[level-membership-for-neurology-category]
Chapter 37 Neuro-otology
Diagnosis and Management of Neuro-otological Disorders
Epidemiology of Vertigo, Dizziness, and Hearing Loss
Specific Disorders Causing Vertigo
Familial Hearing Loss and Vertigo
Common Causes of Nonspecific Dizziness
Common Presentations of Vertigo
Specific Disorders Causing Hearing Loss
Common Presentations of Hearing Loss
Laboratory Investigations in Diagnosis and Management
Historical Background
Accounts of dizziness and vertigo can be found in the writings of ancient Egyptian and Greek physicians. However, prior to the late 19th century, not much was known about the causes of dizziness or hearing loss, and as a result quackery was commonplace. Patients complaining of dizziness or vertigo were usually grouped together with epileptic seizures and stroke under the rubric of “apoplectiform cerebral congestion,” meaning too much blood to the brain. As a result, common treatments included bleeding, leeching, cupping, and purging. In 1861, Prosper Meniere was the first to recognize the association of vertigo with hearing loss and thus to localize the symptom to the inner ear (Baloh, 2001). Although not well received initially, his discovery provided the basis for later studies on the physiology and pathology of the vestibular system.
The advent of modern neuroimaging in the late 1970s and 1980s greatly expanded our understanding of causes of dizziness and vertigo. Prior to this time, stroke was considered an exceedingly rare cause of vertigo (Fisher, 1967). Though it remains a controversial topic even today, infarctions within the cerebellum and brainstem have been identified on imaging studies in patients with isolated vertigo. Imaging studies continue to lead to new discoveries of causes of vertigo, as demonstrated by the recently described disorder of superior canal dehiscence (SCD). But the most common causes of vertigo—Meniere disease, BPPV, and vestibular neuritis—still have no identifiable imaging characteristics.
Over the last 25 years, our understanding of the mechanisms for the common neuro-otological disorders has been greatly enhanced. BPPV can now be readily identified and cured at the bedside with a simple positional maneuver, and variants have also been described (Aw et al., 2005; Fife et al., 2008). The head-thrust test can be used at the bedside to identify a vestibular nerve lesion, and because of this it has particular utility in helping distinguish vestibular neuritis from a posterior fossa stroke (Halmagyi and Curthoys, 1988; Kattah et al., 2009; Newman-Toker et al., 2008; Nuti et al., 2005). Controversies regarding Meniere disease have been clarified, and medical and surgical treatments have improved (Minor et al., 2004). It is now clear that patients with recurrent episodes of vertigo without hearing loss, a condition once called vestibular Meniere disease, do not actually have Meniere disease.
Migraine is now recognized as an important cause of dizziness, even in patients without simultaneous headaches. In fact, benign recurrent vertigo (patients with recurrent episodes of vertigo without accompanying auditory symptoms or other neurological features) is usually a migraine equivalent (Oh et al., 2001b). The disorder of SCD was only recently described and provides important insight into the physiology of the vestibular system (Minor, 2005). A more detailed description of the rotational vertebral artery syndrome has led to appreciation of the high metabolic demands of the inner ear and its susceptibility to ischemia (Choi et al., 2005). Genetic research has identified ion channel dysfunction in disorders such as episodic ataxia and familial hemiplegic migraine, and patients with these disorders also commonly report vertigo (Jen et al., 2004a). It is hoped that identifying specific genes causing vertigo syndromes will lead to a better understanding of the mechanisms and also create the opportunity to develop specific treatments in the future.
Epidemiology of Vertigo, Dizziness, and Hearing Loss
A recent population-based telephone survey in Germany showed nearly 30% of the population had experienced moderate to severe dizziness (Neuhauser et al., 2005). Though most subjects reported nonspecific forms of dizziness, nearly a quarter had true vertigo. Dizziness is more common among females and older people and has important healthcare utilization implications; up to 80% of patients with dizziness seek medical care at some point. In the United States, the National Centers for Health Statistics report 7.5 million annual ambulatory visits to physician offices, hospital outpatient departments, and emergency departments (EDs) for dizziness, making it one of the most common principal complaints (Burt and Schappert, 2004).
Hearing loss affects approximately 16% of adults (age >18 years) in the United States (Lethbridge-Cejku et al., 2006). Men are more commonly affected than women, and the prevalence of hearing loss increases dramatically with age, so that by age 75, nearly 50% of the population reports hearing loss. Hearing loss is an important cause of disability. The most common type of hearing loss is sensorineural, and both idiopathic presbycusis and noise-induced forms are common etiologies. Bothersome tinnitus is less frequent in the U.S. population, with about 3% reporting it, although this increases to about 9% for subjects older than 65 (Adams et al., 1999). The most common type of tinnitus is a high-pitched ringing in both ears.
Normal Anatomy and Physiology
The inner ear is composed of a fluid-filled sac enclosed by a bony capsule with an anterior cochlear part, central chamber (vestibule), and a posterior vestibular part (Fig. 37.1). Endolymph fills up the fluid-filled sac and is separated by a membrane from the perilymph. These fluids primarily differ in their composition of potassium and sodium, with the endolymph resembling intracellular fluid with a high potassium and low sodium content, and perilymph resembling extracellular fluids with a low potassium and high sodium content. Perilymph communicates with the cerebrospinal fluid (CSF) through the cochlear aqueduct.
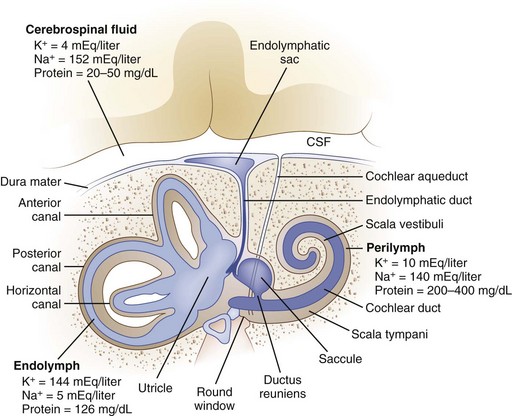
Fig. 37.1 Anatomy of the inner ear. CSF, Cerebrospinal fluid.
(From Baloh, R.W., 1998. Dizziness, Hearing Loss, and Tinnitus. F.A. Davis Company, Philadelphia, Figure 6, p. 16.)
The cochlea senses sound waves after they travel through the external auditory canal and are amplified by the tympanic membrane and ossicles of the middle ear (Baloh and Kerber, 2011). The stapes, the last of three ossicles in the middle ear, contacts the oval window, which directs the forces associated with sound waves along the basilar membrane of the cochlea. These forces stimulate the hair cells, which in turn generate neural signals in the auditory nerve. The auditory nerve enters the lateral brainstem at the pontomedullary junction and synapses in the cochlear nucleus. The trapezoid body is the major decussation of the auditory pathway, but many fibers do not cross to the contralateral side. Signals then travel to the superior olivary complex. Some projections travel from the superior olivary complex to the inferior colliculus through the lateral lemnisci, and others terminate in one of the nuclei of the lateral lemniscus. Next, fibers travel to the ipsilateral medial geniculate body, and then auditory radiations pass through the posterior limb of the internal capsule to reach the auditory cortex of the temporal lobe.
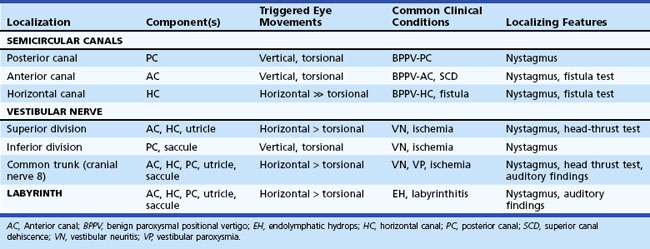
The peripheral vestibular system is composed of three semicircular canals, the utricle and saccule, and the vestibular component of the eighth cranial nerve (Baloh and Kerber, 2011). Each semicircular canal has a sensory epithelium called the crista; the sensory epithelium of the utricle and saccule is called the macule. The semicircular canals sense angular movements, and the utricle and saccule sense linear movements. Two of the semicircular canals (anterior and posterior) are oriented in the vertical plane nearly orthogonal to each other; the third canal is oriented in the horizontal plane (horizontal canal). The crista of each canal is primarily activated by movement occurring in the plane of that canal. When the hair cells of these organs are stimulated, the signal is transferred to the vestibular nuclei via the vestibular portion of cranial nerve VIII. Signals originating from the horizontal semicircular canal then pass via the medial longitudinal fasciculus along the floor of the fourth ventricle to the abducens nuclei in the middle brainstem and the ocular motor complex in the rostral brainstem. The anterior (also referred to as the superior) and posterior canal impulses pass from the vestibular nuclei to the ocular motor nucleus and trochlear nucleus triggering eye movements roughly in the plane of each canal. A key feature is that once vestibular signals leave the vestibular nuclei they divide into vertical, horizontal, and torsional components. As a result, a lesion of central vestibular pathways can cause a pure vertical, pure torsional, or pure horizontal nystagmus.
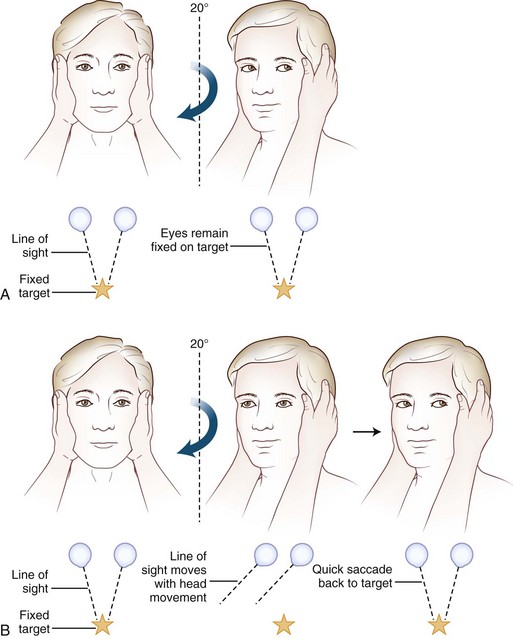
The primary vestibular afferent nerve fibers maintain a constant baseline firing rate of action potentials. When the baseline rate from each ear is symmetrical (or an asymmetry has been centrally compensated), the eyes remain stationary. With an uncompensated asymmetry in the firing rate, either resulting from increased or decreased activity on one side, slow ocular deviation results. By turning the head to the right, the baseline firing rate of the horizontal canal is physiologically altered, causing an increased firing rate on the right side and a decreased firing rate on the left side (Fig. 37.2). The result is a slow deviation of the eyes to the left. In an alert subject, this slow deviation is regularly interrupted by quick movements in the opposite direction (nystagmus) so the eyes do not become pinned to one side. In a comatose patient, only the slow component is seen because the brain cannot generate the corrective fast components.
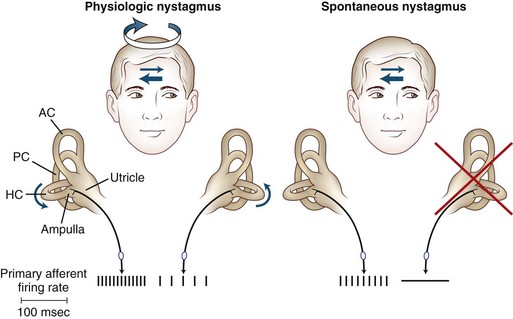
The plane in which the eyes deviate as a result of vestibular stimulation depends on the combination of canals that are stimulated (Table 37.1). If only the posterior semicircular canal on one side is stimulated (as occurs with BPPV), a vertical-torsional deviation of the eyes can be observed, which is followed by a fast corrective response generated by the conscious brain in the opposite direction. However, if the horizontal canal is the source of stimulation (as occurs with the horizontal canal variant of BPPV), a horizontal deviation with a slight torsional component (because this canal is slightly off the horizontal plane) results. If the vestibular nerve is lesioned (vestibular neuritis) or stimulated (vestibular paroxysmia), a horizontal greater than torsional nystagmus is seen that is the vector sum of all three canals—the two vertical canals on one side cancel each other out.
History of Present Illness
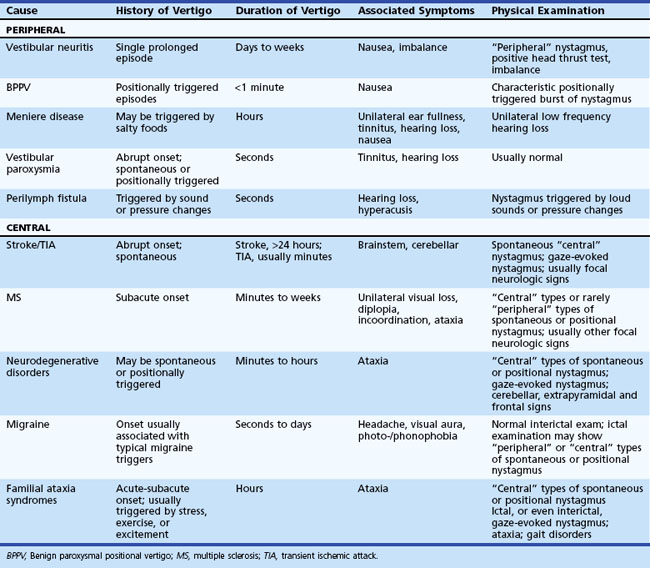
The history and physical examination provide the most important information when evaluating patients complaining of dizziness (Colledge et al., 1996; Lawson et al., 1999). Often, patients have difficulty describing the exact symptom experienced, so the onus is on the clinician to elicit pertinent information. The first step is to define the symptom. No clinician should ever be satisfied to record the complaint simply as “dizziness.” For patients unable to provide a more detailed description of the symptom, the physician can ask the patient to place their symptom into one of the following categories: movement of the environment (vertigo), lightheadedness, or strictly imbalance without an abnormal head sensation. Because patient descriptions about dizziness can be unreliable and inconsistent (Newman-Toker et al., 2007), other details about the symptom become equally important. The physician should also ask the following questions: Is the symptom constant or episodic, are there accompanying symptoms, how did it begin (gradual, sudden, etc.), and were there aggravating or alleviating factors? If episodic, what was the duration and frequency of attacks, and were there triggers? Table 37.2 displays the key distinguishing features of common causes of dizziness. One key point is that any type of dizziness may worsen with position changes, but some disorders such as BPPV only occur after position change.
Physical Examination
General Neurological Examination
The general neurological examination is very important in patients complaining of dizziness, because dizziness can be the earliest symptom of a neurodegenerative disorder (de Lau et al., 2006) and can also be an important symptom of stroke, tumor, demyelination, or other pathologies of the nervous system.
Neuro-otological Examination
Ocular Motor
The first step in assessing ocular motor function is to search for spontaneous involuntary movements of the eyes. The examiner asks the patient to look straight ahead while observing for nystagmus or saccadic intrusions. Nystagmus is characterized by a slow- and fast-phase component and is classified as either spontaneous, gaze-evoked, or positional. The direction of nystagmus is conventionally described by the direction of the fast phase, which is the direction it appears to be “beating” toward. Recording whether the nystagmus is vertical, horizontal, torsional, or a mixture of these provides important localizing information. Spontaneous nystagmus can have either a peripheral or central pattern. Although central lesions can mimic a “peripheral” pattern of nystagmus (Lee and Cho, 2004; Newman-Toker et al., 2008), some very unusual and unlikely circumstances are required for peripheral lesions to cause “central” patterns of nystagmus. A peripheral pattern of spontaneous nystagmus is unidirectional, that is, the eyes beat only to one side ( Video 37.1). Peripheral spontaneous nystagmus never changes direction. It is usually a horizontal greater than torsional pattern because of the physiology of the asymmetry in firing rates within the peripheral vestibular system whereby the vertical canals cancel each other out. The prominent horizontal component results from the unopposed horizontal canal. Other characteristics of peripheral spontaneous nystagmus are suppression with visual fixation, increase in velocity with gaze in the direction of the fast phase, and decrease with gaze in the direction opposite of the fast phase. Some patients are able to suppress this nystagmus so well at the bedside, or have partially recovered from the initiating event, that spontaneous nystagmus may only appear by removing visual fixation. Several simple bedside techniques can be used to remove the patient’s ability to fixate. Frenzel glasses are designed to remove visual fixation by using +30 diopter lenses. An ophthalmoscope can be used to block fixation. While the fundus of one eye is being viewed, the patient is asked to cover the other eye. Probably the simplest technique involves holding a blank sheet of paper close to the patient’s face (so as to block visual fixation) and observing for spontaneous nystagmus from the side.
Video 37.1). Peripheral spontaneous nystagmus never changes direction. It is usually a horizontal greater than torsional pattern because of the physiology of the asymmetry in firing rates within the peripheral vestibular system whereby the vertical canals cancel each other out. The prominent horizontal component results from the unopposed horizontal canal. Other characteristics of peripheral spontaneous nystagmus are suppression with visual fixation, increase in velocity with gaze in the direction of the fast phase, and decrease with gaze in the direction opposite of the fast phase. Some patients are able to suppress this nystagmus so well at the bedside, or have partially recovered from the initiating event, that spontaneous nystagmus may only appear by removing visual fixation. Several simple bedside techniques can be used to remove the patient’s ability to fixate. Frenzel glasses are designed to remove visual fixation by using +30 diopter lenses. An ophthalmoscope can be used to block fixation. While the fundus of one eye is being viewed, the patient is asked to cover the other eye. Probably the simplest technique involves holding a blank sheet of paper close to the patient’s face (so as to block visual fixation) and observing for spontaneous nystagmus from the side.
 Video 37.2). When vertical and/or torsional components are present, the term opsoclonus (or so-called dancing eyes) is used. The eyes make constant random conjugate saccades of unequal amplitude in all directions. Ocular flutter and opsoclonus are pathological findings typically seen in several different types of CNS diseases involving brainstem-cerebellar pathways. Paraneoplastic disorders should be considered in patients presenting with ocular flutter or opsoclonus.
Video 37.2). When vertical and/or torsional components are present, the term opsoclonus (or so-called dancing eyes) is used. The eyes make constant random conjugate saccades of unequal amplitude in all directions. Ocular flutter and opsoclonus are pathological findings typically seen in several different types of CNS diseases involving brainstem-cerebellar pathways. Paraneoplastic disorders should be considered in patients presenting with ocular flutter or opsoclonus.Ocular flutter. Spontaneous back-and-forth saccades without an intersaccadic delay are seen.
Gaze Testing
 Video 37.3). A few beats of unsustained nystagmus with gaze greater than 30 degrees is called end-gaze nystagmus and variably occurs in normal subjects. Gaze-evoked downbeating nystagmus (
Video 37.3). A few beats of unsustained nystagmus with gaze greater than 30 degrees is called end-gaze nystagmus and variably occurs in normal subjects. Gaze-evoked downbeating nystagmus ( Video 37.4), vertical nystagmus that increases on lateral gaze, localizes to the craniocervical junction and midline cerebellum. Gaze testing may also trigger saccadic oscillations.
Video 37.4), vertical nystagmus that increases on lateral gaze, localizes to the craniocervical junction and midline cerebellum. Gaze testing may also trigger saccadic oscillations.Gaze-evoked downbeating nystagmus. Downbeating nystagmus occurs with gaze to either side.
Smooth Pursuit
Smooth pursuit refers to the voluntary movement of the eyes used to track a target moving at a low velocity. It functions to keep the moving object on the fovea to maximize vision. Though characteristically a very smooth movement at low frequency and velocity testing, smooth pursuit inevitably breaks down when tested at high frequencies and velocities. Though smooth pursuit often becomes impaired with advanced age, a recent study found no significant decline in smooth pursuit in a group of healthy elderly individuals (>75 years) tested yearly for at least 9 years (Kerber et al., 2006). Patients with impaired smooth pursuit require frequent small saccades to keep up with the target, thus the term saccadic pursuit is used to describe this finding (see Video 37.3). Abnormalities of smooth pursuit occur as the result of disorders throughout the CNS and with tranquilizing medicines, alcohol, inadequate concentration or vision, and fatigue. Patients with diffuse cortical disease, basal ganglia disease, or diffuse cerebellar disease consistently have bilaterally impaired smooth pursuit. Patients with early or mild cerebellar degenerative disorders may have markedly impaired smooth pursuit with mild or minimal truncal ataxia as the only findings.
 Video 37.5). Undershooting saccades are less specific and often occur in normal subjects.
Video 37.5). Undershooting saccades are less specific and often occur in normal subjects.Vestibular Nerve Examination
Often omitted as part of the cranial nerve examination in general neurology texts, important localizing information can be obtained about the functioning of the vestibular nerve at the bedside. A unilateral or bilateral vestibulopathy can be identified using the head-thrust test (Halmagyi et al., 2008) (Fig. 37.3 and  Video 37.6). To perform the head-thrust test, the physician stands directly in front of the patient, who is seated on the exam table. The patient’s head is held in the examiner’s hands, and the patient is instructed to focus on the examiner’s nose. The head is then quickly moved about 5 to 10 degrees to one side. In patients with normal vestibular function, the VOR results in movement of the eyes in the direction opposite the head movement. Therefore the patient’s eyes remain on the examiner’s nose after the sudden movement. The test is repeated in the opposite direction. If the examiner observes a corrective saccade bringing the patient’s eyes back to the examiner’s nose after the head thrust, impairment of the VOR in the direction of the head movement is identified. Rotating the head slowly back and forth (the doll’s eye test) also induces compensatory eye movements, but both the visual and vestibular systems are activated by this low-velocity test, so a patient with complete vestibular function loss and normal visual pursuit will have normal-appearing compensatory eye movements on the doll’s eye test. This slow rotation of the head, however, is helpful in a comatose patient who is not able to generate voluntary visual tracking eye movements. Slowly rotating the head can also be a helpful test in patients with impairment of the smooth-pursuit system, because smooth movements of the eyes during slow rotation of the head indicates an intact VOR, whereas continued saccadic movements during slow rotation indicates an accompanying deficit of the VOR (Migliaccio et al., 2004).
Video 37.6). To perform the head-thrust test, the physician stands directly in front of the patient, who is seated on the exam table. The patient’s head is held in the examiner’s hands, and the patient is instructed to focus on the examiner’s nose. The head is then quickly moved about 5 to 10 degrees to one side. In patients with normal vestibular function, the VOR results in movement of the eyes in the direction opposite the head movement. Therefore the patient’s eyes remain on the examiner’s nose after the sudden movement. The test is repeated in the opposite direction. If the examiner observes a corrective saccade bringing the patient’s eyes back to the examiner’s nose after the head thrust, impairment of the VOR in the direction of the head movement is identified. Rotating the head slowly back and forth (the doll’s eye test) also induces compensatory eye movements, but both the visual and vestibular systems are activated by this low-velocity test, so a patient with complete vestibular function loss and normal visual pursuit will have normal-appearing compensatory eye movements on the doll’s eye test. This slow rotation of the head, however, is helpful in a comatose patient who is not able to generate voluntary visual tracking eye movements. Slowly rotating the head can also be a helpful test in patients with impairment of the smooth-pursuit system, because smooth movements of the eyes during slow rotation of the head indicates an intact VOR, whereas continued saccadic movements during slow rotation indicates an accompanying deficit of the VOR (Migliaccio et al., 2004).
Positional Testing
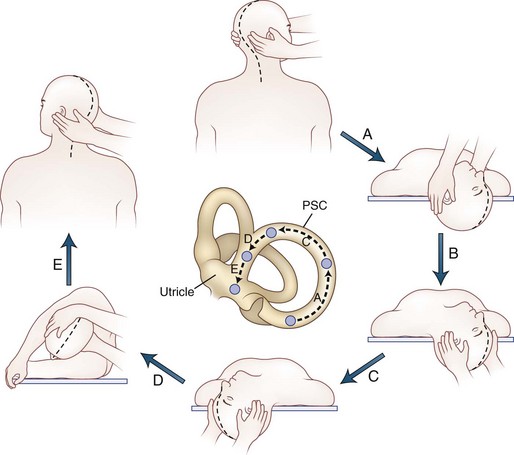
Positional testing can help identify peripheral or central causes of vertigo. The most common positional vertigo, BPPV, is caused by free-floating calcium carbonate debris, usually in the posterior semicircular canal, occasionally in the horizontal canal, or rarely in the anterior canal. The characteristic burst of upbeat torsional nystagmus is triggered in patients with BPPV by a rapid change from an erect sitting position to supine head-hanging left or head-hanging right (the Dix-Hallpike test) ( Video 37.7). When present, the nystagmus is usually only triggered in one of these positions. A burst of nystagmus in the opposite direction (downbeat torsional) occurs when the patient resumes the sitting position. A repositioning maneuver can be used to liberate the clot of debris from the posterior canal. We use the modified Epley maneuver (Fig. 37.4 and
Video 37.7). When present, the nystagmus is usually only triggered in one of these positions. A burst of nystagmus in the opposite direction (downbeat torsional) occurs when the patient resumes the sitting position. A repositioning maneuver can be used to liberate the clot of debris from the posterior canal. We use the modified Epley maneuver (Fig. 37.4 and  Video 37.8), which is more than 80% effective in treating patients with posterior canal BPPV, compared to 10% effectiveness of a sham procedure (Fife et al., 2008). The key feature of this maneuver is the roll across in the plane of the posterior canal so that the clot rotates around the posterior canal and out into the utricle. Once the clot enters the utricle, it may reattach to the membrane, dissolve, or may even remain free-floating in the utricle, but the debris no longer disrupts semicircular canal function. Recurrences are common, however.
Video 37.8), which is more than 80% effective in treating patients with posterior canal BPPV, compared to 10% effectiveness of a sham procedure (Fife et al., 2008). The key feature of this maneuver is the roll across in the plane of the posterior canal so that the clot rotates around the posterior canal and out into the utricle. Once the clot enters the utricle, it may reattach to the membrane, dissolve, or may even remain free-floating in the utricle, but the debris no longer disrupts semicircular canal function. Recurrences are common, however.
If the debris is in the horizontal canal, direction-changing horizontal nystagmus is seen. Patients are tested for the horizontal canal variant of BPPV by turning the head to each side while lying in the supine position. The nystagmus can be either paroxysmal geotropic (beating toward the ground) or persistent apogeotropic nystagmus (beating away from the ground). In the case of geotropic nystagmus, the debris is in the posterior segment (or “long arm”) of the horizontal canal, whereas the debris is in the anterior segment (or “short arm”) when apogeotropic nystagmus is triggered. When geotropic nystagmus is triggered, the side with the stronger nystagmus is the involved side. However, when apogeotropic nystagmus is observed, the involved side is generally opposite the side of the stronger nystagmus. With the geotropic variant, the debris can be removed from the canal by rolling the patient (barbecue fashion) toward the normal side. Other repositioning maneuvers for horizontal canal BPPV include the Gufoni maneuver and the “forced prolonged position” (Fife et al., 2008; Vannucchi et al., 1997). In cases of the apogeotropic variant, performing the barbeque maneuver toward the affected side can convert the nystagmus to geotropic because it moves the particles from the short arm of the canal to the long arm. Once the nystagmus is converted to geotropic, the typical treatments for the geotropic variant are used.
Positional testing can also trigger central types of nystagmus (usually persistent downbeating), which may be the most prominent examination finding in patients with disorders like Chiari malformation or cerebellar ataxia (Kattah and Gujrati, 2005; Kerber et al., 2005a). Central positional nystagmus can mimic the nystagmus of horizontal canal BPPV. Positional nystagmus may also be prominent in patients with migraine-associated dizziness (von Brevern et al., 2005).
Auditory Examination
Finger rubs at different intensities and distances from the ear are a rapid, reliable, and valid screening test for hearing loss in the frequency range of speech (Torres-Russotto et al., 2009). If a patient can hear a faint finger rub stimulus at a distance of 70 cm (approximately one arm’s length) from one ear, then a hearing loss on that side—defined by a gold-standard audiogram threshold of greater than 25 dB at 1000, 2000, and 4000 Hz—is highly unlikely. On the other hand, if a patient cannot hear a strong finger rub stimulus at 70 cm, a hearing loss on that side is highly likely. The whisper test can also be used to assess hearing at the bedside (Bagai et al., 2006). For this test, the examiner stands behind the patient to prevent lip reading and occludes and masks the non–test ear, using a finger to rub and close the external auditory canal. The examiner then whispers a set of three to six random numbers and letters. Overall, the patient is considered to have passed the screening test if they repeat at least 50% of the letters and numbers correctly. The Weber and Rinne tests are commonly used bedside tuning fork tests. To perform these, a tuning fork (256 Hz or 512 Hz) is gently struck on a hard rubber pad, the elbow, or the knee about two-thirds of the way along the tine. To conduct the Weber test, the base of the vibrating fork is placed on the vertex (top or crown of the head), bridge of the nose, upper incisors, or forehead. The patient is asked if the sound is heard and whether it is heard in the middle of the head or in both ears equally, toward the left, or toward the right. In a patient with normal hearing, the tone is heard centrally. In asymmetrical or a unilateral hearing impairment, the tone lateralizes to one side. Lateralization indicates an element of conductive impairment in the ear in which the sound localizes, a sensorineural impairment in the contralateral ear, or both. The Rinne test compares the patient’s hearing by air conduction with that by bone conduction. The fork is first held against the mastoid process until the sound fades. It is then placed 1 inch from the ear. Normal subjects can hear the fork about twice as long by air as by bone conduction. If bone is greater than air conduction, a conductive hearing loss is suggested.
Specific Disorders Causing Vertigo
Peripheral Vestibular Disorders
Peripheral vestibular disorders are important for neurologists to understand because they are common, readily identified at the bedside, and often missed by frontline physicians (see Table 37.2).
Vestibular Neuritis
A common presentation to the ED or outpatient clinic is the rapid onset of severe vertigo, nausea, vomiting, and imbalance. The symptoms gradually resolve over several days, but some symptoms can persist for months. The etiology of this disorder is probably viral, because the course is generally benign and self-limited, it occurs in young healthy individuals, and occasionally occurs in epidemics. Histopathological studies provide evidence of a peripheral vestibular localization and support the etiology of a viral cause. A viral etiology is also the likely cause of most cases of Bell palsy and sudden sensorineural hearing loss. The key to the diagnosis of vestibular neuritis is recognizing the peripheral vestibular pattern of nystagmus and identifying a positive head-thrust test in the setting of a rapid onset of vertigo without other neurological symptoms. Magnetic resonance imaging studies (MRIs) are usually normal in patients with vestibular neuritis (Strupp et al., 1998b). The course of vestibular neuritis is self-limited, and the mainstay of treatment is symptomatic. A recent study of patients with vestibular neuritis showed improvement of peripheral vestibular function, as measured by caloric testing at 1 year, after receiving methylprednisolone within 3 days of onset, compared to placebo (Strupp et al., 2004). A formal vestibular rehabilitation program can help patients compensate for the vestibular lesion (Strupp et al., 1998a).
Benign Paroxysmal Positional Vertigo
Benign paroxysmal positional vertigo may be the most common cause of vertigo in the general population. Patients typically experience brief episodes of vertigo when getting in and out of bed, turning in bed, bending down and straightening up, or extending the head back to look up. As noted earlier, the condition is caused when calcium carbonate debris dislodged from the otoconial membrane inadvertently enters a semicircular canal. The debris can be free-floating within the affected canal (canalithiasis) or stuck against the cupula (cupulolithiasis). Repositioning maneuvers are highly effective in removing the debris from the canal, though recurrence is common (see Fig. 37.4) (Fife et al., 2008). Once the debris is out of the canal, patients are instructed to avoid extreme head positions to prevent the debris from reentering the canal. Patients can also be taught to perform a repositioning maneuver should they have a recurrence of the positional vertigo.
Meniere Disease
Endolymphatic hydrops, or expansion of the endolymph relative to the perilymph, is regarded as the etiology, though the underlying cause is unclear. Additionally, the characteristic histopathological changes of endolymphatic hydrops have been identified in temporal bone specimens of patients with no clinical history of Meniere disease (Merchant et al., 2005). Some patients with well-documented Meniere disease experience abrupt episodes of falling to the ground, without loss of consciousness or associated neurological symptoms. Patients often report the sensation of being pushed or thrown to the ground. The falls are hard and often result in fractures or other injuries. These episodes have been called otolithic catastrophes of Tumarkin because of the suspicion that they represent acute stimulation of the otoliths. The bedside interictal examination of patients with Meniere disease may identify asymmetrical hearing, but the head-thrust test is usually normal. Treatment is initially directed toward an aggressive low-salt diet and diuretics, though the evidence for these treatments is poor. Intratympanic gentamicin injections can be effective and are minimally invasive. Sectioning of the vestibular nerve or destruction of the labyrinth are other procedures (Minor et al., 2004). Autoimmune inner ear disease presents as a fulminate variant of Meniere disease. Another variant is so-called delayed endolymphatic hydrops. Patients with this disorder report recurrent episodes of severe vertigo without auditory symptoms developing years after a severe unilateral hearing loss caused by a viral or bacterial infection.
Vestibular Paroxysmia
Vestibular paroxysmia is characterized by brief (seconds to minutes) episodes of vertigo, occurring suddenly without any apparent trigger (Hufner et al., 2008). The disorder may be analogous to hemifacial spasm and trigeminal neuralgia, which are felt to be due to spontaneous discharges from a partially damaged nerve. In patients with vestibular paroxysmia, unilateral dysfunction can sometimes be identified on vestibular or auditory testing. Like the analogous disorders, it is conceivable that a normal vessel could be compressing the cranial nerve, and surgical removal of the vessel might seem to be a treatment option. However, many asymptomatic subjects have a normal vessel lying on the eighth nerve (usually the anterior inferior cerebellar artery), and most vestibular paroxysmia patients have a favorable course with conservative or medication management (Hufner et al., 2008), so the decision to operate in this delicate region is rarely indicated. Medications associated with a reduction in episodes include carbamazepine, oxcarbazepine, and gabapentin (Hufner et al., 2008; Moon and Hain, 2005).
Vestibular Fistulae
Superior canal dehiscence was first described in 1998 (Minor et al., 1998). As the name implies, dehiscence of the bone overlying the superior canal results in a fistula between the superior canal and the middle cranial fossa. Normally the semicircular canals are enclosed by the rigid bony capsule, so these vestibular structures are unaffected by sound pressure changes. The oval and round windows direct the forces associated with sound waves into the cochlea and along the spiral basilar membrane. A break in the bony capsule of the semicircular canals can redirect some of the sound or pressure to the semicircular canals causing vestibular activation, a phenomenon known as Tullio phenomenon. Prior to the discovery of SCD, fistulas were known to occur with rupture of the oval or round window or erosion into the horizontal semicircular canal from chronic infection. Pressure changes generated by increasing intracranial pressure (ICP) (Valsalva against closed glottis) or increasing middle ear pressure (Valsalva against pinched nostrils or compression of the tragus) triggers brief nystagmus in the plane of the affected canal. Surgically repairing the defect can be attempted if the patient is debilitated by the symptoms, but many patients do well with conservative management. Patients with SCD may have hypersensitivity to bone-conducted sound and bone-conduction thresholds on the audiogram lower than the normal 0 dB hearing levels, even though air conduction thresholds remain normal (Minor, 2005). Other vestibular fistulae can result from trauma or erosion of a cholesteatoma into the horizontal semicircular canal.
Other Peripheral Disorders
There are many other peripheral vestibular causes of vertigo, but most are uncommon. Vertigo often follows a blow to the head, even without a corresponding temporal bone fracture. This so-called labyrinthine concussion results from the susceptibility of the delicate structures of the inner ear to blunt trauma. Vestibular ototoxicity, usually from gentamicin, can cause a vestibulopathy that is usually bilateral but rarely can be unilateral (Waterston and Halmagyi, 1998). A bilateral vestibulopathy can also occur from an immune-mediated disorder (e.g., autoimmune inner ear disease, Cogan syndrome), infectious process (e.g., meningitis, syphilitic labyrinthitis), structural lesion (bilateral acoustic neuroma), or a genetic disorder (e.g., neurodegenerative or isolated vestibular). The bilateral vestibular loss often goes unrecognized because the vestibular symptoms can be overshadowed by auditory or other symptoms. Although the most prominent vestibular symptoms of bilateral vestibulopathy are oscillopsia and imbalance, some nonspecific dizziness and vertigo attacks may occur as well. Acoustic neuromas, vestibular schwannomas, typically present with slowly progressive unilateral hearing loss, but rarely vertigo can occur. Because the tumor growth is slow, the vestibulopathy is compensated by the CNS. Finally, any disorder affecting the skull base, such as sarcoidosis, lymphoma, bacterial and fungal infections, or carcinomatous meningitis, can cause either unilateral or bilateral peripheral vestibular symptoms.
Central Nervous System Disorders
Brainstem or Cerebellar Ischemia/Infarction
Ischemia affecting vestibular pathways within the brainstem or cerebellum often causes vertigo. Brainstem ischemia is normally accompanied by other neurological signs and symptoms, because motor and sensory pathways are in close proximity to vestibular pathways. Vertigo is the most common symptom with Wallenberg syndrome, infarction in the lateral medulla in the territory of the posterior inferior cerebellar artery (PICA), but other neurological symptoms and signs (e.g., diplopia, facial numbness, Horner syndrome) are invariably present. Ischemia of the cerebellum can cause vertigo as the most prominent or only symptom, and a common dilemma is whether the patient with acute-onset vertigo needs an MRI to rule out cerebellar infarction. Computed tomography (CT) scans of the posterior fossa are not a sensitive test for ischemic stroke (Chalela et al., 2007). Abnormal ocular motor findings in patients with brainstem or cerebellar strokes include: (1) spontaneous nystagmus that is purely vertical, horizontal, or torsional, (2) direction-changing gaze-evoked nystagmus (patient looks to the left and has left-beating nystagmus, looks to the right and has right-beating nystagmus), (3) impairment of smooth pursuit, and (4) overshooting saccades. Rarely, central causes of nystagmus can closely mimic the peripheral vestibular pattern of spontaneous nystagmus (Lee et al., 2006b; Newman-Toker et al., 2008). Patients with brainstem or cerebellar infarction need immediate attention because herniation or recurrent stroke can occur. However, because of the rarity of ischemia causing isolated vertigo, MRI need only be considered in patients with significant stroke risk, factors such as older age, known history of stroke, transient ischemic attacks (TIAs), coronary artery disease, or diabetes.
Neurodegenerative Disorders
It is not uncommon for a patient with the main complaint of dizziness to have or later develop typical features of PD, a parkinsonian syndrome (PSP, multiple systems atrophy), or a progressive ataxia disorder (de Lau et al., 2006). However, dizziness in these patients is usually better clarified as imbalance. Positional downbeat nystagmus occurs in patients with spinocerebellar ataxia type 6 (SCA6) and other progressive ataxia disorders (Kattah and Gujrati, 2005; Kerber et al., 2005a).
Vertigo in Inherited Disorders
Migraine
Migraine is a heterogeneous genetic disorder characterized by headaches in addition to many other neurological symptoms. Several rare monogenetic subtypes have been identified. Linkage analysis has identified a number of chromosomal loci in common forms of migraine, but no specific genes have been found. Dizziness has long been known to occur among patients with migraine headaches, and benign recurrent vertigo is usually a migraine equivalent because no other signs or symptoms develop over time, the neurological exam remains normal, and a family or personal history of migraine headaches is common, as are typical migraine triggers. Interestingly, some patients with benign recurrent vertigo (BRV) also report auditory symptoms similar to patients with Meniere disease, and a mild hearing loss may also be seen on the audiogram (Battista, 2004). The key distinguishing factor between migraine and Meniere disease is the lack of progressive unilateral hearing loss in patients with migraine. Other types of dizziness are common in patients with migraine as well, including nonspecific dizziness and positional vertigo (von Brevern et al., 2005). The cause of vertigo in migraine patients is not yet known, but the diagnosis of migraine should be entertained in any patient with chronic recurrent attacks of dizziness of unknown cause. Long-standing motion sensitivity including carsickness, sensitivities to other types of stimuli, and a clear family history of migraine help support the diagnosis. Also, some patients have a typical migraine visual aura or other focal neurological symptoms associated with headache. Though the diagnosis of migraine-associated dizziness remains one of exclusion, little else can cause recurrent episodes without any other symptoms over a long period of time. In a genome-wide linkage scan of BRV patients (20 families) linkage to chromosome 22q12 was found, but genetic heterogeneity was evident (Lee et al., 2006a). Testing linkage using a broader phenotype of BRV and migraine headaches weakened the linkage signal. Thus, no evidence exists at this time that migraine is allelic with BRV, even though migraine has a high prevalence in BRV patients.
Familial Bilateral Vestibulopathy
Familial bilateral vestibulopathy (FBV) patients typically have brief attacks of vertigo (seconds) followed by progressive loss of peripheral vestibular function leading to imbalance and oscillopsia, usually by the fifth decade. The recurrent attacks of vertigo may somehow cause damage to vestibular structures, leading to progressive vestibular loss. Quantitative rotational testing shows gains greater than 2 standard deviations below the normal mean for both sinusoidal and step changes in angular velocity. Caloric testing is insensitive for identifying bilateral vestibulopathy because of the wide range of normal caloric responses. The bedside head-thrust test may show bilateral corrective saccades when vestibulopathy is severe. As the vestibulopathy becomes more severe, attacks of vertigo become less frequent and eventually cease. Despite the high prevalence of familial hearing loss and enormous progress in identifying the genetic basis of deafness, to date no gene mutations have been identified that lead to isolated bilateral vestibulopathy in humans. Only a few FBV families have been described (Brantberg, 2003; Jen et al., 2004b). Given the high prevalence and genetic diversity of familial hearing loss, it seems reasonable to suspect that bilateral vestibulopathy would have a similar prevalence and genetic diversity. The huge disparity in knowledge about genetic deafness and genetic vestibulopathy might stem from our inadequacy to identify vestibulopathy rather than the rareness of the disorder. It is much more straightforward for healthcare providers to identify the symptoms of hearing loss than the symptoms of vestibular loss. Adequate laboratory testing for hearing loss is also much more readily available than it is for vestibular loss. Increased knowledge and use of the bedside head-thrust test, however, has the potential to substantially enhance the identification of bilateral vestibular loss.
Familial Hearing Loss and Vertigo
Familial progressive vestibular-cochlear dysfunction was first identified in 1988. Linkage to chromosome 14q12-13 was later found, and the disorder was designated DFNA9 (DFNA = deafness, familial, non-syndromic, type A [autosomal dominant]) (Manolis et al., 1996). Using an organ-specific approach, mutations within COCH were found to cause DFNA9 (Robertson et al., 1998). This disorder of progressive hearing loss is unique because no other autosomal dominant genetic hearing loss syndromes have vertigo as a common symptom. Progressive hearing loss is the most prominent symptom of DFNA9. Vertigo occurs in about 50% of DFNA9 patients. When present, vertigo may be spontaneous in onset or positionally triggered (Lemaire et al., 2003). Age of onset is variable, with some patients developing symptoms in the second to third decade and others developing symptoms later. Vertigo attacks last minutes to hours and can be accompanied by worsening of hearing, aural fullness, or tinnitus, thus closely mimicking Meniere syndrome. Vertigo episodes can precede or accompany onset of hearing loss. In addition to severe progressive hearing loss, eventually DFNA9 patients develop progressive loss of vestibular function and corresponding symptoms of imbalance and oscillopsia. Because some patients have attacks closely resembling Meniere syndrome, the COCH gene was screened for mutations in idiopathic Meniere disease patients, but none were found. No studies report the use of effective treatments for vertigo attacks, but like FBV patients, these attacks generally only last a few years and then become less frequent, presumably due to loss of vestibular function. Of the many autosomal dominant genes that cause hearing loss, DFNA11 is the only other one associated with vestibulopathy.
Enlarged vestibular aqueduct syndrome (EVA), designated DFNB4 (DFNB = deafness, familial, non-syndromic, type B [autosomal recessive]), is characterized by early-onset hearing loss with enlargement of the vestibular aqueduct best seen on temporal bone CT. Normally, the vestibular aqueduct is less than 1.5 mm in diameter, but in EVA it is much larger. The mechanism leading to hearing loss and vertigo is unclear. The vestibular aqueduct contains the endolymphatic duct, which connects the medial wall of the vestibule to the endolymphatic sac and is an important structure in the exchange of endolymph. Enlargement may cause increased transmission of ICPs to the inner ear structures. However, the Valsalva maneuver—which increases ICP—does not trigger symptoms in EVA patients. Vertigo attacks last 15 minutes to 3 hours and are not associated with changes in hearing. Vertigo attacks may begin at the onset of hearing loss (early childhood) or years later and can be triggered by blows to the head or vigorous spinning (Oh et al., 2001a). Quantitative vestibular testing may be normal in EVA patients or reveal mild to moderate loss of vestibular function. Enlargement of the vestibular aqueduct has also been observed in Pendred syndrome (PS), branchio-oto-renal syndrome, CHARGE (coloboma of the eye, heart defects, atresia choanae, retardation of growth or development, genitourinary anomalies, and ear abnormalities or hearing impairment), Waardenburg syndrome, and distal renal tubular acidosis with deafness. EVA syndrome is allelic to PS, which is characterized by developmental abnormalities of the cochlea in combination with thyroid dysfunction and goiter.
Familial Ataxia Syndromes
Vestibular symptoms and signs are common with several of the hereditary ataxia syndromes including SCA types 1, 2, 3, 6, and 7, Friedreich ataxia, Refsum disease, and episodic ataxia (EA) types 2, 3, 4, and 5. In most of these disorders, the symptoms are slowly progressive, with the cerebellar ataxia and incoordination overshadowing the vestibular symptoms. Head movement–induced oscillopsia commonly occurs because the patient is unable to suppress the VOR with fixation. Attacks of vertigo may occur in up to half of patients with SCA6 (Takahashi et al., 2004), many of which are positionally triggered (Jen et al., 1998). Persistent downbeating nystagmus is often seen after placing patients into the head-hanging position; the positional vertigo and nystagmus can even be the initial symptom in these patients. Most of the episodic ataxia syndromes have onset before the age of 20 (Jen et al., 2004a). The attacks are characterized by extreme incoordination leading to severe difficulty walking during attacks. Vertigo can occur as part of these attacks, and migraine headaches are common in these patients as well. In fact EA2, SCA6, and familial hemiplegic migraine type 1 are all caused by mutations with the same gene, CACNA1A. An additional feature of EA2 and EA4 is the eventual development of interictal nystagmus and progressive ataxia. Patients with EA2 often have a dramatic response to treatment with acetazolamide.
Common Causes of Nonspecific Dizziness
Patients with nonspecific dizziness are probably referred to neurologists more frequently than patients with true vertigo. These patients are usually bothered by lightheadedness (wooziness), presyncope, imbalance, motion sensitivity, or anxiety. Side effects or toxicity from medications are common causes of nonspecific dizziness. Bothersome lightheadedness can be a direct effect of the medication itself or the result of lowering of the patient’s blood pressure. Ataxia can be caused by antiepileptic medications and is usually reversible once the medication is decreased or stopped. Patients with peripheral neuropathy causing dizziness report significant worsening of their balance in poor lighting and also the sensation that they are walking on cushions. Drops in blood pressure can be caused by dehydration, vasovagal attacks, or as part of an autonomic neuropathy. Patients with panic attacks can present with nonspecific dizziness, but their spells are invariably accompanied by other symptoms such as sense of fear or doom, palpitations, sweating, shortness of breath, or paresthesias. Other medical conditions such as cardiac arrhythmias or metabolic disturbances can also cause nonspecific dizziness. In the elderly, confluent white matter hyperintensities have a strong association with dizziness and balance problems. Presumably the result of small vessel arteriosclerosis, decreased cerebral perfusion (Marstrand et al., 2002) has been identified in these patients even when blood pressure taken at the arm is normal. Patients with dizziness related to white matter hyperintensities on MRI usually feel better sitting or lying down and typically have impairment of tandem gait. Since many elderly patients are taking blood pressure medications, at least a trial of lowering or discontinuing these medications is warranted.
Common Presentations of Vertigo
Acute Severe Vertigo
The patient presenting with new-onset severe vertigo probably has vestibular neuritis but stroke should also be a concern. An abrupt onset and accompanying focal neurological symptoms, particularly those that can be related to the posterior circulation, suggests an ischemic stroke. If no significant abnormalities are noted on the general neurological examination, attention should focus on the neuro-otological evaluation. If no spontaneous nystagmus is observed, a technique to block visual fixation should be applied. The direction of the nystagmus should be noted and the effect of gaze assessed. If a peripheral vestibular pattern of nystagmus is identified, a positive head-thrust test in the direction opposite the fast phase of nystagmus is highly localizing to the vestibular nerve. By far the most common cause of this presentation is vestibular neuritis. A central vestibular lesion (e.g., ischemic stroke) becomes a serious concern if there are “red flags” such as other central signs or symptoms, direction-changing nystagmus, vertical nystagmus, a negative head-thrust test (i.e., no corrective saccade after the head-thrust test to the direction opposite the fast phase of spontaneous nystagmus), or substantial stroke risk factors (Kattah et al., 2009; Lee et al., 2006b). Vertebral artery dissection can lead to an acute vertigo presentation, but the most common symptom is severe, sudden-onset occipital or neck pain, with additional neurological signs and symptoms (de Bray et al., 1997). If hearing loss accompanies the episode, labyrinthitis is the most likely diagnosis, but auditory involvement does not exclude the possibility of a vascular cause, because the anterior inferior cerebellar artery supplies both the inner ear and brain. When hearing loss and facial weakness accompany the acute onset of vertigo, one should closely inspect the outer ear for vesicles characteristic of herpes zoster (Ramsay Hunt syndrome). An acoustic neuroma is a slow-growing tumor, so only rarely is it associated with acute-onset vertigo. Migraine can mimic vestibular neuritis, though the diagnosis of migraine-associated vertigo hinges on recurrent episodes and lack of progressive auditory symptoms.
Recurrent Attacks of Vertigo
In patients with recurrent attacks of vertigo, the key diagnostic information lies in the details of the attacks. Meniere disease is the likely cause in patients with recurrent vertigo lasting longer than 20 minutes and associated with unilateral auditory symptoms. If the Meniere-like attacks present in a fulminate fashion, the diagnosis of autoimmune inner ear disease should be considered. Transient ischemic attacks (TIA) should be suspected in patients having brief episodes (minutes) of vertigo, particularly when vascular risk factors are present and other neurological symptoms are reported. Case series of patients with rotational vertebral artery syndrome demonstrate that the inner ear and possibly central vestibular pathways have high energy requirements and are therefore susceptible to levels of ischemia tolerated by other parts of the brain (Choi et al., 2005). Crescendo TIAs can be the harbinger of impending stroke or basilar artery occlusion. As with acute severe vertigo, accompanying auditory symptoms do not exclude the possibility of an ischemic disorder. Migraine and the migraine equivalent, BRV, are characterized by a history of similar symptoms, a normal examination, family or personal history of migraine headaches and/or BRV, other migraine characteristics, and typical triggers. Attacks are otherwise highly variable, lasting anywhere from seconds to days. If the attacks are consistently seconds in duration, the diagnosis of vestibular paroxysmia should be considered. Multiple sclerosis may be the cause when patients have recurrent episodes of vertigo and a history of other attacks of neurological symptoms, particularly when fixed deficits such as an afferent pupillary defect or internuclear ophthalmoplegia are identified on the examination.
Recurrent Positional Vertigo
Positional vertigo is defined by the symptom being triggered, not simply worsened, by certain positional changes. Vestibular neuritis is often confused with BPPV because vestibular neuritis patients can often settle into a relatively comfortable position and then experience dramatic worsening with movement. The patient complaining of recurrent episodes of vertigo triggered by certain head movements likely has BPPV, but this is not the only possibility. BPPV can be identified and treated at the bedside, so positional testing should be performed in any patient with this complaint. Positional testing can also uncover the other causes of positionally triggered dizziness (Bertholon et al., 2002). The history strongly suggests the diagnosis of BPPV when the positional vertigo is brief (<1 minute), has typical triggers, and is unaccompanied by other neurological symptoms. A burst of vertical torsional nystagmus is specific for BPPV of the posterior canal (Aw et al., 2005). If the Dix-Hallpike test is negative, the examiner should search for the horizontal canal variant of BPPV. Central positional nystagmus occurs as the result of disorders affecting the posterior fossa, including tumors, cerebellar degeneration, Chiari malformation, or MS. The nystagmus of these disorders is typically downbeating and persistent, though a pure torsional nystagmus may occur as well. Patients with loss of one vertebral artery may develop vertigo or significant dizziness after head turns to the direction opposite the intact artery because the bony structures of the spinal column can pinch off the remaining vertebral artery (Choi et al., 2005). Central types of nystagmus develop as a result, and vertigo can be the most prominent symptom. Finally, migraine can also closely mimic BPPV and central positional nystagmus (von Brevern et al., 2005). Patients with migraine as the cause typically report a longer duration of symptoms once the positional vertigo is triggered, and the nystagmus may be of a central or peripheral type. The mechanisms are not clear, but the disorder is benign because it is usually self-limited and not progressive. Associations between migraine and typical BPPV have also been made, but the link between these disorders is unclear.
Specific Disorders Causing Hearing Loss
Meniere Disease
Auditory symptoms in Meniere disease consist of a fluctuating sense of fullness and pressure along with tinnitus and decreased hearing in one ear. In the early stages, the hearing loss is completely reversible, but in later stages a residual hearing loss remains. Tinnitus may persist between episodes but usually increases in intensity immediately before or during the acute episode. It is typically described as a roaring sound like the sound of the ocean or a hollow seashell. The hearing loss on the audiogram appears in the early stages as a low-frequency loss. However, as the disorder progresses, a more complete hearing loss occurs. In a small number of patients, the disorder becomes bilateral. Eventually, severe permanent hearing loss develops, and the episodic nature spontaneously disappears. When the progression of hearing loss (particularly when bilateral) is fulminant and rapidly progressive, the diagnosis of autoimmune inner ear disease should be considered. Also see the section on Meniere disease under Specific Disorders Causing Vertigo.
Superior Canal Dehiscence
Patients with SCD may experience conductive hyperacusis (hearing their eye move or the impact of their feet during walking or running) and autophony (hearing their own breath and voice sounds) in the affected ear. An air/bone gap is often identified on standard audiograms. The Weber tuning fork test typically lateralizes to the affected ear, and the Rinne turning fork test may show bone conduction greater than air conduction (see Specific Disorders Causing Vertigo).
Common Presentations of Hearing Loss
Asymmetrical Sensorineural Hearing Loss
Evaluation of patients identified as having an asymmetrical sensorineural hearing loss is primarily the search for a tumor in the area of the internal auditory canal or cerebellopontine angle, or more rarely other lesions of the temporal bone or brain. With an asymmetry of hearing defined as 15 dB or greater in two or more frequencies or a 15% or more asymmetry in speech discrimination scores, approximately 10% of patients will have lesions identified on MRI (Cueva, 2004). Acoustic neuromas are by far the most common abnormality found. Other causative lesions may include glomus jugulare tumors, ectatic basilar artery with brainstem compression, or petrous apex cholesterol granuloma. Auditory brainstem response testing shows a sensitivity and specificity around 70%, with a false-positive rate of 77%, but a false-negative rate of 29% (Cueva, 2004).
Sudden Sensorineural Hearing Loss
The etiology of sudden sensorineural hearing loss is similar to both Bell palsy and vestibular neuritis in that a viral cause is presumed in the majority of cases, but proof of a viral pathophysiology in a given case is difficult to obtain. The hearing loss can abruptly develop or evolve over several hours. Acoustic neuromas may be found in around 5% of patients with this presentation (Aarnisalo et al., 2004), but one should also be aware of false-positive MRIs, particularly for lesions smaller than 6 mm (Arriaga et al., 1995). Focal ischemia to the cochlea, cochlear nerve, or the root entry zone can also cause an abrupt loss of hearing over several minutes. In the setting of a patient at risk for stroke, this cause should be considered early, because it can be the harbinger of basilar artery occlusion (Toyoda et al., 2002). Sudden-onset bilateral hearing loss can rarely result from bilateral lesions of the primary auditory cortex in the transverse temporal gyri of Heschl. Deficits can range from auditory agnosia for speech or nonspeech sounds, with relatively normal hearing thresholds, to rare cases of cortical deafness characterized by markedly elevated pure-tone thresholds.
Laboratory Investigations in Diagnosis and Management
Dizziness and Vertigo
The history and physical examination should determine what diagnostic tests if any are necessary in patients presenting with dizziness or vertigo. Studies have repeatedly shown that MRI, audiogram, and vestibular tests are no different in unselected patients complaining of dizziness when compared to age-matched controls (Colledge et al., 1996; Colledge et al., 2002; Hajioff et al., 2002; Lawson et al., 1999; Yardley et al., 1998). Many disorders causing dizziness can be diagnosed and even treated at the bedside, with no further diagnostic tests indicated.
Imaging
Brain imaging is commonly ordered in patients complaining of dizziness. Though a CT scan can rule out a large mass, smaller lesions can not be excluded because of artifact and poor resolution in the posterior fossa (Chalela et al., 2007). MRI is the imaging modality of choice but is expensive and generally a much less practical test than CT. Determining what patients should have an MRI can be difficult, which is why an understanding of the common peripheral vestibular disorders is important. Patients identified as having BPPV, vestibular neuritis, or Meniere disease do not require an imaging study. Additionally, patients with normal neurological and neuro-otological examinations reporting episodes of dizziness dating back more than several months are highly unlikely to have a relevant abnormality on MRI. Though studies show improved hearing preservation after surgery in patients with acoustic neuromas when diagnosed early, this does not mean that every patient complaining of dizziness requires an MRI to exclude this cause. Acoustic neuromas are rare, whereas dizziness and vertigo are extremely common. On the other hand, for any patient experiencing focal neurological symptoms or having unexplained neurological deficits or an otherwise rapid, unexplained progression of symptoms, an MRI should be strongly considered to rule out a mass lesion, stroke, structural abnormality, or MS. In dizzy patients with gradually progressive hearing loss, MRI may also be helpful.
Vestibular Testing
Eye Movement Recording
Methods of Recording Eye Movements
The earliest measures of eye movement recording were made using electro-oculography (EOG). This technique utilizes the potential difference between the cornea and retina, known as the corneal-retinal potential, which acts as an electric dipole oriented in the direction of the long axis of the eye (Baloh and Kerber, 2011). When electrodes are placed in the vicinity of the eyes, it becomes more positive when the eye rotates toward it and less positive when it rotates in the opposite direction. Recordings are made with a three-electrode system using differential amplifiers. Two of the (active) electrodes are placed on either side of the eye, and the reference (ground) electrode is placed somewhere remote from the eye. The two active electrodes measure a potential change of equal amplitude but opposite in direction. The difference in potential between these electrodes is amplified and used to control the displacement of a pen-writing recorder or similar device to produce a permanent record. Because the emphasis was initially on recording nystagmus, this test is also referred to as electronystagmography (ENG).
More recently, newer techniques for recording eye movements have been developed. One of these, video-oculography (VOG), is becoming popular in clinical use, while the other, the magnetic search coil technique, remains primarily a research tool. Each of these techniques has advantages and disadvantages. EOG is still probably the most widely used technique for recording eye movements. The equipment is less expensive to purchase, and the test provides reliable clinical information when individuals with proper training use it (Furman et al., 1996). Artifacts from lid movements or muscle action potentials are common, thus the “noise” is greater in ENG than either VOG or the magnetic search coil technique, and measuring torsional eye movements is difficult. Probably the main clinical advantage of VOG is the ability to go back and observe the actual video recording of the eye movements. This becomes particularly helpful when trying to distinguish an actual eye movement from an artifact such as a blink. Measuring torsional eye movements is also possible with VOG, although systems for doing so are still being developed. Disadvantages of VOG are the inability to measure eye movements with the eyes closed and difficulties stabilizing the head gear. The magnetic search coil technique uses a contact lens embedded with two coils of wire. One coil is wound in the frontal plane to sense horizontal and vertical movements and the other is wound in the sagittal plane to sense torsional eye movements. When the subject sits in a magnetic field, voltages are induced in these search coils that can be used to measure eye position. The magnetic search coil technique is the gold standard for measuring eye movements because it allows measurement of eye rotations around all three axes, with high sensitivity and low noise. This technique can be particularly helpful in obtaining accurate measures in patients who cannot reliably direct their gaze at calibration targets, since the device can be precalibrated. The main disadvantage of the magnetic search coil technique is the invasive nature of using the contact lens, which can cause discomfort for the patient or (rarely) corneal abrasion. Because of these factors, the magnetic search coil technique is generally only used in research laboratories. Newer versions of VOG systems with high-frequency sampling rates now rival the degree of precision of the magnetic search coil technique (Houben et al., 2006) and are thus being incorporated into more research laboratories. Though increased precision of VOG and the magnetic search coil technique is a clear advantage over EOG in research studies, it remains to be shown whether these tests enhance clinical diagnostic capabilities or change the management of patients when compared to EOG (Schmid-Priscoveanu and Allum, 1999).
Eye Movement Subtests
A standard test battery includes a search for pathological nystagmus or saccadic intrusions with fixation and with eyes open in the darkness, tests of visual ocular control (saccades, smooth pursuit, OKN), and the bithermal caloric test (Baloh and Kerber, 2011). Administration of these subtests usually proceeds in a manner similar to the bedside evaluation. By convention, for horizontal recordings, eye movements to the right result in an upward deflection of the tracing and those to the left result in downward deflection. For vertical recordings, upward and downward eye movements produce upward and downward deflections.
Visual Ocular Motor Control
Smooth Pursuit
Recordings of smooth pursuit are made by having the patient follow a target back and forth. By convention, most laboratories do this using a target that moves in a sinusoidal fashion. The accuracy of smooth pursuit is quantified by repeatedly sampling eye and target velocities and plotting the two velocities against each other. A computer algorithm makes the comparison between eye and target velocity after saccade waveforms have been removed. The slope of this eye/target velocity relationship represents the gain of the smooth pursuit system, which depends on the velocity and frequency of the target movements. Higher velocity and frequency testing results in lower gains. Though each laboratory must establish normal values, typically normal subjects have very high mean gains (0.92 ± 0.05 at 0.2 Hz, 22.6 degrees/sec). Though a patient’s age is typically considered when interpreting results, a recent study shows smooth-pursuit gains can be well maintained in subjects well into their ninth decade (Kerber et al., 2006).
Optokinetic Nystagmus
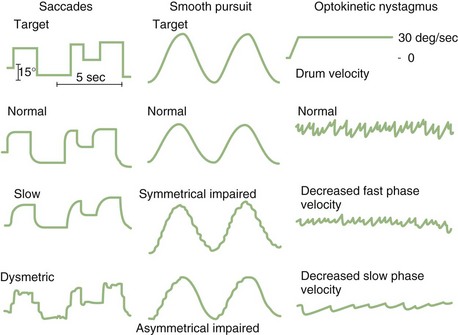
Laboratory testing of OKN uses a full-field visual stimulus (typically a patterned drum) that moves at a constant velocity and frequency around the subject, who is either instructed to follow the target (resulting in large-amplitude nystagmus) or stare through it (resulting in small-amplitude nystagmus). This stimulus also causes a sensation of self-rotation called circular vection even though the peripheral vestibular system is not being stimulated. The OKN gain is measured by comparing the slow-component velocity of the eye movement to the target velocity. As with smooth-pursuit testing, gain drops off with increasing frequency and velocity of the target in normal subjects. The normal and abnormal EOG appearance of saccades, smooth pursuit, and OKN are demonstrated in (Fig. 37.5).
Bithermal Caloric Testing
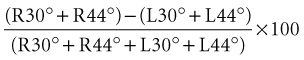
Directional preponderance is also calculated using the following formula:
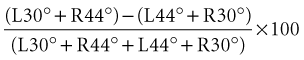
Dividing by the total response normalizes the measurements to remove the large variability in absolute magnitude of normal caloric responses. Typically, the finding of significant vestibular paresis of 25% to 30% with bithermal caloric stimulation suggests a lesion in the vestibular system that is located anywhere from the end-organ to the vestibular nerve root entry zone in the brainstem. This finding is a strong indicator of a unilateral peripheral lesion, but it must be placed in the context of the patient’s clinical history and bedside examination; a caloric paresis can also occur in central vestibular disorders. A recent study found a high rate of significant vestibular paresis (as measured by the caloric test) in patients with acute vertigo presentations caused by stroke (Newman-Toker et al., 2008). It is also common to find a caloric asymmetry in control subjects without dizziness (particularly those with migraine or diabetes) who undergo this test. A significant directional preponderance on caloric testing (>30%) indicates an imbalance in the vestibular system but is nonlocalizing, occurring with both peripheral and central lesions.
Rotational Testing
Rotational testing requires use of a motorized chair. The patient is placed in the chair that rotates under the control of a computer, and the patient’s head and body move in unison with the chair. The chair is in a dark room, so fixation is removed. Eye movements induced by the vestibular system stimulating movements of the patient’s head and body within the chair are recorded using any of the previously mentioned eye recording techniques. The computer precisely controls the velocity and frequency of rotations so that the VOR can be measured at multiple frequencies in a single session. Sinusoidal and step (impulse) changes in angular velocity are routinely used (Fig. 37.6). In clinical testing, generally only rotations about the vertical axis are used, which maximally stimulates the horizontal canals. Off-vertical rotation can be used to measure the function of the vertical semicircular canals and otoliths as well, but typically this is only done in research studies. For sinusoidal rotations, results are reported as gain (peak slow-component eye velocity divided by peak chair velocity) and phase (timing between the peak velocity of eye and head) at different frequencies.
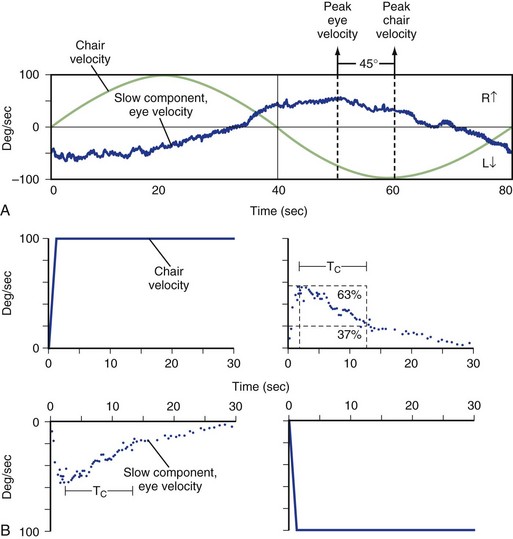
Because both inner ears are stimulated at the typically low velocities and frequencies used, rotational testing is most effective at determining a bilateral peripheral vestibular hypofunction that leads to a decreased gain and increased phase. Unilateral vestibular hypofunction can be suggested by a normal gain with increased phase on standard testing or a decreased unilateral gain with shortened time constant on impulse (rapid movement) testing. Normal rotational testing results in gains around 0.5 at low-frequency rotation (0.05 Hz), with gains approaching 1.0 at higher-frequency rotations (>1 Hz). Even patients with partial loss of bilateral vestibular function may have gains in the normal range at the higher frequency rotations, probably owing to the contribution of additional sensory systems (Jen et al., 2005; Wiest et al., 2001). The main disadvantage of rotational chair testing is the expense associated with setting it up. As a result, this vestibular test is typically only available at large academic centers. Because of this, portable devices using either passive (examiner-generated) head rotations or active (patient-generated) head turns have been developed, but the quality of evidence to support the use of these tests is low (Fife et al., 2000).
Posturography
Posturography is a method for quantifying balance. It is not a diagnostic test and is of little use for localizing a lesion. It can be helpful for following the course of a patient and may serve as a quantitative measure of the response to therapy or in research studies. As noted earlier, sway increases in older people, and several studies have shown that the frequency of falls increases as sway increases, suggesting that posturography may be a clinical tool for identifying older people at risk for falling, though whether it is better at this than a careful clinical assessment is unclear (Piirtola and Era, 2006). Posturography may be helpful in identifying patients with factitious balance disorders (Gianoli et al., 2000).
Vestibular Evoked Myogenic Potentials
It has long been known that the sacculus, which during the course of its evolution functioned as an organ of hearing and still does in primitive vertebrates, can be stimulated by loud sounds. As a result of this stimulation, a signal travels via the inferior trunk of the vestibular nerve to cranial nerve VIII and into the brainstem. From there, inhibitory postsynaptic potentials travel to the ipsilateral sternocleidomastoid muscle (SCM), essentially allowing the individual to reflexively turn towards the sound. To generate this vestibular evoked myogenic potential (VEMP) response, intense clicks of about 95 to 100 dB above normal hearing level (NHL) are required (Welgampola and Colebatch, 2005). The response is measured from an activated ipsilateral SCM. Tonic contraction of the muscle is required to demonstrate the inhibitory response. The amplitude of the response and also the threshold needed to generate it are measured. Because the absolute amplitudes vary greatly from patient to patient, the more reliable abnormality is detecting a side-to-side difference in an individual. Additionally, responses are unreliable in subjects older than 60 years and in patients with middle ear abnormalities. Abnormal VEMP responses can be detected in most disorders affecting the peripheral vestibular system, but this test can be particularly helpful in identifying patients with vestibular neuritis selectively affecting the inferior vestibular nerve (Halmagyi et al., 2002) or SCD (Minor, 2005). Because caloric and rotational testing mainly stimulate the horizontal semicircular canal (which sends afferent responses via the superior vestibular nerve), the rare disorder affecting only the inferior vestibular nerve will not be identified with these tests. In patients with SCD, VEMP testing leads to increased amplitudes and lowered thresholds due to the low-impedance pathway created by the third window.
Hearing Loss and Tinnitus
Auditory Testing
Audiological assessment is the basis for quantifying auditory impairment. Most neurologists rely on bedside assessments of hearing. In defining an auditory abnormality, tuning forks are no substitute for a complete audiological battery. Audiological testing is most reliable in defining peripheral or cochlear auditory disturbances and often may provide useful information, based on subtests, to diagnose retrocochlear disorders such as an acoustic neuroma. Tests for central auditory dysfunction are more difficult and poorly understood. Detailed descriptions of audiological tests, both peripheral and central, are provided in standard texts (Katz et al., 2009).
Pure-Tone Testing
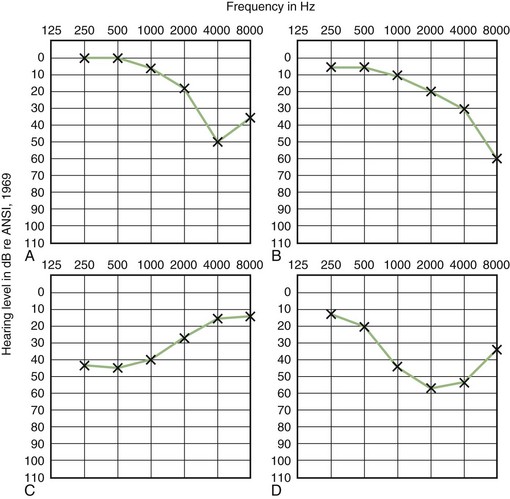
Comparison of air- and bone-conduction thresholds establishes the type of hearing loss. Conductive loss results from disorders in the outer or middle ear. The audiogram of patients with SCD may also have an air/bone gap, even though there is no abnormality of the outer or middle ear. This exception results from the third window created by the dehiscence which increases bone conduction. Sensorineural loss is associated with disorders of the cochlear and eighth cranial nerves. Mixed loss is a conductive and sensorineural loss coexisting in the same ear. Typical audiogram pure-tone pattern seen in patients with four common causes of sensorineural hearing loss are shown in (Fig. 37.7).
Acoustic Reflex Testing
Acoustic reflex measures the contraction of the stapedius muscle (innervated by the seventh cranial nerve) in response to a loud sound. The afferent limb of the reflex arch is through the auditory portion of the eighth cranial nerve, and the efferent portion of the reflex arch is through the seventh cranial nerve. The stapedius muscle normally contracts on both sides when an adequate sound is presented in one ear. As a result of contraction of the stapedius muscle, the tympanic membrane tightens or stiffens, thereby increasing the impedance or resistance of the eardrum to acoustic energy and resulting in a slight attenuation of sound transmitted through the middle ear system. In a normal subject, the acoustic reflex occurs in response to a pure tone between 70 and 100 dB above hearing level or when a white noise stimulus is presented at 65 dB above hearing level. Patients with conductive hearing loss do not have reflexes because the lesion prevents a change in compliance with stapedius muscle contraction. With cochlear lesions, the acoustic reflex may be present at sensation levels less than 60 dB above the auditory pure-tone threshold, which is a form of abnormal loudness growth or recruitment. Cochlear hearing losses must be moderate or severe before the acoustic reflex is lost. In contrast, patients with retrocochlear or eighth cranial nerve lesions often have abnormal acoustic reflexes with normal hearing. The reflex may be absent or exhibit an elevated threshold or abnormal decay. Reflex decay is present if the amplitude of the reflex decreases to half its original size within 10 seconds of stimulation at 1000 Hz, 10 dB above reflex threshold. This abnormality occurs in approximately 80% of patients with acoustic neuromas. Observation of the pattern of acoustic reflex testing, along with hearing evaluation, permits inferences to support the presence of a cochlear, conductive, or retrocochlear lesion of the seventh or eighth cranial nerves (Table 37.3).
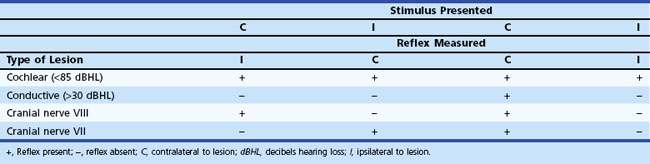
Evoked Potentials
Abnormal interwave latencies (I-III or I-V) are seen with retrocochlear lesions (cerebellopontine angle tumors) and can even be seen when only mild or no hearing loss is detected on pure tone audiometry. However, compared to brain MRI with gadolinium, the sensitivity of the ABR test is low, particularly with small tumors (Cueva, 2004). The least specific finding is the absence of all waves. This occurs in some patients with acoustic neuroma and in some with cerebellopontine angle meningiomas. Such patients often have marked hearing deficits with poor discrimination, suggesting retrocochlear disease. The absence of all waves should not occur unless a severe hearing loss exists.
Management of Patients with Vertigo
Treatments of Specific Disorders
The treatment of patients with dizziness and vertigo hinges on making a specific diagnosis. BPPV can be diagnosed and treated at the bedside, requiring no further treatment. Once repositioning is confirmed to be successful (see Fig. 37.4), patients are instructed to avoid head-hanging positions such as those used by dentists and hairdressers. These positions can cause the particles to reaccumulate in the posterior semicircular canals. For patients with horizontal canal BPPV, the “barbeque” rotation, Gufoni maneuver, or forced prolonged position can be used (Fife et al., 2008; Tirelli and Russolo, 2004; Vannucchi et al., 1997) can be used. The management of patients with vestibular neuritis is primarily symptomatic. After a couple of days, patients should be encouraged to move around and begin vestibular rehabilitation exercises that force the brain to compensate. Improved vestibular recovery measured by caloric responses has been shown in patients treated within 3 days for vestibular neuritis with methylprednisolone (Strupp et al., 2004), though it is not known whether this translates to improved functional or symptomatic recovery. Prolonged use of sedating medications to treat symptoms is not recommended, because it can slow down the vestibular compensation process. The early treatment of Meniere disease continues to be a low-salt diet and diuretics, though the evidence for these is not great (Minor et al., 2004). Minimally invasive intratympanic gentamicin injections can be used for patients with debilitating symptoms. Surgical ablation of the labyrinth or sectioning of the vestibular nerve are other options. Patients with vestibular paroxysmia may benefit from carbamazepine or a similar antiepileptic medication (Hufner et al., 2008). The third window in patients with SCD can be surgically repaired, but this is usually only recommended in patients debilitated by the symptoms (Minor, 2005).
Patients identified as having a small infarction in the posterior fossa should be closely monitored, as herniation or recurrent stroke can occur. Patients identified within 3 hours of an infarction are candidates for intravenous tissue plasminogen activator (tPA). There are some case reports of patients with recurrent vertigo caused by severe stenosis of the basilar artery who are cured by endovascular stenting (Kerber et al., 2005b), but this remains an experimental procedure. Patients identified with demyelinating lesions may be candidates for disease-modifying treatments even after presenting with a clinically isolated syndrome. Patients with episodic ataxia are typically highly responsive to treatment with acetazolamide, and there is anecdotal evidence of benefit of the use of acetazolamide in patients with BRV, a migraine equivalent. Patients with migraine-associated dizziness should first attempt to identify and eliminate triggers of their symptoms and also obtain adequate sleep and cardiovascular exercise. If these general measures are not adequate in controlling symptoms, a migraine prophylactic medication could be tried. Small trials of triptan medications in patients with migrainous vertigo suggest safety of these medicines but no significant benefit (Neuhauser et al., 2003).
Symptomatic Treatment of Vertigo
The commonly used antivertiginous drugs and their dosages are listed in Table 37.4 (Huppert et al., 2011). It is often difficult to predict which drugs or combinations of drugs will be most effective in individual patients, and large trials are lacking. Additionally, the mechanisms of these medications are not specific to the vestibular system, so side effects are common. Anticholinergic or antihistamine drug are usually effective in treating patients with mild to moderate vertigo, and sedation is minimal. If the patient is particularly bothered by nausea, the antiemetics prochlorperazine and metoclopramide can be effective and combined with other antivertiginous medications. For severe vertigo, sedation is often desirable, and drugs such as promethazine and diazepam are particularly useful, though prolonged use is not recommended.
| Class | Dosage† |
|---|---|
| ANTIHISTAMINES | |
| Meclizine | 25 mg PO q 4-6 h |
| Dimenhydrinate | 50 mg PO or IM q 4-6 h, or 100 mg suppository q 8 h |
| Promethazine | 25-50 mg PO or IM or as a suppository q 4-6 h |
| ANTICHOLINERGIC AGENT | |
| Scopolamine | 0.2 mg PO q 4-6 h, or 0.5 mg transdermally q 3 days |
| BENZODIAZEPINES | |
| Diazepam | 5 or 10 mg PO, IM, IV q 4-6 h |
| Lorazepam | 0.5-2 mg PO, IM, IV q 6-8 h |
| PHENOTHIAZINE | |
| Prochlorperazine | 5 or 10 mg PO or IM q 6 h, or 25 mg suppository q 12 h |
| BENZAMIDE | |
| Metoclopramide | 5 or 10 mg PO, IM, or IV q 4-6 h |
IM, intramuscular; IV intravenous; PO, oral.
† Usual adult starting dosage; maintenance dosage can be increased by a factor of 2-3. The most common side effect is drowsiness.
Aarnisalo A.A., Suoranta H., Ylikoski J. Magnetic resonance imaging findings in the auditory pathway of patients with sudden deafness. Otol Neurotol. 2004;25:245-249.
Adams P.F., Hendershot G.E., Marano M.A. Current estimates from the National Health Interview Survey, 1996. Vital Health Stat. 1999;10:1-203.
Arriaga M.A., Carrier D., Houston G.D. False-positive magnetic resonance imaging of small internal auditory canal tumors: a clinical, radiologic, and pathologic correlation study. Otolaryngol Head Neck Surg. 1995;113:61-70.
Aw S.T., Todd M.J., Aw G.E., et al. Benign positional nystagmus: a study of its three-dimensional spatio-temporal characteristics. Neurology. 2005;64:1897-1905.
Bagai A., Thavendiranathan P., Detsky A.S. Does this patient have hearing impairment? JAMA. 2006;295:416-428.
Baloh R.W. Prosper Meniere and his disease. Arch Neurol. 2001;58:1151-1156.
Baloh R.W., Kerber K.A. Clinical Neurophysiology of the Vestibular System, fourth ed. Philadelphia: Oxford University Press; 2011.
Battista R.A. Audiometric findings of patients with migraine-associated dizziness. Otol Neurotol. 2004;25:987-992.
Bertholon P., Bronstein A.M., Davies R.A., et al. Positional down beating nystagmus in 50 patients: cerebellar disorders and possible anterior semicircular canalithiasis. J Neurol Neurosurg Psychiatry. 2002;72:366-372.
Brantberg K. Familial early-onset progressive vestibulopathy without hearing impairment. Acta Otolaryngol. 2003;123:713-717.
Burt C.W., Schappert S.M. Ambulatory care visits to physician offices, hospital outpatient departments, and emergency departments: United States, 1999-2000. Vital Health Stat. 2004;13:1-70.
Chalela J.A., Kidwell C.S., Nentwich L.M., et al. Magnetic resonance imaging and computed tomography in emergency assessment of patients with suspected acute stroke: a prospective comparison. Lancet. 2007;369:293-298.
Choi K.D., Shin H.Y., Kim J.S., et al. Rotational vertebral artery syndrome: oculographic analysis of nystagmus. Neurology. 2005;65:1287-1290.
Colledge N., Lewis S., Mead G., et al. Magnetic resonance brain imaging in people with dizziness: a comparison with non-dizzy people. J Neurol Neurosurg Psychiatry. 2002;72:587-589.
Colledge N.R., Barr-Hamilton R.M., Lewis S.J., et al. Evaluation of investigations to diagnose the cause of dizziness in elderly people: a community based controlled study. BMJ. 1996;313:788-792.
Cueva R.A. Auditory brainstem response versus magnetic resonance imaging for the evaluation of asymmetric sensorineural hearing loss. Laryngoscope. 2004;114:1686-1692.
de Bray J.M., Penisson-Besnier I., Dubas F., et al. Extracranial and intracranial vertebrobasilar dissections: diagnosis and prognosis. J Neurol Neurosurg Psychiatry. 1997;63:46-51.
de Lau L.M., Koudstaal P.J., Hofman A., et al. Subjective complaints precede Parkinson disease: the Rotterdam study. Arch Neurol. 2006;63:362-365.
Fife T.D., Iverson D.J., Lempert T., et al. Practice parameter: therapies for benign paroxysmal positional vertigo (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70:2067-2074.
Fife T.D., Tusa R.J., Furman J.M., et al. Assessment: vestibular testing techniques in adults and children: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2000;55:1431-1441.
Fisher C.M. Vertigo in cerebrovascular disease. Arch Otolaryngol. 1967;85:529-534.
Furman J.M., Baloh R.W., Hain T.C., et al. Assessment: electronystagmography. Report of the Therapeutics and Technology Assessment Subcommittee. Neurology. 1996;46:1763-1766.
Gianoli G., McWilliams S., Soileau J., et al. Posturographic performance in patients with the potential for secondary gain. Otolaryngol Head Neck Surg. 2000;122:11-18.
Hajioff D., Barr-Hamilton R.M., Colledge N.R., et al. Is electronystagmography of diagnostic value in the elderly? Clin Otolaryngol Allied Sci. 2002;27:27-31.
Halmagyi G.M., Aw S.T., Karlberg M., et al. Inferior vestibular neuritis. Ann N Y Acad Sci. 2002;956:306-313.
Halmagyi G.M., Curthoys I.S. A clinical sign of canal paresis. Arch Neurol. 1988;45:737-739.
Halmagyi G.M., Weber K.P., Aw S.T., et al. Impulsive testing of semicircular canal function. Prog Brain Res. 2008;171:187-194.
Houben M.M., Goumans J., van der Steen J. Recording three-dimensional eye movements: scleral search coils versus video oculography. Invest Ophthalmol Vis Sci. 2006;47:179-187.
Hufner K., Barresi D., Glaser M., et al. Vestibular paroxysmia: diagnostic features and medical treatment. Neurology. 2008;71:1006-1014.
Huppert D., Strupp M., Muckter H., et al. Which medication do I need to manage dizzy patients? Acta Otolaryngol. 2011. In Press
Jen J., Baloh R.H., Ishiyama A., et al. Dejerine-Sottas syndrome and vestibular loss due to a point mutation in the PMP22 gene. J Neurol Sci. 2005;237:21-24.
Jen J., Kim G.W., Baloh R.W. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62:17-22.
Jen J.C., Wang H., Lee H., et al. Suggestive linkage to chromosome 6q in families with bilateral vestibulopathy. Neurology. 2004;63:2376-2379.
Jen J.C., Yue Q., Karrim J., et al. Spinocerebellar ataxia type 6 with positional vertigo and acetazolamide responsive episodic ataxia. J Neurol Neurosurg Psychiatry. 1998;65:565-568.
Kattah J.C., Gujrati M. Familial positional downbeat nystagmus and cerebellar ataxia: clinical and pathologic findings. Ann N Y Acad Sci. 2005;1039:540-543.
Kattah J.C., Talkad A.V., Wang D.Z., et al. HINTS to diagnose stroke in the acute vestibular syndrome: three-step bedside oculomotor examination more sensitive than early MRI diffusion-weighted imaging. Stroke. 2009;40:3504-3510.
Katz J., Medwetsky L., Burkard R.F., et al. Handbook of clinical audiology, sixth ed, Philadelphia: Lippincott Williams & Wilkins, 2009.
Kerber K.A., Ishiyama G.P., Baloh R.W. A longitudinal study of oculomotor function in normal older people. Neurobiol Aging. 2006;27:1346-1353.
Kerber K.A., Jen J.C., Perlman S., et al. Late-onset pure cerebellar ataxia: differentiating those with and without identifiable mutations. J Neurol Sci. 2005;238:41-45.
Kerber K.A., Rasmussen P.A., Masaryk T.J., et al. Recurrent vertigo attacks cured by stenting a basilar artery stenosis. Neurology. 2005;65:962.
Lawson J., Fitzgerald J., Birchall J., et al. Diagnosis of geriatric patients with severe dizziness. J Am Geriatr Soc. 1999;47:12-17.
Lee H., Cho Y.W. A case of isolated nodulus infarction presenting as a vestibular neuritis. J Neurol Sci. 2004;221:117-119.
Lee H., Jen J.C., Wang H., et al. A genome-wide linkage scan of familial benign recurrent vertigo: linkage to 22q12 with evidence of heterogeneity. Hum Mol Genet. 2006;15:251-258.
Lee H., Sohn S.I., Cho Y.W., et al. Cerebellar infarction presenting isolated vertigo: frequency and vascular topographical patterns. Neurology. 2006;67:1178-1183.
Lemaire F.X., Feenstra L., Huygen P.L., et al. Progressive late-onset sensorineural hearing loss and vestibular impairment with vertigo (DFNA9/COCH): longitudinal analyses in a Belgian family. Otol Neurotol. 2003;24:743-748.
Lethbridge-Cejku M., Rose D., Vickerie J. Summary statistics for U.S. Adults: National Health Interview Survey, 2004. Vital Health Stat. 10, 2006.
Manolis E.N., Yandavi N., Nadol J.B.Jr., et al. A gene for non-syndromic autosomal dominant progressive postlingual sensorineural hearing loss maps to chromosome 14q12-13. Hum Mol Genet. 1996;5:1047-1050.
Marstrand J.R., Garde E., Rostrup E., et al. Cerebral perfusion and cerebrovascular reactivity are reduced in white matter hyperintensities. Stroke. 2002;33:972-976.
Merchant S.N., Adams J.C., Nadol J.B.Jr. Pathophysiology of Meniere’s syndrome: are symptoms caused by endolymphatic hydrops? Otol Neurotol. 2005;26:74-81.
Migliaccio A.A., Halmagyi G.M., McGarvie L.A., et al. Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain. 2004;127:280-293.
Minor L.B. Clinical manifestations of superior semicircular canal dehiscence. Laryngoscope. 2005;115:1717-1727.
Minor L.B., Schessel D.A., Carey J.P. Meniere’s disease. Curr Opin Neurol. 2004;17:9-16.
Minor L.B., Solomon D., Zinreich J.S., et al. Sound- and/or pressure-induced vertigo due to bone dehiscence of the superior semicircular canal. Arch Otolaryngol Head Neck Surg. 1998;124:249-258.
Moon I.S., Hain T.C. Delayed quick spins after vestibular nerve section respond to anticonvulsant therapy. Otol Neurotol. 2005;26:82-85.
Neuhauser H., Radtke A., von Brevern M., et al. Zolmitriptan for treatment of migrainous vertigo: a pilot randomized placebo-controlled trial. Neurology. 2003;60:882-883.
Neuhauser H.K., von Brevern M., Radtke A., et al. Epidemiology of vestibular vertigo: a neurotologic survey of the general population. Neurology. 2005;65:898-904.
Newman-Toker D.E., Cannon L.M., Stofferahn M.E., et al. Imprecision in patient reports of dizziness symptom quality: a cross-sectional study conducted in an acute care setting. Mayo Clin Proc. 2007;82:1329-1340.
Newman-Toker D.E., Kattah J.C., Alvernia J.E., et al. Normal head impulse test differentiates acute cerebellar strokes from vestibular neuritis. Neurology. 2008;70:2378-2385.
Nuti D., Mandala M., Broman A.T., et al. Acute vestibular neuritis: prognosis based upon bedside clinical tests (thrusts and heaves). Ann N Y Acad Sci. 2005;1039:359-367.
Oh A.K., Ishiyama A., Baloh R.W. Vertigo and the enlarged vestibular aqueduct syndrome. J Neurol. 2001;248:971-974.
Oh A.K., Lee H., Jen J.C., et al. Familial benign recurrent vertigo. Am J Med Genet. 2001;100:287-291.
Piirtola M., Era P. Force platform measurements as predictors of falls among older people–a review. Gerontology. 2006;52:1-16.
Robertson N.G., Lu L., Heller S., et al. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet. 1998;20:299-303.
Schmid-Priscoveanu A., Allum J.H. Infrared and video oculography–alternatives to electrooculography. HNO. 1999;47:472-478.
Strupp M., Arbusow V., Maag K.P., et al. Vestibular exercises improve central vestibulospinal compensation after vestibular neuritis. Neurology. 1998;51:838-844.
Strupp M., Jager L., Muller-Lisse U., et al. High resolution Gd-DTPA MR imaging of the inner ear in 60 patients with idiopathic vestibular neuritis: no evidence for contrast enhancement of the labyrinth or vestibular nerve. J Vestib Res. 1998;8:427-433.
Strupp M., Zingler V.C., Arbusow V., et al. Methylprednisolone, valacyclovir, or the combination for vestibular neuritis. N Engl J Med. 2004;351:354-361.
Takahashi H., Ishikawa K., Tsutsumi T., et al. A clinical and genetic study in a large cohort of patients with spinocerebellar ataxia type 6. J Hum Genet. 2004;49:256-264.
Tirelli G., Russolo M. 360-Degree canalith repositioning procedure for the horizontal canal. Otolaryngol Head Neck Surg. 2004;131:740-746.
Torres-Russotto D., Landau W.M., Harding G.W., et al. Calibrated finger rub auditory screening test (CALFRAST). Neurology. 2009;72:1595-1600.
Toyoda K., Hirano T., Kumai Y., et al. Bilateral deafness as a prodromal symptom of basilar artery occlusion. J Neurol Sci. 2002;193:147-150.
Vannucchi P., Giannoni B., Pagnini P. Treatment of horizontal semicircular canal benign paroxysmal positional vertigo. J Vestib Res. 1997;7:1-6.
von Brevern M., Zeise D., Neuhauser H., et al. Acute migrainous vertigo: clinical and oculographic findings. Brain. 2005;128:365-374.
Waterston J.A., Halmagyi G.M. Unilateral vestibulotoxicity due to systemic gentamicin therapy. Acta Otolaryngol. 1998;118:474-478.
Welgampola M.S., Colebatch J.G. Characteristics and clinical applications of vestibular-evoked myogenic potentials. Neurology. 2005;64:1682-1688.
Wiest G., Demer J.L., Tian J., et al. Vestibular function in severe bilateral vestibulopathy. J Neurol Neurosurg Psychiatry. 2001;71:53-57.
Yardley L., Burgneay J., Nazareth I., et al. Neuro-otological and psychiatric abnormalities in a community sample of people with dizziness: a blind, controlled investigation. J Neurol Neurosurg Psychiatry. 1998;65:679-684.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 37 Neuro-otology
Diagnosis and Management of Neuro-otological Disorders
Epidemiology of Vertigo, Dizziness, and Hearing Loss
Specific Disorders Causing Vertigo
Familial Hearing Loss and Vertigo
Common Causes of Nonspecific Dizziness
Common Presentations of Vertigo
Specific Disorders Causing Hearing Loss
Common Presentations of Hearing Loss
Laboratory Investigations in Diagnosis and Management
Historical Background
Accounts of dizziness and vertigo can be found in the writings of ancient Egyptian and Greek physicians. However, prior to the late 19th century, not much was known about the causes of dizziness or hearing loss, and as a result quackery was commonplace. Patients complaining of dizziness or vertigo were usually grouped together with epileptic seizures and stroke under the rubric of “apoplectiform cerebral congestion,” meaning too much blood to the brain. As a result, common treatments included bleeding, leeching, cupping, and purging. In 1861, Prosper Meniere was the first to recognize the association of vertigo with hearing loss and thus to localize the symptom to the inner ear (Baloh, 2001). Although not well received initially, his discovery provided the basis for later studies on the physiology and pathology of the vestibular system.
The advent of modern neuroimaging in the late 1970s and 1980s greatly expanded our understanding of causes of dizziness and vertigo. Prior to this time, stroke was considered an exceedingly rare cause of vertigo (Fisher, 1967). Though it remains a controversial topic even today, infarctions within the cerebellum and brainstem have been identified on imaging studies in patients with isolated vertigo. Imaging studies continue to lead to new discoveries of causes of vertigo, as demonstrated by the recently described disorder of superior canal dehiscence (SCD). But the most common causes of vertigo—Meniere disease, BPPV, and vestibular neuritis—still have no identifiable imaging characteristics.
Over the last 25 years, our understanding of the mechanisms for the common neuro-otological disorders has been greatly enhanced. BPPV can now be readily identified and cured at the bedside with a simple positional maneuver, and variants have also been described (Aw et al., 2005; Fife et al., 2008). The head-thrust test can be used at the bedside to identify a vestibular nerve lesion, and because of this it has particular utility in helping distinguish vestibular neuritis from a posterior fossa stroke (Halmagyi and Curthoys, 1988; Kattah et al., 2009; Newman-Toker et al., 2008; Nuti et al., 2005). Controversies regarding Meniere disease have been clarified, and medical and surgical treatments have improved (Minor et al., 2004). It is now clear that patients with recurrent episodes of vertigo without hearing loss, a condition once called vestibular Meniere disease, do not actually have Meniere disease.
Migraine is now recognized as an important cause of dizziness, even in patients without simultaneous headaches. In fact, benign recurrent vertigo (patients with recurrent episodes of vertigo without accompanying auditory symptoms or other neurological features) is usually a migraine equivalent (Oh et al., 2001b). The disorder of SCD was only recently described and provides important insight into the physiology of the vestibular system (Minor, 2005). A more detailed description of the rotational vertebral artery syndrome has led to appreciation of the high metabolic demands of the inner ear and its susceptibility to ischemia (Choi et al., 2005). Genetic research has identified ion channel dysfunction in disorders such as episodic ataxia and familial hemiplegic migraine, and patients with these disorders also commonly report vertigo (Jen et al., 2004a). It is hoped that identifying specific genes causing vertigo syndromes will lead to a better understanding of the mechanisms and also create the opportunity to develop specific treatments in the future.
Epidemiology of Vertigo, Dizziness, and Hearing Loss
A recent population-based telephone survey in Germany showed nearly 30% of the population had experienced moderate to severe dizziness (Neuhauser et al., 2005). Though most subjects reported nonspecific forms of dizziness, nearly a quarter had true vertigo. Dizziness is more common among females and older people and has important healthcare utilization implications; up to 80% of patients with dizziness seek medical care at some point. In the United States, the National Centers for Health Statistics report 7.5 million annual ambulatory visits to physician offices, hospital outpatient departments, and emergency departments (EDs) for dizziness, making it one of the most common principal complaints (Burt and Schappert, 2004).
Hearing loss affects approximately 16% of adults (age >18 years) in the United States (Lethbridge-Cejku et al., 2006). Men are more commonly affected than women, and the prevalence of hearing loss increases dramatically with age, so that by age 75, nearly 50% of the population reports hearing loss. Hearing loss is an important cause of disability. The most common type of hearing loss is sensorineural, and both idiopathic presbycusis and noise-induced forms are common etiologies. Bothersome tinnitus is less frequent in the U.S. population, with about 3% reporting it, although this increases to about 9% for subjects older than 65 (Adams et al., 1999). The most common type of tinnitus is a high-pitched ringing in both ears.
Normal Anatomy and Physiology
The inner ear is composed of a fluid-filled sac enclosed by a bony capsule with an anterior cochlear part, central chamber (vestibule), and a posterior vestibular part (Fig. 37.1). Endolymph fills up the fluid-filled sac and is separated by a membrane from the perilymph. These fluids primarily differ in their composition of potassium and sodium, with the endolymph resembling intracellular fluid with a high potassium and low sodium content, and perilymph resembling extracellular fluids with a low potassium and high sodium content. Perilymph communicates with the cerebrospinal fluid (CSF) through the cochlear aqueduct.
Fig. 37.1 Anatomy of the inner ear. CSF, Cerebrospinal fluid.
(From Baloh, R.W., 1998. Dizziness, Hearing Loss, and Tinnitus. F.A. Davis Company, Philadelphia, Figure 6, p. 16.)
The cochlea senses sound waves after they travel through the external auditory canal and are amplified by the tympanic membrane and ossicles of the middle ear (Baloh and Kerber, 2011). The stapes, the last of three ossicles in the middle ear, contacts the oval window, which directs the forces associated with sound waves along the basilar membrane of the cochlea. These forces stimulate the hair cells, which in turn generate neural signals in the auditory nerve. The auditory nerve enters the lateral brainstem at the pontomedullary junction and synapses in the cochlear nucleus. The trapezoid body is the major decussation of the auditory pathway, but many fibers do not cross to the contralateral side. Signals then travel to the superior olivary complex. Some projections travel from the superior olivary complex to the inferior colliculus through the lateral lemnisci, and others terminate in one of the nuclei of the lateral lemniscus. Next, fibers travel to the ipsilateral medial geniculate body, and then auditory radiations pass through the posterior limb of the internal capsule to reach the auditory cortex of the temporal lobe.
The peripheral vestibular system is composed of three semicircular canals, the utricle and saccule, and the vestibular component of the eighth cranial nerve (Baloh and Kerber, 2011). Each semicircular canal has a sensory epithelium called the crista; the sensory epithelium of the utricle and saccule is called the macule. The semicircular canals sense angular movements, and the utricle and saccule sense linear movements. Two of the semicircular canals (anterior and posterior) are oriented in the vertical plane nearly orthogonal to each other; the third canal is oriented in the horizontal plane (horizontal canal). The crista of each canal is primarily activated by movement occurring in the plane of that canal. When the hair cells of these organs are stimulated, the signal is transferred to the vestibular nuclei via the vestibular portion of cranial nerve VIII. Signals originating from the horizontal semicircular canal then pass via the medial longitudinal fasciculus along the floor of the fourth ventricle to the abducens nuclei in the middle brainstem and the ocular motor complex in the rostral brainstem. The anterior (also referred to as the superior) and posterior canal impulses pass from the vestibular nuclei to the ocular motor nucleus and trochlear nucleus triggering eye movements roughly in the plane of each canal. A key feature is that once vestibular signals leave the vestibular nuclei they divide into vertical, horizontal, and torsional components. As a result, a lesion of central vestibular pathways can cause a pure vertical, pure torsional, or pure horizontal nystagmus.
The primary vestibular afferent nerve fibers maintain a constant baseline firing rate of action potentials. When the baseline rate from each ear is symmetrical (or an asymmetry has been centrally compensated), the eyes remain stationary. With an uncompensated asymmetry in the firing rate, either resulting from increased or decreased activity on one side, slow ocular deviation results. By turning the head to the right, the baseline firing rate of the horizontal canal is physiologically altered, causing an increased firing rate on the right side and a decreased firing rate on the left side (Fig. 37.2). The result is a slow deviation of the eyes to the left. In an alert subject, this slow deviation is regularly interrupted by quick movements in the opposite direction (nystagmus) so the eyes do not become pinned to one side. In a comatose patient, only the slow component is seen because the brain cannot generate the corrective fast components.
The plane in which the eyes deviate as a result of vestibular stimulation depends on the combination of canals that are stimulated (Table 37.1). If only the posterior semicircular canal on one side is stimulated (as occurs with BPPV), a vertical-torsional deviation of the eyes can be observed, which is followed by a fast corrective response generated by the conscious brain in the opposite direction. However, if the horizontal canal is the source of stimulation (as occurs with the horizontal canal variant of BPPV), a horizontal deviation with a slight torsional component (because this canal is slightly off the horizontal plane) results. If the vestibular nerve is lesioned (vestibular neuritis) or stimulated (vestibular paroxysmia), a horizontal greater than torsional nystagmus is seen that is the vector sum of all three canals—the two vertical canals on one side cancel each other out.
History of Present Illness
The history and physical examination provide the most important information when evaluating patients complaining of dizziness (Colledge et al., 1996; Lawson et al., 1999). Often, patients have difficulty describing the exact symptom experienced, so the onus is on the clinician to elicit pertinent information. The first step is to define the symptom. No clinician should ever be satisfied to record the complaint simply as “dizziness.” For patients unable to provide a more detailed description of the symptom, the physician can ask the patient to place their symptom into one of the following categories: movement of the environment (vertigo), lightheadedness, or strictly imbalance without an abnormal head sensation. Because patient descriptions about dizziness can be unreliable and inconsistent (Newman-Toker et al., 2007), other details about the symptom become equally important. The physician should also ask the following questions: Is the symptom constant or episodic, are there accompanying symptoms, how did it begin (gradual, sudden, etc.), and were there aggravating or alleviating factors? If episodic, what was the duration and frequency of attacks, and were there triggers? Table 37.2 displays the key distinguishing features of common causes of dizziness. One key point is that any type of dizziness may worsen with position changes, but some disorders such as BPPV only occur after position change.
Physical Examination
General Neurological Examination
The general neurological examination is very important in patients complaining of dizziness, because dizziness can be the earliest symptom of a neurodegenerative disorder (de Lau et al., 2006) and can also be an important symptom of stroke, tumor, demyelination, or other pathologies of the nervous system.
Neuro-otological Examination
Ocular Motor
The first step in assessing ocular motor function is to search for spontaneous involuntary movements of the eyes. The examiner asks the patient to look straight ahead while observing for nystagmus or saccadic intrusions. Nystagmus is characterized by a slow- and fast-phase component and is classified as either spontaneous, gaze-evoked, or positional. The direction of nystagmus is conventionally described by the direction of the fast phase, which is the direction it appears to be “beating” toward. Recording whether the nystagmus is vertical, horizontal, torsional, or a mixture of these provides important localizing information. Spontaneous nystagmus can have either a peripheral or central pattern. Although central lesions can mimic a “peripheral” pattern of nystagmus (Lee and Cho, 2004; Newman-Toker et al., 2008), some very unusual and unlikely circumstances are required for peripheral lesions to cause “central” patterns of nystagmus. A peripheral pattern of spontaneous nystagmus is unidirectional, that is, the eyes beat only to one side (Video 37.1). Peripheral spontaneous nystagmus never changes direction. It is usually a horizontal greater than torsional pattern because of the physiology of the asymmetry in firing rates within the peripheral vestibular system whereby the vertical canals cancel each other out. The prominent horizontal component results from the unopposed horizontal canal. Other characteristics of peripheral spontaneous nystagmus are suppression with visual fixation, increase in velocity with gaze in the direction of the fast phase, and decrease with gaze in the direction opposite of the fast phase. Some patients are able to suppress this nystagmus so well at the bedside, or have partially recovered from the initiating event, that spontaneous nystagmus may only appear by removing visual fixation. Several simple bedside techniques can be used to remove the patient’s ability to fixate. Frenzel glasses are designed to remove visual fixation by using +30 diopter lenses. An ophthalmoscope can be used to block fixation. While the fundus of one eye is being viewed, the patient is asked to cover the other eye. Probably the simplest technique involves holding a blank sheet of paper close to the patient’s face (so as to block visual fixation) and observing for spontaneous nystagmus from the side.
Ocular flutter. Spontaneous back-and-forth saccades without an intersaccadic delay are seen.
Gaze Testing
Gaze-evoked downbeating nystagmus. Downbeating nystagmus occurs with gaze to either side.
Smooth Pursuit
Smooth pursuit refers to the voluntary movement of the eyes used to track a target moving at a low velocity. It functions to keep the moving object on the fovea to maximize vision. Though characteristically a very smooth movement at low frequency and velocity testing, smooth pursuit inevitably breaks down when tested at high frequencies and velocities. Though smooth pursuit often becomes impaired with advanced age, a recent study found no significant decline in smooth pursuit in a group of healthy elderly individuals (>75 years) tested yearly for at least 9 years (Kerber et al., 2006). Patients with impaired smooth pursuit require frequent small saccades to keep up with the target, thus the term saccadic pursuit is used to describe this finding (see Video 37.3). Abnormalities of smooth pursuit occur as the result of disorders throughout the CNS and with tranquilizing medicines, alcohol, inadequate concentration or vision, and fatigue. Patients with diffuse cortical disease, basal ganglia disease, or diffuse cerebellar disease consistently have bilaterally impaired smooth pursuit. Patients with early or mild cerebellar degenerative disorders may have markedly impaired smooth pursuit with mild or minimal truncal ataxia as the only findings.
Vestibular Nerve Examination
Often omitted as part of the cranial nerve examination in general neurology texts, important localizing information can be obtained about the functioning of the vestibular nerve at the bedside. A unilateral or bilateral vestibulopathy can be identified using the head-thrust test (Halmagyi et al., 2008) (Fig. 37.3 and Video 37.6). To perform the head-thrust test, the physician stands directly in front of the patient, who is seated on the exam table. The patient’s head is held in the examiner’s hands, and the patient is instructed to focus on the examiner’s nose. The head is then quickly moved about 5 to 10 degrees to one side. In patients with normal vestibular function, the VOR results in movement of the eyes in the direction opposite the head movement. Therefore the patient’s eyes remain on the examiner’s nose after the sudden movement. The test is repeated in the opposite direction. If the examiner observes a corrective saccade bringing the patient’s eyes back to the examiner’s nose after the head thrust, impairment of the VOR in the direction of the head movement is identified. Rotating the head slowly back and forth (the doll’s eye test) also induces compensatory eye movements, but both the visual and vestibular systems are activated by this low-velocity test, so a patient with complete vestibular function loss and normal visual pursuit will have normal-appearing compensatory eye movements on the doll’s eye test. This slow rotation of the head, however, is helpful in a comatose patient who is not able to generate voluntary visual tracking eye movements. Slowly rotating the head can also be a helpful test in patients with impairment of the smooth-pursuit system, because smooth movements of the eyes during slow rotation of the head indicates an intact VOR, whereas continued saccadic movements during slow rotation indicates an accompanying deficit of the VOR (Migliaccio et al., 2004).
Positional Testing
Positional testing can help identify peripheral or central causes of vertigo. The most common positional vertigo, BPPV, is caused by free-floating calcium carbonate debris, usually in the posterior semicircular canal, occasionally in the horizontal canal, or rarely in the anterior canal. The characteristic burst of upbeat torsional nystagmus is triggered in patients with BPPV by a rapid change from an erect sitting position to supine head-hanging left or head-hanging right (the Dix-Hallpike test) (Video 37.7). When present, the nystagmus is usually only triggered in one of these positions. A burst of nystagmus in the opposite direction (downbeat torsional) occurs when the patient resumes the sitting position. A repositioning maneuver can be used to liberate the clot of debris from the posterior canal. We use the modified Epley maneuver (Fig. 37.4 and Video 37.8), which is more than 80% effective in treating patients with posterior canal BPPV, compared to 10% effectiveness of a sham procedure (Fife et al., 2008). The key feature of this maneuver is the roll across in the plane of the posterior canal so that the clot rotates around the posterior canal and out into the utricle. Once the clot enters the utricle, it may reattach to the membrane, dissolve, or may even remain free-floating in the utricle, but the debris no longer disrupts semicircular canal function. Recurrences are common, however.
If the debris is in the horizontal canal, direction-changing horizontal nystagmus is seen. Patients are tested for the horizontal canal variant of BPPV by turning the head to each side while lying in the supine position. The nystagmus can be either paroxysmal geotropic (beating toward the ground) or persistent apogeotropic nystagmus (beating away from the ground). In the case of geotropic nystagmus, the debris is in the posterior segment (or “long arm”) of the horizontal canal, whereas the debris is in the anterior segment (or “short arm”) when apogeotropic nystagmus is triggered. When geotropic nystagmus is triggered, the side with the stronger nystagmus is the involved side. However, when apogeotropic nystagmus is observed, the involved side is generally opposite the side of the stronger nystagmus. With the geotropic variant, the debris can be removed from the canal by rolling the patient (barbecue fashion) toward the normal side. Other repositioning maneuvers for horizontal canal BPPV include the Gufoni maneuver and the “forced prolonged position” (Fife et al., 2008; Vannucchi et al., 1997). In cases of the apogeotropic variant, performing the barbeque maneuver toward the affected side can convert the nystagmus to geotropic because it moves the particles from the short arm of the canal to the long arm. Once the nystagmus is converted to geotropic, the typical treatments for the geotropic variant are used.
Positional testing can also trigger central types of nystagmus (usually persistent downbeating), which may be the most prominent examination finding in patients with disorders like Chiari malformation or cerebellar ataxia (Kattah and Gujrati, 2005; Kerber et al., 2005a). Central positional nystagmus can mimic the nystagmus of horizontal canal BPPV. Positional nystagmus may also be prominent in patients with migraine-associated dizziness (von Brevern et al., 2005).
Auditory Examination
Finger rubs at different intensities and distances from the ear are a rapid, reliable, and valid screening test for hearing loss in the frequency range of speech (Torres-Russotto et al., 2009). If a patient can hear a faint finger rub stimulus at a distance of 70 cm (approximately one arm’s length) from one ear, then a hearing loss on that side—defined by a gold-standard audiogram threshold of greater than 25 dB at 1000, 2000, and 4000 Hz—is highly unlikely. On the other hand, if a patient cannot hear a strong finger rub stimulus at 70 cm, a hearing loss on that side is highly likely. The whisper test can also be used to assess hearing at the bedside (Bagai et al., 2006). For this test, the examiner stands behind the patient to prevent lip reading and occludes and masks the non–test ear, using a finger to rub and close the external auditory canal. The examiner then whispers a set of three to six random numbers and letters. Overall, the patient is considered to have passed the screening test if they repeat at least 50% of the letters and numbers correctly. The Weber and Rinne tests are commonly used bedside tuning fork tests. To perform these, a tuning fork (256 Hz or 512 Hz) is gently struck on a hard rubber pad, the elbow, or the knee about two-thirds of the way along the tine. To conduct the Weber test, the base of the vibrating fork is placed on the vertex (top or crown of the head), bridge of the nose, upper incisors, or forehead. The patient is asked if the sound is heard and whether it is heard in the middle of the head or in both ears equally, toward the left, or toward the right. In a patient with normal hearing, the tone is heard centrally. In asymmetrical or a unilateral hearing impairment, the tone lateralizes to one side. Lateralization indicates an element of conductive impairment in the ear in which the sound localizes, a sensorineural impairment in the contralateral ear, or both. The Rinne test compares the patient’s hearing by air conduction with that by bone conduction. The fork is first held against the mastoid process until the sound fades. It is then placed 1 inch from the ear. Normal subjects can hear the fork about twice as long by air as by bone conduction. If bone is greater than air conduction, a conductive hearing loss is suggested.
Specific Disorders Causing Vertigo
Peripheral Vestibular Disorders
Peripheral vestibular disorders are important for neurologists to understand because they are common, readily identified at the bedside, and often missed by frontline physicians (see Table 37.2).
Vestibular Neuritis
A common presentation to the ED or outpatient clinic is the rapid onset of severe vertigo, nausea, vomiting, and imbalance. The symptoms gradually resolve over several days, but some symptoms can persist for months. The etiology of this disorder is probably viral, because the course is generally benign and self-limited, it occurs in young healthy individuals, and occasionally occurs in epidemics. Histopathological studies provide evidence of a peripheral vestibular localization and support the etiology of a viral cause. A viral etiology is also the likely cause of most cases of Bell palsy and sudden sensorineural hearing loss. The key to the diagnosis of vestibular neuritis is recognizing the peripheral vestibular pattern of nystagmus and identifying a positive head-thrust test in the setting of a rapid onset of vertigo without other neurological symptoms. Magnetic resonance imaging studies (MRIs) are usually normal in patients with vestibular neuritis (Strupp et al., 1998b). The course of vestibular neuritis is self-limited, and the mainstay of treatment is symptomatic. A recent study of patients with vestibular neuritis showed improvement of peripheral vestibular function, as measured by caloric testing at 1 year, after receiving methylprednisolone within 3 days of onset, compared to placebo (Strupp et al., 2004). A formal vestibular rehabilitation program can help patients compensate for the vestibular lesion (Strupp et al., 1998a).
