta Paediatr. 1995;84:1173.
8 Woods WG, Tuchman M, Robison LL, et al. A population-based study of the usefulness of screening for neuroblastoma. Lancet. 1996;348:1682.
9 Woods WG, Gao RN, Shuster JJ, et al. Screening of infants and mortality due to neuroblastoma. N Engl J Med. 2002;346:1041.
10 Schilling FH, Spix C, Berthold F, et al. Neuroblastoma screening at one year of age. N Engl J Med. 2002;346:1047.
11 Oue T, Inoue M, Yoneda A, et al. Profile of neuroblastoma detected by mass screening, resected after observation without treatment: results of the Wait and See pilot study. J Pediatr Surg. 2005;40:359.
12 De Roos AJ, Olshan AF, Teschke K, et al. Parental occupational exposures to chemicals and incidence of neuroblastoma in offspring. Am J Epidemiol. 2001;154:106.
13 De Roos AJ, Teschke K, Savitz DA, et al. Parental occupational exposures to electromagnetic fields and radiation and the incidence of neuroblastoma in offspring. Epidemiology. 2001;12:508.
14 Munzer C, Menegaux F, Lacour B, et al. Birth-related characteristics, congenital malformation, maternal reproductive history and neuroblastoma: the ESCALE study (SFCE). Int J Cancer. 2008;122:2315.
15 Kushner BH, Gilbert F, Helson L. Familial neuroblastoma: Case reports, literature review, and etiologic considerations. Cancer. 1986;57:1887.
16 Shojaei-Brosseau T, Chompret A, Abel A, et al. Genetic epidemiology of neuroblastoma: a study of 426 cases at the Institut Gustave-Roussy in France. Pediatr Blood Cancer. 2004;42:99.
17 Maris JM, Weiss MJ, Mosse Y, et al. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12-13. Cancer Res. 2002;62:6651.
18 Mosse YP, Laudenslager M, Khazi D, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004;75:727.
19 Trochet D, Bourdeaut F, Janoueix-Lerosey I, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74:761.
20 Raabe EH, Laudenslager M, Winter C, et al. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. 2008;27:469.
21 Biegel JA, White PS, Marshall HN, et al. Constitutional 1p36 deletion in a child with neuroblastoma. Am J Hum Genet. 1993;52:176.
22 Mosse Y, Greshock J, King A, et al. Identification and high-resolution mapping of a constitutional 11q deletion in an infant with multifocal neuroblastoma. Lancet Oncol. 2003;4:769.
23 Satge D, Moore SW, Stiller CA, et al. Abnormal constitutional karyotypes in patients with neuroblastoma: a report of four new cases and review of 47 others in the literature. Cancer Genet Cytogenet. 2003;147:89.
24 Maris JM, Mosse YP, Bradfield JP, et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N Engl J Med. 2008;358:2585.
25 Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930.
26 Fong CT, Dracopoli NC, White PS, et al. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: correlation with N-myc amplification. Proc Natl Acad Sci USA. 1989;86:3753.
27 Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264.
28 Brodeur GM, Seeger RC, Schwab M, et al. Amplification of N-myc in untreated neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121.
29 Weiss WA, Aldape K, Mohapatra G, et al. Targeted expression of MYCN causes neuroblastoma in transgenic mice. Embo J. 1997;16:2985.
30 Katzenstein HM, Bowman LC, Brodeur GM, et al. Prognostic significance of age, MYCN oncogene amplification, tumor cell ploidy, and histology in 110 infants with stage D S neuroblastoma: the Pediatric Oncology Group experience—a Pediatric Oncology Group study. J Clin Oncol. 1998;16:2007.
31 George RE, London WB, Cohn SL, et al. Hyperdiploidy plus nonamplified MYCN confers a favorable prognosis in children 12 to 18 months old with disseminated neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol. 2005;23:6466.
32 Shimada H, Ambros IM, Dehner LP, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999;86:364.
33 Sano H, Bonadio J, Gerbing RB, et al. International neuroblastoma pathology classification adds independent prognostic information beyond the prognostic contribution of age. Eur J Cancer. 2006;42:1113.
34 Delellis RA. The adrenal glands. In: Sternbert SS, editor. Diagnostic surgical pathology, vol 1. New York: Raven Press; 1989:445.
35 Cohn SL, Pearson ADJ, London WB, et al. The International Neuroblastoma Rsk Group (INRG) classification system. J Clin Oncol. 2009;27:289.
36 Qualman SJ, O’Dorisio MS, Fleshman DJ, et al. Neuroblastoma. Correlation of neuropeptide expression in tumor tissue with other prognostic factors. Cancer. 1992;70:2005.
37 Matthay KK, Blaes F, Hero B, et al. Opsoclonus myoclonus syndrome in neuroblastoma, a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. Cancer Lett. 2005;228:275.
38 McCluskey AG, Boyd M, Ross SC, et al. [131I]meta-iodobenzylguanidine and topotecan combination treatment of tumors expressing the noradrenaline transporter. Clin Cancer Res. 2005;11:7929.
39 Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466.
40 Hero B, Hunneman DH, Gahr M, et al. Evaluation of catecholamine metabolites, mIBG scan, and bone marrow cytology as response markers in stage 4 neuroblastoma. Med Pediatr Oncol. 2001;36:220.
41 Shulkin BL, Hutchinson RJ, Castle VP, et al. Neuroblastoma: positron emission tomography with 2-[fluorine-18]-fluoro-2-deoxy-D-glucose compared with metaiodobenzylguanidine scintigraphy. Radiology. 1996;199:743.
42 Kushner BH, Yeung HW, Larson SM, et al. Extending positron emission tomography scan utility to high-risk neuroblastoma: Fluorine-18 fluorodeoxyglucose positron emission tomography as sole imaging modality in follow-up of patients. J Clin Oncol. 2001;19:3397.
43 LaBrosse EH, Comoy E, Bohuan C, et al. Catecholamine metabolism in neuroblastoma. J Natl Cancer Inst. 1976;57:633.
44 Evans AE, D’Angio G, Randolph J. A proposed staging for children with neuroblastoma. Children’s Cancer Study Group A. Cancer. 1971;27:374.
45 Nitschke R, Smith EI, Shochat S, et al. Localized neuroblastoma treated by surgery. A Pediatric Oncology Group Study. J Clin Oncol. 1988;6:1271.
46 Hayes FA, Green A, Hustu HO, et al. Surgicopathologic staging of neuroblastoma: prognostic significance of regional lymph node metastases. J Pediatr. 1983;102:59.
47 Cecchetto G, Mosseri V, De Bernardi B, et al. Surgical risk factors in primary surgery for localized neuroblastoma: The LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. J Clin Oncol. 2005;23:8483.
48 Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system. J Clin Oncol. 2009;27:298.
49 Matthay KK, Edeline V, Lumbroso J, et al. Correlation of early metastatic response by 123I-metaiodobenzylguanidine scintigraphy with overall response and event-free survival in stage IV neuroblastoma. J Clin Oncol. 2003;21:2486.
50 Messina JA, Cheng SC, Franc BL, et al. Evaluation of semi-quantitative scoring system for metaiodobenzylguanidine (mIBG) scans in patients with relapsed neuroblastoma. Pediatr Blood Cancer. 2006;47:865.
51 Seeger RC, Reynolds CP, Gallego R, et al. Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children’s Cancer Group Study. J Clin Oncol. 2000;18:4067.
52 Beiske K, Ambros PF, Burchill SA, et al. Detecting minimal residual disease in neuroblastoma patients—the present state of the art. Cancer Lett. 2005;228:229.
53 London WB, Castleberry RP, Matthay KK, et al. Evidence for an age cut-off greater than 365 days for Neuroblastoma Risk Group Stratification in the Children’s Oncology Group (COG). J Clin Oncol. 2005;23:6459.
54 Cecchetto G, Mosseri V, De Bernardi B, et al. Surgical risk factors in primary surgery for localized neuroblastoma: the LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. J Clin Oncol. 2005;23:8483.
55 Shamberger RC, Allarde-Segundo A, Kozakewich HP, et al. Surgical management of stage III and IV neuroblastoma: resection before or after chemotherapy? J Pediatr Surg. 1991;26:1113. discussion 1117
56 Von Allmen D, Grupp S, Diller L, et al. Aggressive surgical therapy and radiotherapy for patients with high-risk neuroblastoma treated with rapid sequence tandem transplant. J Pediatr Surg. 2005;40:936. discussion 941
57 La Quaglia MP, Kushner BH, Su W, et al. The impact of gross total resection on local control and survival in high-risk neuroblastoma. J Pediatr Surg. 2004;39:412. discussion 412
58 Wheldon TE, O’Donoghue JA, Gregor A. Radiobiological rationale for hyperfractionation in the radiotherapy of neuroblastoma. Int J Radiat Oncol Biol Phys. 1987;13:1430.
59 Livingstone A, Maris RJ, Russell J, et al. N-myc gene copy number in neuroblastoma cell lines and resistance to experimental treatment. Eur J Cancer. 1994;30A:382.
60 Paulino AC, Mayr NA, Simon JH, et al. Locoregional control in infants with neuroblastoma: Role of radiation therapy and late toxicity. Int J Radiat Oncol Biol Phys. 2002;52:1025.
61 Halperin EC. Long-term results of therapy for stage C neuroblastoma. J Surg Oncol. 1996;63:172.
62 Wolden SL, Barker CA, Kushner BH, et al. Brain-sparing radiotherapy for neuroblastoma skull metastases. Pediatr Blood Cancer. 2008;50:1163.
63 Jacobson GM, Sause WT, O’Brien RT. Dose response analysis of pediatric neuroblastoma to megavoltage radiation. Am J Clin Oncol. 1984;7:693.
64 Kreissman SG, Villablanca JG, Seeger RC, et al. A randomized Phase 3 trial of myeloablative autologous peripheral blood stem cell (PBSC) transplant (ASCT) for high-risk neuroblastoma (HR-NB) employing immunomagnetic purged (P) versus unpurged (UP) PBSC: a Children’s Oncology Group study. Proc ASCO. 2008;26:541s. [abstract]
65 COG study (ANBL0532) Phase III Randomized Trial of Single vs. Tandem Myeloblative Consolidation Therapy for High-Risk Neuroblastoma.
66 McWilliams NB. Neuroblastoma in infancy. In: Pochedly C, editor. Neuroblastoma: Tumor biology and therapy. Boca Raton, Fla: CRC Press; 1990:229.
67 Hayes FA, Green AA, O’Connor DM. Chemotherapeutic management of epidural neuroblastoma. Med Pediatr Oncol. 1989;17:6.
68 Hoover M, Bowman LC, Crawford SE, et al. Long-term outcome of patients with intraspinal neuroblastoma. Med Pediatr Oncol. 1999;32:353.
69 Plantaz D, Rubie H, Michon J, et al. The treatment of neuroblastoma with intraspinal extension with chemotherapy followed by surgical removal of residual disease. A prospective study of 42 patients—results of the NBL 90 Study of the French Society of Pediatric Oncology. Cancer. 1996;78:311.
70 De Barnardi B, Pianca C, Pistamiglio P, et al. Neuroblastoma with symptomatic spinal cord compression at diagnosis: treatment and results with 76 cases. J Clin Oncol. 2001;19:183.
71 August CS, Serota FT, Koch PA, et al. Treatment of advanced neuroblastoma with supralethal chemotherapy, radiation, and allogeneic autologous marrow reconstitution. J Clin Oncol. 1984;2:609.
72 Croog VJ, Kramer K, Kushner BH, et al. Whole neuraxis irradiation to address central nervous system relapse in high-risk neuroblastoma. Int J Radiat Oncol Biol Phys. 2009. in press
73 Castleberry RP, Cantor AB, Green AA, et al. Phase II investigational window using carboplatin, iproplatin, ifosfamide, and epirubicin in children with untreated disseminated neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol. 1994;12:1616.
74 Kretschmar CS, Kletzel K, et al. Phase II therapy with taxol and topotecan in untreated children (>365 days) with disseminated (INSS stage 4) neuroblastoma (NB). A POG study. Med Pediatr Oncol. 1995;24:243.
75 Matthay KK, Villablanca JG, Seeger RC, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341:1165.
76 Kushner BH, Kramer K, Meyers PA, et al. Pilot study of topotecan and high-dose cyclophosphamide for resistant pediatric solid tumors. Med Pediatr Oncol. 2000;35:468.
77 Pearson AD, Pinkerton CR, Lewis IJ, et al. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: A randomised trial. Lancet Oncol. 2008;9:247.
78 Wagner LM, Crews KR, Iacono LC, et al. Phase I trial of temozolomide and protracted irinotecan in pediatric patients with refractory solid tumors. Clin Cancer Res. 2004;10:840.
79 Kushner BH, Kramer K, Modak S, et al. Irinotecan plus temozolomide for relapsed or refractory neuroblastoma. J Clin Oncol. 2006;24:5271.
80 Rubie H, Chisholm J, Defachelles AS, et al. Phase II study of temozolomide in relapsed or refractory high-risk neuroblastoma: a joint Societe Francaise des Cancers de l’Enfant and United Kingdom Children Cancer Study Group—New Agents Group Study. J Clin Oncol. 2006;24:5259.
81 Perez CA, Matthay KK, Atkinson JB, et al. Biologic variables in the outcome of stages I and II neuroblastoma treated with surgery as primary therapy: a children’s cancer study group study. J Clin Oncol. 2000;18:1260.
82 Strother D, London WB, Yap J, et al. Surgery and restricted use of chemotherapy as treatment of low-risk neuroblastoma: Preliminary results of Children’s Oncology Group Protocol 9641. Pediatr Blood Cancer. 2006;47:384.
83 Cohn SL, Look AT, Joshi VV, et al. Lack of correlation of N-myc gene amplification with prognosis in localized neuroblastoma: a Pediatric Oncology Group study. Cancer Res. 1995;55:721.
84 Bagatell R, Beck-Popovic M, London WB, et al. Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol. 2009;27:365.
85 Nickerson HJ, Matthay KK, Seeger RC, et al. Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children’s cancer group study. J Clin Oncol. 2000;18:477.
86 Schleiermacher G, Rubie H, Hartmann O, et al. Treatment of stage 4s neuroblastoma—Report of 10 years’ experience of the French Society of Paediatric Oncology (SFOP). Br J Cancer. 2003;89:470.
87 Guglielmi M, de Bernardi B, Rizzo A. Resection of primary tumor at diagnosis in stage IV-S neuroblastoma: does it affect the clinical course? J Clin Oncol. 1996;14:1537.
88 Matthay KK, Perez C, Seeger RC, et al. Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children’s cancer group study. J Clin Oncol. 1998;16:1256.
89 Schmidt ML, Lukens JN, Seeger RC, et al. Biologic factors determine prognosis in infants with stage IV neuroblastoma: a prospective Children’s Cancer Group study. J Clin Oncol. 2000;18:1260.
90 Castleberry RP, Kun LE, Shuster JJ, et al. Radiotherapy improves the outlook for patients older than 1 year with Pediatric Oncology Group stage C neuroblastoma. J Clin Oncol. 1991;9:789.
91 de Bernardi B, Rogers D, Carli M, et al. Localized neuroblastoma. Surgical and pathologic staging. Cancer. 1987;60:1066.
92 Baker DL, Schmidt M, Cohn S, et al. A Phase III trial of biologically-based therapy reduction for intermediate risk neuroblastoma. Proc ASCO. 2007;25:527s.
93 Schmidt ML, Lal A, Seeger RC, et al. Favorable prognosis for patients ages 12-18 months with stage 4 MYCN-non-amplified neuroblastoma: a Children’s Cancer Group study. J Clin Oncol. 2005;23:6474.
94 Matthay KK. Neuroblastoma: biology and therapy. Oncology. 1997;11:1857. discussion 1869, 1875
95 Matthay KK, Reynolds CP, Shimada H, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a Children’s Oncology Group study. J Clin Oncol. 2009;27:1007.
96 Berthold F, Boos J, Burdach S, et al. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol. 2005;6:649.
97 Pritchard J, Cotterill SJ, Germond SM, et al. High dose melphalan in the treatment of advanced neuroblastoma: results of a randomised trial (ENSG-1) by the European Neuroblastoma Study Group. Pediatr Blood Cancer. 2005;44:348.
98 Kreissman SG, Villablanca JG, Seeger RC, et al. A randomized Phase 3 trial of myeloablative autologous peripheral blood stem cell (PBSC) transplant (ASCT) for high-risk neuroblastoma (HR-NB) employing immunomagnetic purged (P) versus unpurged (UP) PBSC: a Children’s Oncology Group study. Proc ASCO. 2008;26:541s.
99 Cheung NV, Heller G. Chemotherapy dose intensity correlates strongly with response, median survival, and median progression-free survival in metastatic neuroblastoma. J Clin Oncol. 1991;9:1050.
100 Castleberry RP, Cantor AB, Green AA, et al. Phase II investigational window using carboplatin, iproplatin, ifosfamide, and epirubicin in children with untreated disseminated neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol. 1994;12:1616.
101 Matthay KK, Castleberry RP. Treatment of advanced neuroblastoma: the U.S. experience. In: Brodeur GM, Sawada T, Tsuchida Y, et al, editors. Neuroblastoma. Amsterdam: Elsevier Science; 2000:437.
102 Valteau-Couanet D, Michon J, Boneu A, et al. Results of induction chemotherapy in children older than 1 year with a stage 4 neuroblastoma treated with the NB 97 French Society of Pediatric Oncology (SFOP) protocol. J Clin Oncol. 2005;23:532.
103 Pearson AD, Pinkerton CR, Lewis IJ, et al. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: a randomised trial. Lancet Oncol. 2008;9:247.
104 Pole JG, Casper J, Elfenbein G, et al. High-dose chemotherapy supported by marrow infusions for advanced neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol. 1991;9:152.
105 Stram DO, Matthay KK, O’Leary M, et al. Consolidation chemoradiotherapy and autologous bone marrow transplantation versus continued chemotherapy for metastatic neuroblastoma: a report of two concurrent Children’s Cancer Group studies. J Clin Oncol. 1996;14:2417.
106 Villablanca JG, Matthay KK, Swift PS, et al. Phase I trial of carboplatin, etoposide, melphalan and local irradiation (CEM-LI) with purged autologous bone marrow transplantation for children with high-risk neuroblastoma. Med Pediatr Oncol. 1999;33:170.
107 George RE, Li S, Madeiros-Nancarrow C, et al. High-risk neuroblastoma treated with tandem autologous peripheral-blood stem cell-supported transplantation: long-term survival update. J Clin Oncol. 2006;24:2891.
108 Haas-Kogan DA, Swift PS, Selch M, et al. Impact of radiotherapy for high-risk neuroblastoma: a Children’s Cancer Group study. Int J Radiat Oncol Biol Phys. 2003;56:28.
109 Wolden SL, Gollamudi SV, Kushner BH, et al. Local control with multi-modality therapy for stage 4 neuroblastoma. Int J Radiat Oncol Biol Phys. 2000;46:969.
110 Kushner BH, Wolden S, La Quaglia MP, et al. Hyperfractionated low-dose radiotherapy for high-risk neuroblastoma after intensive chemotherapy and surgery. J Clin Oncol. 2001;19:2821.
111 Cheung NK, Kushner BH, LaQuaglia M, et al. N7: A novel multi-modality therapy of high risk neuroblastoma (NB) in children diagnosed over 1 year of age. Med Pediatr Oncol. 2001;36:227.
112 Klingebiel T, Berthold F, Treuner J, et al. Metaiodobenzylguanidine (mIBG) in treatment of 47 patients with neuroblastoma: results of the German Neuroblastoma Trial. Med Pediatr Oncol. 1991;19:84.
113 Lashford LS, Lewis IJ, Fielding SL, et al. Phase I/II study of iodine 131 metaiodobenzylguanidine in chemoresistant neuroblastoma: A United Kingdom Children’s Cancer Study Group investigation. J Clin Oncol. 1992;10:1889.
114 Matthay KK, Yanik G, Messina J, et al. Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. J Clin Oncol. 2007;25:1054.
115 Matthay KK, DeSantes K, Hasegawa B, et al. Phase I dose escalation of 131I-metaiodobenzylguanidine with autologous bone marrow support in refractory neuroblastoma. J Clin Oncol. 1998;16:229.
116 Yanik GA, Levine JE, Matthay KK, et al. Pilot study of iodine-131-metaiodobenzylguanidine in combination with myeloablative chemotherapy and autologous stem-cell support for the treatment of neuroblastoma. J Clin Oncol. 2002;20:2142.
117 Matthay KK, Tan JC, Villablanca JG, et al. Phase I dose escalation of iodine-131-metaiodobenzylguanidine with myeloablative chemotherapy and autologous stem-cell transplantation in refractory neuroblastoma: a new approach to Neuroblastoma Therapy Consortium Study. J Clin Oncol. 2006;24:50.
118 Haase GM, Meagher DPJr, McNeely LK, et al. Electron beam intraoperative radiation therapy for pediatric neoplasms. Cancer. 1994;74:740.
119 Leavy PJ, Odom LF, Poole M, et al. Intra-operative radiation therapy in pediatric neuroblastoma. Med Pediatr Oncol. 1997;28:424.
120 Aitken DR, Hopkins GA, Archambeau JO, et al. Intraoperative radiation therapy in the treatment of neuroblastoma: report of a pilot study. Ann Surg Oncol. 1995;2:343.
121 Goodman KA, Wolden SL, LaQuaglia MP, et al. Intraoperative high-dose-rate brachytherapy for pediatric solid tumors: a 10-year experience. Brachytherapy. 2003;2:139.
122 Gillis AM, Sutton E, Dewitt KD, et al. Long-term outcome and toxicities of intraoperative radiotherapy for high-risk neuroblastoma. Int J Radiat Oncol Biol Phys. 2007;69:858.
123 Kushner BH, Kramer K, Cheung NK, et al. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J Clin Oncol. 2001;19:4189.
124 Yu AL, Uttenreuther-Fischer MM, Huang CS, et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol. 1998;16:2169.
125 Furman WL, Stewart CF, Poquette CA, et al. Direct translation of a protracted irinotecan schedule from a xenograft model to a phase I trial in children. J Clin Oncol. 1999;17:1815.
126 Pratt CB, Stuart C, Santana VM, et al. Phase I study of topotecan for pediatric patients with malignant solid tumors. J Clin Oncol. 1994;12:539.
127 Kretschmar CS, Kletzel M, Murray K, et al. Phase II therapy with Taxol and topotecan in untreated children >365 days with disseminated neuroblastoma. A POG study. Med Pediatr Oncol. 1995;24:243.
128 Nitschke R, Parkhurst J, Sullivan J, et al. Topotecan in pediatric patients with recurrent and progressive solid tumors: a Pediatric Oncology Group phase II study. J Pediatr Hematol Oncol. 1998;20:315.
129 Saylors RLIII, Stewart CF, Zamboni WC, et al. Phase I study of topotecan in combination with cyclophosphamide in pediatric patients with malignant solid tumors: A Pediatric Oncology Group Study. J Clin Oncol. 1998;16:945.
130 Wagner LM, Crews KR, Iacono LC, et al. Phase I trial of temozolomide and protracted irinotecan in pediatric patients with refractory solid tumors. Clin Cancer Res. 2004;10:840.
131 Wagner LM, McLendon RE, Yoon KJ, et al. Targeting methylguanine-DNA methyltransferase in the treatment of neuroblastoma. Clin Cancer Res. 2007;13(18 Pt 1):5418.
132 Mariotti A, Marcora E, Bunone G, et al. N-(4-hydroxyphenyl)-retinamide: a potent inducer of apoptosis in human neuroblastoma cells. J Natl Cancer Inst. 1994;86:1245.
133 Maurer BJ, Metelitsa LS, Seeger RC, et al. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxy phenyl)-retinamide in neuroblastoma cell lines. J Natl Cancer Inst. 1999;91:1138.
134 Garaventa A, Luksch R, Lo Piccolo MS, et al. Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin Cancer Res. 2003;9:2032.
135 Children’s Oncology Group (CCG 09709)Villablanca JG, Krailo MD, et al. Phase I trial of oral fenretinide in children with high-risk solid tumors: A report from the Children’s Oncology Group (CCG 09709). J Clin Oncol. 2006;24:3423.
136 Maurer BJ, Kalous O, Yesair DW, et al. Improved oral delivery of N-(4-hydroxyphenyl)retinamide with a novel LYM-X-SORB organized lipid complex. Clin Cancer Res. 2007;13:3079.
137 Orienti I, Zuccari G, Bergamante V, et al. Fenretinide-polyvinylalcohol conjugates: New systems allowing fenretinide intravenous administration. Biomacromolecules. 2007;8:3258.
138 Ozkaynak MF, Sondel PM, Krailo MD, et al. Phase I study of chimeric human/murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: a Children’s Cancer Group Study. J Clin Oncol. 2000;18:4077.
139 Yu AL, Batova A, Alvarada C, et al. Usefulness of a chimeric anti-GD2 (ch14.18) and GM-CSF for refractory neuroblastoma: a POG phase II study. Proc Am Soc Clin Oncol. 1997;16:513a.
140 Cheung NK, Kushner BH, Yeh SDJ, et al. 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: A phase II study. Int J Oncol. 1998;12:1299.
141 Osenga KL, Hank JA, Albertini MR, et al. a phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the Children’s Oncology Group. Clin Cancer Res. 2006;12:1750.
142 Keshelava N, Davicioni E, Wan Z, et al. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst. 2007;99:1107.
143 Dickson PV, Hamner JB, Sims TL, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13:3942.
144 Meyer GE, Chesler L, Liu D, et al. Nordihydroguaiaretic acid inhibits insulin-like growth factor signaling, growth, and survival in human neuroblastoma cells. J Cell Biochem. 2007;102:1529.
145 Mayfield JK, Riseborough EJ, Jaffe N, et al. Spinal deformity in children treated for neuroblastoma. J Bone Joint Surg [Am]. 1981;63a:183.
146 Peschel RE, Chen M, Seashore J. The treatment of massive hepatomegaly in stage IV-S neuroblastoma. Int J Radiat Oncol Biol Phys. 1981;7:549.