Chapter 492 Neuroblastoma
Pathogenesis
The etiology of neuroblastoma in most cases remains unknown. Familial neuroblastoma accounts for 1-2% of all cases, is associated with a younger age at diagnosis, and has been linked to mutations in the Phox2B and ALK genes. Neuroblastoma is associated with other neural crest disorders, including Hirschsprung disease, central hypoventilation syndrome, and neurofibromatosis type I, and potentially congenital cardiovascular malformations (Table 492-1). Children with Beckwith-Wiedemann syndrome and hemihypertrophy also have a higher incidence of neuroblastoma. Increased incidence of neuroblastoma is associated with some maternal and paternal occupational chemical exposures, farming, and work related to electronics, although no single environmental exposure has been shown to cause neuroblastomas.
| EPONYM | FEATURES |
|---|---|
| Pepper syndrome | Massive involvement of the liver with metastatic disease with or without respiratory distress. |
| Horner syndrome | Unilateral ptosis, myosis, and anhidrosis associated with a thoracic or cervical primary tumor. Symptoms do not resolve with tumor resection. |
| Hutchinson syndrome | Limping and irritability in young child associated with bone and bone marrow metastases. |
| Opsoclonus-myoclonus-ataxia syndrome | Myoclonic jerking and random eye movement with or without cerebellar ataxia. Often associated with a biologically favorable and differentiated tumor. The condition is likely immune mediated, may not resolve with tumor removal, and often exhibits progressive neuropsychological sequelae. |
| Kerner-Morrison syndrome | Intractable secretory diarrhea due to tumor secretion of vasointestinal peptides. Tumors are generally biologically favorable. |
| Neurocristopathy syndrome | Neuroblastoma associated with other neural crest disorders, including congenital hypoventilation syndrome or Hirschsprung disease. Germline mutations in the paired homeobox gene PHOX2B have been identified in a subset of patients with this disease. |
From Park JR, Eggert A, Caron H: Neuroblastoma: biology, prognosis, and treatment, Pediatr Clin North Am 55:97-120, 2008.
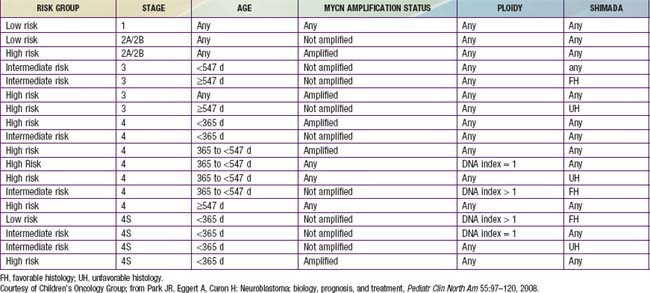
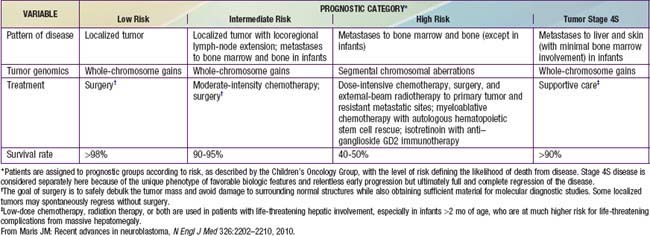
Genetic characteristics of neuroblastoma tumor tissue that are of prognostic importance include amplification of the MYCN (N-myc) proto-oncogene and tumor cell DNA content, or ploidy (Tables 492-2 to 492-4). Amplification of MYCN is strongly associated with advanced tumor stage and poor outcomes. Hyperdiploidy confers better prognosis if the child is <1 yr of age at diagnosis. Other chromosomal abnormalities, including loss of heterozygosity (LOH) of 1p, 11q, and 14q, and gain of 17q, are commonly found in neuroblastoma tumors and are associated with worse outcomes. In addition, many other biologic factors have been shown to be associated with neuroblastoma outcomes, including tumor histology and vascularity and the expression levels of nerve growth factor receptor (TrkA, TrkB), ferritin, lactate dehydrogenase, ganglioside GD2, neuropeptide Y, chromogranin A, CD44, multidrug resistance–associated protein, and telomerase. These factors and many others are under investigation in clinical trials to determine whether they can be used to reduce therapy for children predicted to fare well with minimal therapy and to intensify therapy for those predicted to be at high risk for relapse.
Clinical Manifestations
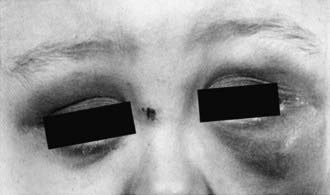
Neuroblastoma can mimic many other disorders and may be difficult to diagnose. The signs and symptoms of neuroblastoma reflect the tumor site and extent of disease. Metastatic disease can cause a variety of signs and symptoms, including fever, irritability, failure to thrive, bone pain, cytopenias, bluish subcutaneous nodules, orbital proptosis, and periorbital ecchymoses (Fig. 492-1). Localized disease can manifest as an asymptomatic mass or as mass-related symptoms, including spinal cord compression, bowel obstruction, and superior vena cava syndrome.
Diagnosis
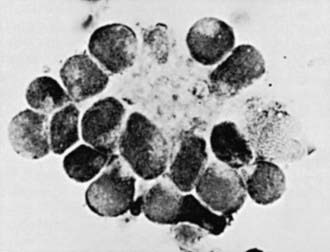
Neuroblastoma is usually discovered as a mass or multiple masses on plain radiography, CT, or MRI (Fig. 492-2). On plain radiography or CT, the mass often contains calcification and hemorrhage. Prenatal diagnosis of neuroblastoma on maternal ultrasound scans is sometimes possible. Tumor markers, including catecholamine metabolites homovanillic acid (HVA) and vanillylmandelic acid (VMA) in urine, are elevated in 95% of cases and help to confirm the diagnosis. A pathologic diagnosis is established from tumor tissue obtained by biopsy. Neuroblastoma can be diagnosed without a primary tumor biopsy if small round blue tumor cells are observed in bone marrow samples (Fig. 492-3) and an elevation of VMA or HVA is found in the urine.
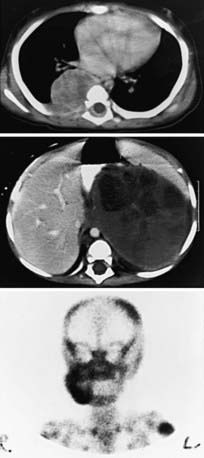
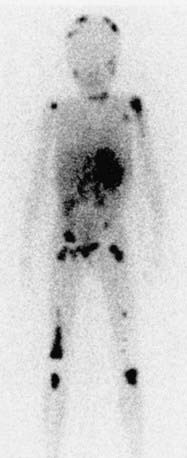
Evaluations for metastatic disease should include CT or MRI of the chest and abdomen, bone scans to detect cortical bone involvement, and at least two independent bone marrow aspirations and biopsies to evaluate for marrow disease. Iodine-123 meta-iodobenzylguanidine (123I-MIBG) studies may also be used to better define the extent of disease (Fig. 492-4). MRI of the spine should be performed in cases with suspected or potential spinal cord compression, but imaging of the brain with either CT or MRI is not routinely performed unless dictated by the clinical presentation.
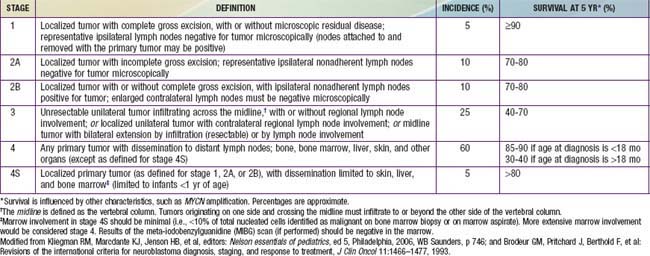
The International Neuroblastoma Staging System (INSS) now is currently used to stage patients with neuroblastoma after initial surgical resection (see Table 492-3). INSS stage 1 tumors are confined to the organ or structure of origin and are completely resected. INSS stage 2 tumors extend beyond the structure of origin but not across the midline, either with (stage 2B) or without (stage 2A) ipsilateral lymph node involvement. INSS stage 3 tumors extend beyond the midline, with or without bilateral lymph node involvement, whereas INSS stage 4 tumors are disseminated, with metastases to bones, bone marrow, liver, distant lymph nodes, and other organs. INSS stage 4S refers to neuroblastoma in children less <1 yr of age with dissemination to liver, skin, and/or bone marrow without bone involvement and with a primary tumor that would otherwise be staged as INSS stage 1 or 2. A new International Neuroblastoma Risk Group Staging System (INRGSS) is currently being developed to allow for more effective comparisons of treatments and outcomes worldwide.
Treatment
Treatment strategies for neuroblastoma have changed dramatically over the past 20 years, with significant reduction in treatment intensity for children who have localized, low-risk tumors and continued increased treatment intensity and addition of new agents for treatment of children who have high-risk neuroblastoma. Currently the patient’s age and tumor stage are combined with cytogenetic and molecular features of the tumor to determine the treatment risk group and estimated prognosis for each patient (see Tables 492-2 to 492-4). The usual treatment for low-risk neuroblastoma is surgery for stages 1 and 2 and observation for stage 4S with cure rates generally >90% without further therapy. Treatment with chemotherapy or radiation for the rare child with local recurrence can still be curative. Children with spinal cord compression at diagnosis also may require urgent treatment with chemotherapy, surgery, or irradiation to avoid neurologic damage. Stage 4S neuroblastomas have a very favorable prognosis, because many regress spontaneously. Chemotherapy or resection of the primary tumor does not improve survival rates, but for infants with massive liver involvement and respiratory compromise, small doses of cyclophosphamide or low-dose hepatic irradiation may alleviate symptoms. For children with stage 4S neuroblastoma who require treatment for symptoms, the survival rate is 81%.
Attiyeh EF, London WB, Mosse YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353:2243-2253.
Baker DL, Schmidt ML, Cohn SL, et al. Outcome after reduced chemotherapy for immediate-risk neuroblastoma. N Engl J Med. 2010;363(14):1313-1322.
Capasso M, Devoto M, Hou C, et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat Genet. 2009;41:718-723.
Cohn SL, Pearson AD, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG task force report. J Clin Oncol. 2009;27:289-297.
George RE, Lipshultz SE, Lipsitz SR, et al. Association between congenital cardiovascular malformations and neuroblastoma. J Pediatr. 2004;144:444-448.
George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975-978.
Kushner BH, Cheung NKV. Neuroblastoma—linking a common allele to a rare disease. N Engl J Med. 2008;358:2635-2637.
London WB, Bone L, Simon T, et al. The role of age in neuroblastoma risk stratification: the German, Italian, and Children’s Oncology Group perspectives. Canc Lett. 2005;228:257-266.
Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202-2210.
Maris JM, Hogarty MD, Bagatell R, et al. Neuroblastoma. Lancet. 2007;369:2106-2120.
Maris JM, Mosse YP, Bradfield JP, et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N Engl J Med. 2008;358:2585-2593.
Matthay KK, Reynolds CP, Seeger RC, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a Children’s Oncology Group study. J Clin Oncol. 2009;27:1007-1013.
Mosse YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930-936.
Mullassery D, Dominici C, Jesudason EC, et al. Neuroblastoma: contemporary management. Arch Dis Child Educ Pract Ed. 2009;94:177-185.
Nickerson HJ, Matthay KK, Seeger RC, et al. Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children’s Cancer Group study. J Clin Oncol. 2000;18:477-486.
Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am. 2008;55:97-120.
Rudnick E, Khakoo Y, Antunes NL, et al. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies: a report from the Children’s Cancer Group study. Med Pediatr Oncol. 2001;36:612-622.
Swarbrick A, Woods SL, Shaw A, et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat Med. 2010;16(10):1134-1140.
Yu AL, Gilman AL, Ozkaynak F, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363(14):1324-1334.
Zage PE, Kletzel M, Murray K, et al. Outcomes of the POG 9340/9341/9342 trials for children with high-risk neuroblastoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2008;51:747-753.





