[level-membership-for-internal-medicine-category]
14 Nervous system
The neurological history
The essentials of history taking have been covered elsewhere, but there are some points particularly salient to neurological conditions. The repertoire of neurological symptoms is actually quite limited, though perhaps larger than that of other specialties. Box 14.1 lists the usual ones, and each should be specifically enquired about in taking a neurological history.
Box 14.1 Neurological symptoms
 Cognitive symptoms, especially memory impairment
Cognitive symptoms, especially memory impairment



 Alteration of perception, including déjà vu
Alteration of perception, including déjà vu








 More than double vision (polyopia, palinopsia)
More than double vision (polyopia, palinopsia)
 Oscillopsia (a visual sensation of stationary objects swaying back and forth)
Oscillopsia (a visual sensation of stationary objects swaying back and forth)





 Abnormal movements of muscles (including cramp, fasciculations)
Abnormal movements of muscles (including cramp, fasciculations)
 Abnormal movements of parts of the body (including tremor, dystonia, myoclonus)
Abnormal movements of parts of the body (including tremor, dystonia, myoclonus)

 Impairment of control of limbs
Impairment of control of limbs







The three most common causes of attacks of reduced consciousness or awareness that lead to neurological consultations are: neurocardiogenic syncope, epilepsy and psychogenic non-epileptic attacks. It is far more informative to hear a description from a witness than to request potentially misleading and inappropriate investigations. In the setting of the emergency department, obtaining the witness’s account may involve contact by telephone; this is time well invested. Table 14.1 summarizes some points which help to distinguish syncope from seizures.
Table 14.1 Points which help to distinguish syncope from seizures involving loss of consciousness. Minor injury, incontinence and sleepiness after the event do not distinguish well
The symptoms of focal seizure activity which precede a secondarily generalized attack may or may not be remembered after the attack is over
Vocalization at onset of generalized convulsion (GC)
Postictal amnesia – paramedical staff present when patient ‘comes round’, or patient ‘comes round’ in hospital (although he walked to the ambulance)
The most frequent symptom leading to neurological referral is headache. It is also a common reason for attending an emergency department. The vast majority of headaches are not caused by life-threatening disorders such as aneurysmal subarachnoid haemorrhage, meningitis or brain tumour, but are caused by common primary headache syndromes, particularly migraine. Table 14.2 outlines some of the features which may help to distinguish headaches of different sorts.
Table 14.2 Headaches: points to consider in the history
| Aspect of history | Feature | Diagnosis |
|---|---|---|
| Region/location | Focal, retro-orbital | Cluster headache |
| Migraine | ||
| Retro-orbital lesion | ||
| Focal, frontal | Sinus pathology | |
| Unilateral | Migraine | |
| Chronic paroxysmal hemicrania (CPH) | ||
| Hemicrania continua | ||
| Generalized | Migraine | |
| Tension-type headache | ||
| Temporal aspects | >50% of days | Chronic daily headache (chronic migraine; tension-type headache) |
| Attacks of hours/days | Migraine | |
| Attacks of up to 1 hour | Cluster headache | |
| Many attacks, lasting minutes | CPH | |
| Attacks of seconds | Trigeminal neuralgia | |
| Worse on waking | Raised intracranial pressure | |
| Sleep apnoea | ||
| New, acute/subacute onset | Meningitis | |
| Abscess | ||
| Encephalitis | ||
| Explosive onset, severe | Subarachnoid haemorrhage (SAH) | |
| Character and severity | Tight band around head, bland, featureless, not very severe | Tension-type headache |
| Throbbing, moderately severe | Migraine | |
| Extremely severe, constant | Cluster headache | |
| Severe, stabbing, lancinating | Trigeminal neuralgia | |
| Provoking/relieving factors | Provoked by alcohol | Migraine |
| Cluster headache | ||
| Occur at night, start in sleep | Migraine | |
| Cluster headache | ||
| Hypnic headache | ||
| Relieved by sleep | Migraine | |
| Triggered by touching or moving the face | Trigeminal neuralgia | |
| Caused by cough | Chiari malformation | |
| Idiopathic cough headache | ||
| Exertion | Exacerbates migraine | |
| Benign exertional headache | ||
| Orgasm | Benign coital headache (SAH has to be excluded in a severe single attack) | |
| Associated symptoms | Nausea, vomiting, photophobia, phonophobia | Migraine |
| Migraine aura (visual, sensory, etc.) | Migraine | |
| Neck stiffness, photophobia, vomiting, symptoms of fever | Meningitis | |
| Tear production ipsilateral to a unilateral headache | Migraine | |
| Cluster headache | ||
| SUNCT* | ||
| Ipsilateral conjunctival injection | Cluster headache | |
| SUNCT* | ||
| Visual obscurations | Raised intracranial pressure with papilloedema | |
| Persistent focal neurological symptoms | Intracranial lesion | |
| General health | Indications of systemic neoplasia | Metastasis |
| Polymyalgia and weight loss | Giant cell arteritis |
* SUNCT, short-lasting, unilateral, neuralgiform headache attacks with conjunctival injection and tearing (a rare disorder).
Most organic neurological disorders which give rise to sensory symptoms involve structural or functional damage to nerves somewhere, whether it be in peripheral nerves, nerve roots, spinal cord or brain. Hypersensitivity (hyperaesthesia) to all modalities of sensation is therefore improbable or impossible. Patients who appear to have very sensitive skin as a result of a lesion (e.g. herpes zoster radiculitis) have combinations of paraesthesia, hypoaesthesia, dysaesthesia, allodynia, hyperalgesia and hyperpathia. Some of these terms are not without their ambiguities. Table 14.3 provides a definition for each.
Table 14.3 Nomenclature of cutaneous sensory symptoms
| Hypoaesthesia | Reduced cutaneous sensation of any modality |
| Paraesthesia | Spontaneous abnormal sensation including, tingling, pins and needles and pain |
| Neuralgia | Pain in the distribution of a nerve or nerve root |
| Dysaesthesia | An abnormal perception of a sensory stimulus, e.g. touch causes tingling or pain |
| Allodynia | Pain caused by a stimulus that does not normally cause pain |
| Hyperalgesia | An abnormally intense perception of a mildly painful stimulus |
| Hyperpathia | Perseveration, augmentation and, on occasion, spread of pain |
| The pain threshold is normal or sometimes high | |
| In this, the threshold for perceiving pain may be raised and there may be delay in perceiving a painful stimulus, but once perceived, the pain is severe and prolonged and may spread | |
| Hyperaesthesia | An ambiguous term, best avoided |
Cranial nerve examination
The optic (II) nerves
The optic nerve runs from the back of the globe of the eye to the apex of the orbit and into the skull through the optic canal to the optic chiasm, where it is joined by the optic nerve from the other eye. Directly above the optic chiasm is the hypothalamus. Directly below is the pituitary gland. The pituitary stalk runs from the hypothalamus to the pituitary gland just behind the optic chiasm, between the optic tracts. Sensory afferents from all points of the retina run in the nerve-fibre layer on the inner surface of the retina to enter the optic nerve at the optic disc. Fibres from the temporal retina (nasal visual half-field) are placed laterally while those from the nasal retina (temporal visual half-field) are medial. Fibres from the upper half of the retina run in the upper half of the optic nerve. At the optic chiasm, fibres from the nasal half of the retina (temporal visual half-field) cross (decussate) to the contralateral optic tract, while the fibres from the temporal half of the retina do not cross but proceed posteriorly into the ipsilateral optic tract (Fig. 14.1). Starting within the chiasm and continuing further posteriorly within the optic tract, fibres which convey matching information from each eye (i.e. homonymous fibres, representing equivalent parts in the temporal retina for one eye, nasal retina for the other eye) become aligned with each other. Thus, each optic nerve conveys information from its respective eye, but every part of the sensory visual system behind the optic chiasm on each side deals with vision for the contralateral binocular visual half-field.
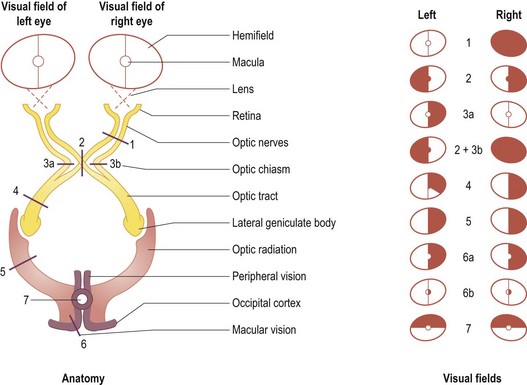
2: Optic chiasm lesion (bitemporal hemianopia).
3a or 3b: Uniocular nasal hemianopia (rare).
3a plus 3b: Binasal hemianopia (very rare).
4: Optic tract lesion (incongruous homonymous hemianopia).
5: Visual radiation (homonymous quadrantanopia or hemianopia).
6a: Occipital cortex lesion sparing the occipital pole (homonymous hemianopia with macular sparing).
6b: Occipital pole lesion (homonymous paracentral hemiscotoma).
7: A bilateral occipital cortex lesion (homonymous altitudinal hemianopia).
The pupils
Other common pupillary abnormalities of neurological relevance




The oculomotor (III), trochlear (IV) and abducens (VI) nerves – eye movements
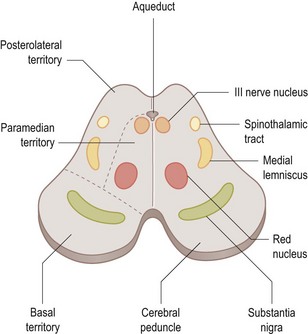
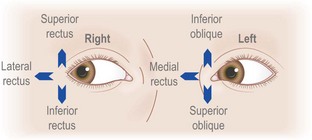
The nucleus for the third cranial nerve is in the midbrain (Fig. 14.2) and emerges ventrally (anteriorly), medial to the cerebral peduncle, passing forward through the cavernous sinus to the superior orbital fissure. In the orbit, the superior ramus supplies superior rectus and levator palpebrae superioris. The inferior ramus supplies inferior rectus, inferior oblique and medial rectus and parasympathetic fibres from the inferior ramus pass to the ciliary ganglion and thence to the ciliary muscle and the pupil sphincter.
The sixth nerve nucleus is beneath the floor of the fourth ventricle in the pons (Fig. 14.3). Nerve fibres run forward (ventrally) through the pons emerging at its lower border, then up the skull base and forward through the cavernous sinus to the superior orbital fissure and into the orbit to supply the lateral rectus muscle. The nerve is long, thin and very susceptible to dysfunction, most notably in the setting of raised intracranial pressure of any aetiology, which may give rise to either unilateral or bilateral sixth nerve lesions. This is referred to as a ‘false localizing sign’, since a focal mass lesion causing raised intracranial pressure may be remote from the sixth nerves and their nuclei or there may be no focal cause of the raised pressure at all (e.g. idiopathic intracranial hypertension).
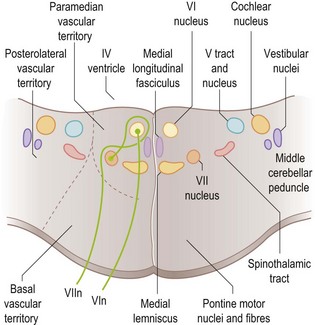
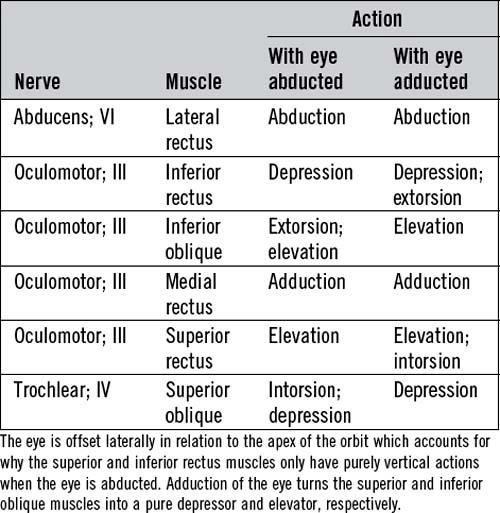
Table 14.4 and Figure 14.4 outline the actions of each eye muscle.
Examination of eye movements
Diplopia testing
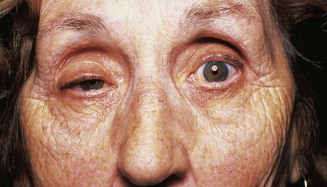
If a patient complains of double vision, first establish that it is true diplopia and not monocular diplopia. In patients who have obvious, easily visible paresis of movement of one or both eyes, the reason for diplopia is self-evident (Fig. 14.5).
Diplopia develops with even very subtle misalignment of the eyes, which cannot be seen on simple inspection. In this situation, if the ophthalmoparesis affects just one eye, it is possible to work out which eye muscles are underactive by diplopia testing. The true image is that generated by the eye with normal movements. The false image is that generated by the eye with the paretic muscle or muscles. For example, if a patient develops double vision on looking to the right, with horizontal separation of the images, the false image will be the one further out to the right. This is true whether it is the right eye which does not abduct adequately (right lateral rectus weakness) or if it is the left eye which does not adduct adequately (left medial rectus weakness). If this does not seem immediately clear, consider the extreme case: one eye moves, the other does not. An image (an examiner’s finger or a white pinhead) moves to the patient’s right. The image remains in the middle of the field of the eye which moves, but moves progressively to the right of the field of the eye which does not. The same rule applies in all directions of gaze. Diplopia is always maximal in the direction in which the weak muscle has its purest action (see Table 14.4).
The severity of the diplopia should be assessed in eight positions: looking to left and right, up and down, and obliquely up and down to the left and obliquely up and down to the right. To work out which muscle is underactive, where the diplopia is maximal, cover each eye in turn and get the patient to tell you which of the two images disappears. Inconsistent answers are common, however, and the assistance of an ophthalmologist or optometrist is frequently desirable. The features of lesions of the third, fourth and sixth cranial nerves are summarized in Table 14.5.
Table 14.5 The effects of lesions of the oculomotor (III), trochlear (IV) and abducens (VI) nerves
| Affected nerve | Signs | Comment |
|---|---|---|
| Oculomotor | Paresis of adduction (medial rectus) | The eye becomes abducted because of unopposed action of lateral rectus, and slightly depressed because of action of superior oblique |
| Paresis of elevation (superior rectus and inferior oblique) | The pure depressor action of superior oblique cannot be tested because the eye cannot be adducted | |
| Intorsion of the eye on attempted down gaze indicates intact trochlear nerve and superior oblique function | ||
| Paresis of depression (inferior rectus) | ||
| Ptosis due to paresis of levator palpebrae superioris | With complete ptosis, there is of course no diplopia | |
| Dilated, unreactive pupil | This feature is not present in pupil-sparing lesions (microvascular lesions of nucleus or nerve) | |
| Trochlear | Paresis of superior oblique | Extorsion of the eye due to unopposed action of inferior oblique leads to diplopia such that a vertical line looks V-shaped |
| The patient compensates with a head tilt to the side opposite the lesion, intact intorsion on that side tending to correct the diplopia | ||
| Abducens | Paresis of lateral rectus | Horizontal diplopia |
In assessing patients who have double vision, it is best first to establish which muscles appear to be weak, and then try to decide what the nature of the problem is likely to be, taking into consideration all the physical signs. Thus, impairment of eye movements in one eye in combination with proptosis of that eye may occur because of mechanical restriction of eye movements by an intraorbital lesion. Weakness of muscles in both eyes with different patterns of involvement of the muscles in the two eyes is likely to be due to a disorder of the muscles themselves (orbital myositis, thyroid eye disease) or due to ocular or generalized myasthenia. The pupils will not be involved. Bilateral, asymmetrical combinations of cranial nerve lesions are relatively uncommon (neoplastic infiltration, cranial polyneuritis). Bilateral sixth cranial nerve lesions are common, usually but not exclusively as a feature of raised intracranial pressure. Multiple oculomotor neuropathies in one eye direct attention to the superior orbital fissure and the cavernous sinus (see Box 14.2).
Box 14.2 Cranial nerve involvement in lesions of the cavernous sinus and superior orbital fissure
1 Lesion of the cavernous sinus (e.g. internal carotid aneurysm; internal carotid artery dissection; meningioma):
2 Lesion of the superior orbital fissure (e.g. Tolosa Hunt syndrome):
Horizontal gaze paresis; internuclear ophthalmoparesis
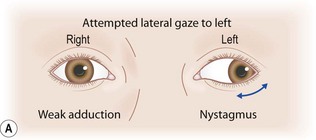
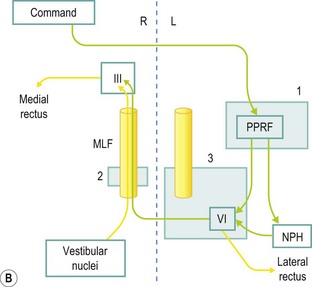
Neural control of voluntary lateral gaze to the right starts in the left cerebral hemisphere, such that a large left cerebral hemisphere lesion may be associated with failure of right gaze and a tendency for the eyes to deviate to the left (the side of the lesion). Output runs to the right paramedian pontine reticular formation (PPRF); hence,a right-sided pontine lesion may involve a right gaze paresis. Output from the right PPRF goes to the right sixth nerve nucleus, resulting in right eye abduction, and across, via the left medial longitudinal fasciculus (MLF), to the left third nerve nucleus, resulting in simultaneous left eye adduction. Attempted right gaze in the setting of a left MLF lesion results in abduction of the right eye, but failure of adduction of the left eye – an internuclear ophthalmoparesis (INO) (see Fig. 14.6). Bilateral MLF lesions give rise to bilateral INO, in which case, with lateral gaze in either direction, only the abducting eye moves normally. Commonly, nystagmus is seen in the abducting eye. The pathway for adducting both eyes for near vision is separate, and sometimes in bilateral INO preservation of adduction of the eyes for near vision can be demonstrated, proving that the problem is not bilateral medial rectus weakness.
Non-paralytic strabismus
Neurologists need to be able to recognize non-paralytic strabismus. Decompensation of a longstanding squint may be a cause of acquired diplopia, but this is an ophthalmological problem and it is covered in Chapter 19.
The trigeminal (V) nerve
Sensory component of the trigeminal nerve
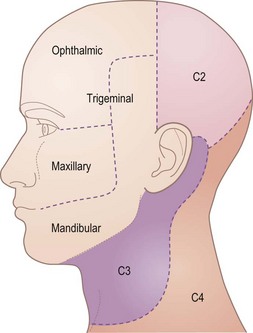
The primary sensory neurones are in the trigeminal ganglion, just behind the cavernous sinus at the apex of the petrous bone. Central projections run in the trigeminal nerve into the pons. Figure 14.7 shows the cutaneous distribution of the three divisions of the trigeminal nerve: ophthalmic (V1), maxillary (V2) and mandibular (V3). These nerves also mediate general sensation inside the mouth and nose and proprioception. The ophthalmic nerve passes through the cavernous sinus and superior orbital fissure. The maxillary nerve also passes through the cavernous sinus, but leaves the inside of the skull through the foramen rotundum. The mandibular nerve passes through the foramen ovale.
The facial (VII) nerve
The facial nerve nucleus is in the caudal pons, lying ventrolateral to the sixth cranial nerve nucleus (see Fig. 14.3). It receives upper motor neurone input from both cerebral hemispheres. Lower motor neurone fibres from the facial nucleus first pass round the sixth nerve nucleus, then emerge from the pons to form the facial nerve. From here, it travels laterally, adjacent to the eighth cranial nerve, to the internal auditory meatus, thence to the facial canal which has a relatively long and tortuous course through the skull, emerging at the stylomastoid foramen. The nerve then passes forward into the parotid gland and divides into branches which supply all the facial muscles and the platysma muscle on one side. In the facial canal, a branch of the facial nerve supplies the stapedius muscle.
The vestibular and cochlear (VIII) nerves
Testing the vestibular and cochlear nerves
The assessment of deafness is covered in detail elsewhere (Ch. 20). At the bedside, a crude assessment of hearing can be achieved by rubbing your index finger and thumb together close to the patient’s ear, or by whispering numbers close to his ear, with the contralateral ear occluded. Rinne’s test is good for distinguishing between conduction and sensorineural deafness, as long as you use the appropriate tuning fork (frequency 512 – not the lower frequency tuning fork (128) used for testing vibration sense).
The disorder variously called labyrinthitis and acute vestibular neuritis (the latter is regarded as being the more accurate designation) causes vertigo and peripheral vestibular nystagmus (see p. 293). A common symptom is positioning vertigo, most commonly benign paroxysmal positioning vertigo. Positioning vertigo and nystagmus are best elicited and tested by the Dix-Hallpike test (see Ch. 20).
The glossopharyngeal (IX) nerve
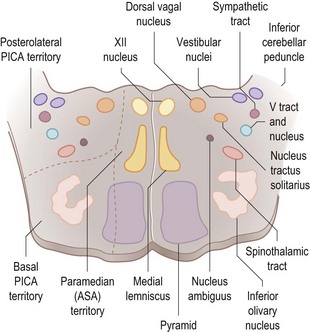
There are anatomical and functional relationships between the glossopharyngeal nerve, the vagus nerve and the cranial component of the accessory nerve. The nucleus ambiguus in the medulla (Fig. 14.8) contains the motor neurones which innervate striated muscle of the palate, pharynx, larynx and upper oesophagus, fibres running partly in the glossopharyngeal nerve, mainly in the vagus nerve and partly in the cranial portion of the accessory nerve. Situated more dorsally in the medulla, the dorsal motor nucleus of the vagus and the inferior salivatory nucleus (whose fibres join the glossopharyngeal nerve) contain preganglionic parasympathetic neurones which control glands and smooth muscle. Special visceral afferents (i.e. taste fibres from the intermediate nerve and the glossopharyngeal nerve) enter the solitary tract to end in the nucleus of the solitary tract in the medulla. General somatic sensory afferents in the glossopharyngeal and vagus nerves join trigeminal sensory nuclei.
The accessory (XI) nerve
Testing the accessory nerve
The left sternocleidomastoid muscle contributes to rotation of the head to the right and vice versa. Weakness of the left sternocleidomastoid is therefore assessed by asking the patient to turn his head to the right with force, while the examiner opposes the rotation with the left hand, pushing carefully against the right side of the face. Failure of contraction of the muscle can be seen. Weakness of the upper part of trapezius is one of the causes of winging of the scapula, the other being weakness of serratus anterior due to a lesion of the long thoracic nerve (see p. 305). Scapular winging due to trapezius weakness is most commonly caused by a lesion of the accessory nerve in the posterior triangle of the neck, often iatrogenic related to surgical excision of a lymph node.
The hypoglossal (XII) nerve
Testing the hypoglossal nerve
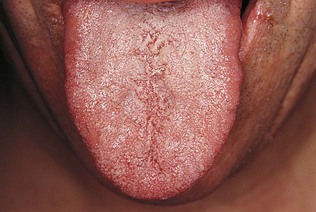
A unilateral hypoglossal nerve lesion in the acute stage leads to weakness of tongue muscles on the affected side. Attempted protrusion of the tongue results in deviation of the tongue toward the weak side. A chronic lesion will be associated with visible muscle atrophy of the affected side of the tongue. Bilateral wasting and weakness of the tongue with fasciculation is usually due to the progressive bulbar atrophy of motor neurone disease (Fig. 14.9).
Speech
Cerebellar (ataxic) dysarthria
Look carefully for nystagmus and other eye movement disorders, and for limb and gait ataxia.
Motor system
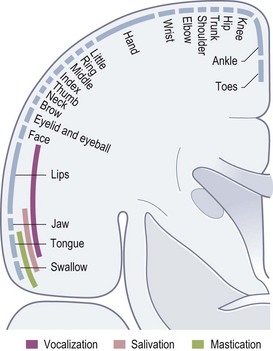
For the purposes of clinical neurological examination and interpretation of neurological signs, the anatomical considerations can be kept simple. The motor neurones whose axons terminate at neuromuscular junctions and which activate voluntary striated muscle contraction are situated in the anterior horns of the spinal cord grey matter and in motor cranial nerve nuclei. These are the lower motor neurones. Upper motor neurones are those situated in the cerebral cortex whose axons leave the cortex to control, directly or indirectly, the lower motor neurones. Some of these are in the somatotopically organized primary motor cortex (Fig. 14.10) in the precentral gyrus at the back of the frontal lobe, but the distribution of upper motor neurones in the cortex is much wider than just the primary motor cortex, and includes the supplementary motor area and premotor cortex in the frontal lobes. The cerebral control of posture and movement involves widely distributed cortical motor systems. Crucial also are circuits involving the basal ganglia and cerebellum. Output from the cortex is either direct to motor neurones via corticospinal and corticobulbar tracts or indirect via projections to the brainstem which has major influences by various pathways.
Motor system examination
Inspection
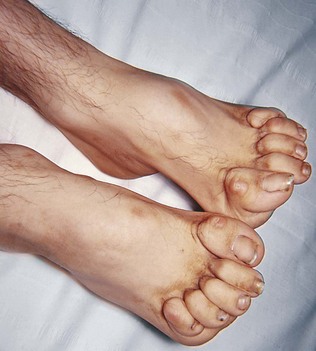
Pes cavus (Fig. 14.11) with clawing of the toes may be idiopathic or an indication of longstanding imbalance of muscle power in the feet (mainly weakness of intrinsic foot muscles), caused commonly by Charcot-Marie-Tooth disease, but also by other hereditary or congenital disorders of peripheral nerve or spinal cord. Contractures of joints can be a consequence of muscle disease, peripheral neuropathy or CNS disease. Scoliosis is common in neurofibromatosis, but may be a complication of other neurological disorders such as syringomyelia and Friedreich’s ataxia.
Testing muscle power
Clinical methods of recording the severity of muscle weakness have considerable shortcomings, but in neurological practice it is desirable to chart muscle strength in order to document progression or recovery and to gauge the effectiveness of therapeutic interventions. The Medical Research Council scale is widely used (Box 14.3), but a problem with this scale is that the vast majority of muscle weakness one will encounter is grade 4. It also performs badly for fingers and toes where gravity has scarcely any influence, so that grades 1, 2 and 3 are more or less identical. Many clinicians find a scale of normal power, mild, moderate and severe weakness and paralysis is practical for everyday use. Accurate quantitative myometry ought to be a way forward, but has hardly entered routine clinical practice.
Box 14.3 The Medical Research Council scale for grading muscle strength






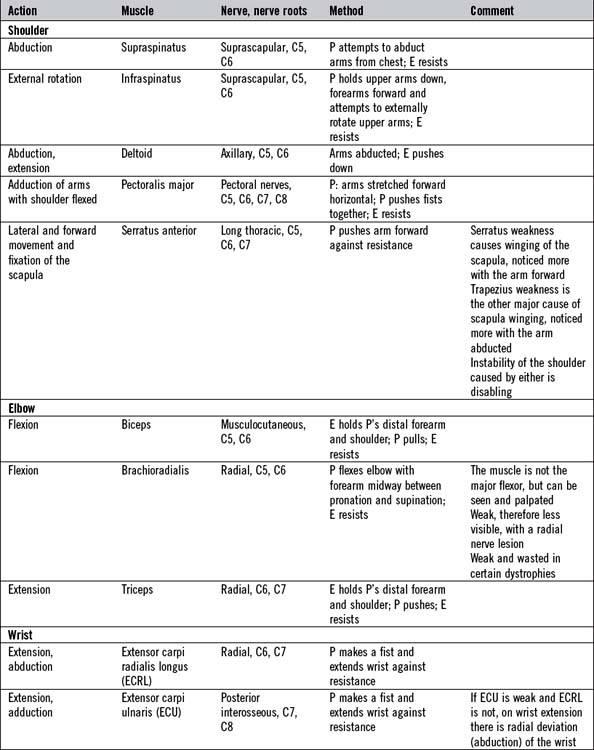
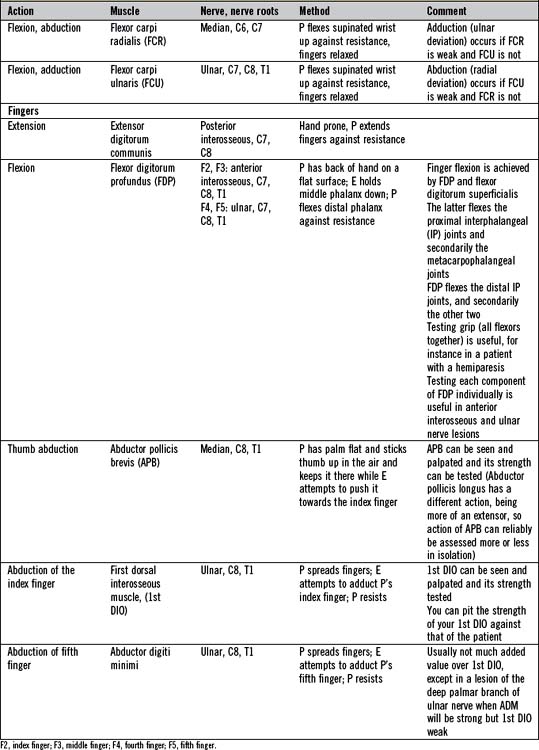
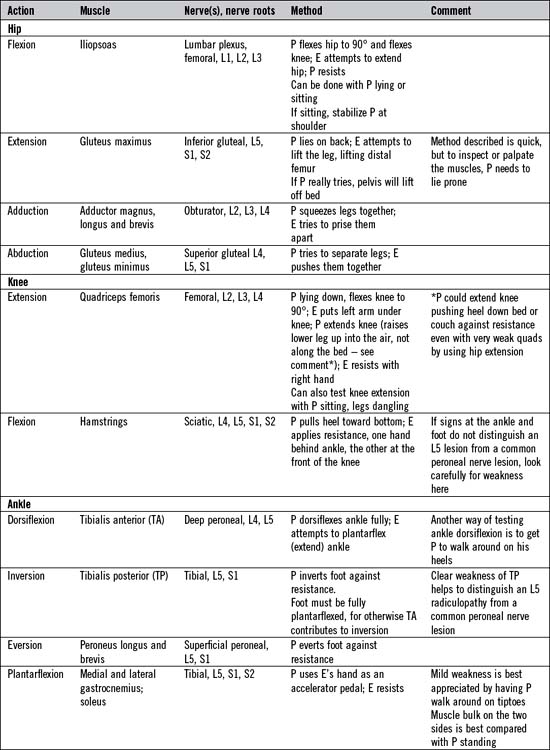
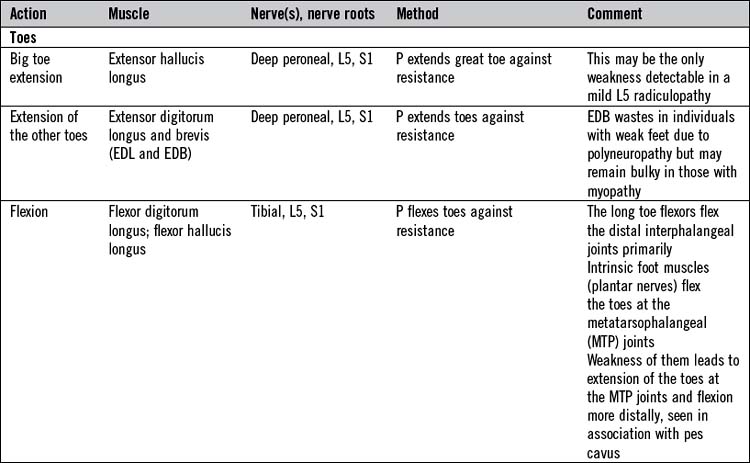
There is not the space here to describe in detail the testing of all the important individual muscles and muscle groups. Tables 14.6 and 14.7 represent a summary of commonly tested muscles and their actions, but a number of specific points are important:
 There are two tests in which muscle strength is routinely tested isotonically (i.e. the muscle shortens while lifting a constant load) as opposed to isometrically. Abdominal trunk muscles are tested by asking the patient to sit up from lying flat with his arms folded on his chest. Standing up from a sitting position or rising from a crouched position are good tests of hip and knee extension and spine extension muscles, and are useful in testing patients with myopathies. Normally these actions can be accomplished without use of the arms. Weak patients often have to resort to using their arms to push themselves up, either using surrounding furniture or, failing that,pushing themselves up with their arms on their own legs (Gowers’ sign; Fig. 14.12).
There are two tests in which muscle strength is routinely tested isotonically (i.e. the muscle shortens while lifting a constant load) as opposed to isometrically. Abdominal trunk muscles are tested by asking the patient to sit up from lying flat with his arms folded on his chest. Standing up from a sitting position or rising from a crouched position are good tests of hip and knee extension and spine extension muscles, and are useful in testing patients with myopathies. Normally these actions can be accomplished without use of the arms. Weak patients often have to resort to using their arms to push themselves up, either using surrounding furniture or, failing that,pushing themselves up with their arms on their own legs (Gowers’ sign; Fig. 14.12).





Cerebellar system
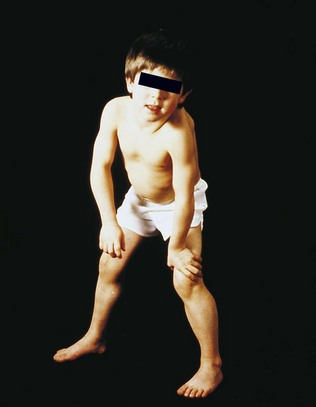
Reflexes
The tendon reflexes
Testing the tendon reflexes
Eliciting tendon reflexes is not completely straightforward and requires practice. The aim is to ensure that the tendon of the relevant muscle being tested is briskly and effectively struck. Accordingly, sufficient force must be delivered. For the biceps reflex, place your own finger over the tendon and strike your finger. Similarly the brachioradialis reflex can be elicited by applying the tendon hammer to your own fingers positioned just above the wrist on the lateral aspect of the radius bone. The other standard reflexes (triceps, knee (quadriceps) and ankle (gastrocnemii and soleus)) involve directly striking the tendon with the tendon hammer. The standard tendon reflexes examined are summarized in Table 14.8.
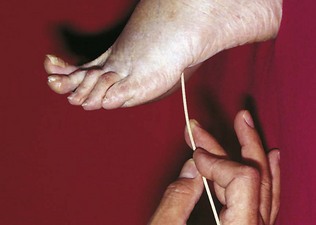
The plantar reflex
Testing the plantar reflex
The ideal instrument to use is an orange stick (Fig. 14.13). Hold the patient’s foot. Apply the orange stick to the sole of the foot on the lateral border just in front of the heel. Draw it forward towards the base of the fifth toe. This may suffice. Some continue round across the ball of the foot. It is best to press fairly firmly, warning the patient that this is not a pleasant test. Try to do the test effectively once or twice, not a large number of times. If you remain in doubt as to whether a patient’s response is extensor or not, then later, when testing pinprick sensation, test the dorsum of the toe in question. Involuntary extension of the toe towards the pin is an extensor response.
Movement disorders
Tremor
The common tremor disorders include enhanced physiological tremor, essential tremor, parkinsonian tremor, dystonic tremor, cerebellar (‘intention’) tremor, drug-induced tremor and psychogenic tremor (Table 14.9). Combinations of tremor and myoclonus are seen in encephalopathies, mainly in hospitalized patients.
| Tremor variety | Comment |
|---|---|
| Physiological |
Parkinson’s disease
The main features of PD are summarized in Box 14.4. Rigidity and tremor have been discussed above. Posture and gait are discussed below.







Impaired facial expression and generalized bradykinesia or akinesia are observations, but bradykinesia can also be tested formally, elements of the Unified Parkinson’s Disease Rating Scale being readily incorporated into routine clinical practice (see Box 14.5). Postural stability is tested by the pull test, done after testing gait.
Box 14.5 Tests for bradykinesia in Parkinson’s disease
3 Pronation-supination movements of the arms:
In this test, asymmetry with impaired amplitude on the more affected side is readily observed.
Other movement disorders (hyperkinetic movement disorders)
Generally these disorders are identified simply by history and by observation rather than any other component of examination. They are summarized in Table 14.10.
| Movement disorder | Characteristics |
|---|---|
| Parkinsonism | See text |
| Dystonia |
Chorea denotes irregular, brief, jerky, unrepetitive, unintentional movements, affecting differing parts randomly (unilateral in hemichorea)
In mild cases, the patient just looks fidgety
Many causes including metabolic, drug induced, autoimmune, neurodegenerative
Athetosis denotes slower, more writhing movements than chorea
The two often coexist, particularly in levodopa-induced dyskinesia
Sensation
The cell bodies of primary sensory neurones are in dorsal root ganglia and equivalent ganglia on cranial nerves (particularly the trigeminal ganglion). Their peripheral axons convey sensory information from sensory end organs in skin, muscle spindles and tendons. Their central axons project into the spinal cord or brainstem. Large diameter myelinated axons transmit proprioception, vibration and touch. Small diameter myelinated and unmyelinated axons transmit pain and temperature. The centrally projecting axons for proprioception and vibration sense enter the spinal cord and turn immediately to head rostrally in the posterior (dorsal) columns ipsilaterally, to synapse in the gracile and cuneate nuclei at the cervicomedullary junction. Axons from these nuclei cross in the medulla and proceed to the thalamus. In contrast, axons for pain and temperature, having entered the spinal cord, synapse in the dorsal horns. The axons of second-order sensory neurones cross over to the contralateral anteriorly (ventrally) situated spinothalamic tracts and run rostrally all the way to the thalamus. Touch sensation travels by both routes. Sensory pathways from the trigeminal sensory nuclei cross in the brainstem to project to the contralateral thalamus. From the thalamus, third-order neurones project via the posterior limb of the internal capsule to the primary sensory cortex in the parietal lobe, which is somatotopically organized (Fig. 14.14), akin to the primary motor cortex.
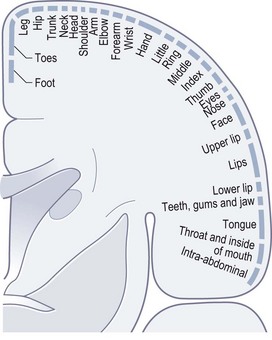
Cutaneous sensory examination
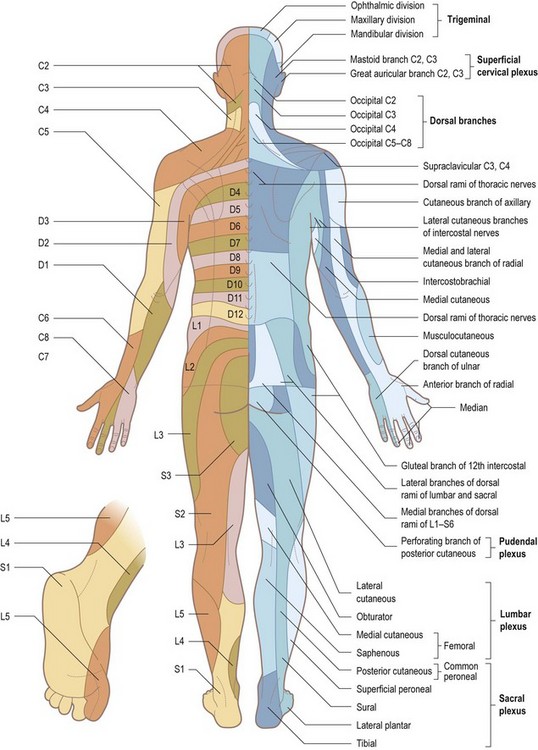
Disorganized sensory testing tends to confuse both patient and examiner. With cutaneous sensory testing, the goal is to see whether any hypoaesthesia conforms to a meaningful pattern (see Table 14.13 and Figs 14.15 and 14.16). Defining an area of hypoaesthesia is almost invariably better achieved by starting testing within the area of hypoaesthesia and moving the stimulus out into areas where sensation is normally perceived.
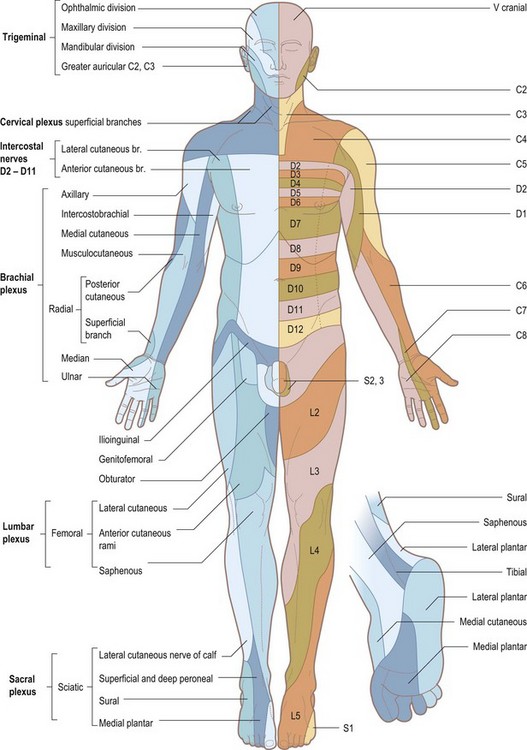
Figure 14.15 Anterior view to show the segmental innervation of the skin (left) and the peripheral nerve supply (right).
Proprioception
Testing joint position sense
Pseudoathetosis
Patients with impaired joint position sense in their fingers may exhibit so-called pseudoathetosis: outstretched fingers drift up or down or to the side when the patient has his eyes closed. This is because, proprioception having failed, vision is the only means by which the patient knows where his fingers are and can keep them in one place (Fig. 14.17).

Posture and gait
First, simply observe the patient standing. Look for abnormalities of posture. Established PD gives a kyphotic posture (Fig. 14.18). Neck extension weakness causes forward flexion of the neck; in extreme cases, a state of drop head is apparent. Axial dystonia may be observed. Muscular dystrophy may be associated with hyperlordosis. Check the patient’s balance and do Romberg’s test (see above). Then ask the patient to walk a short distance, turn round and walk back again. Mild gait ataxia can be brought out by getting the patient to walk heel to toe. If relevant, test for retropulsion (the pull test; see p. 312).

Figure 14.18 Parkinson’s disease, showing the typical rigid, flexed posture involving the trunk and limbs. The face is impassive.
Table 14.11 provides a summary of important kinds of neurological gait disorders. Sometimes it can be difficult to be certain that a patient with a frontal gait disorder does not also have PD. Patients with severe osteoarthritis of the hips sometimes have a gait disorder which looks deceptively like a myopathic gait. Finally, as in other domains, a functional gait disorder does not necessarily mean that there is no organic disease.
| Gait | Characteristics |
|---|---|
| Myopathic (proximal, symmetrical myopathy) |
A consequence of foot drop (unilateral or bilateral) – common peroneal nerve lesion, motor polyneuropathy, e.g. Charcot-Marie-Tooth disease, distal myopathy
The leg has to be lifted abnormally high to make a step
With mild ankle dorsiflexion weakness, the foot slaps onto the ground as weight is transferred onto the heel
Midline cerebellar lesion (cerebellar vermis): wide-based unsteady gait with irregular sized paces and potential to topple in any direction; there may be little or no limb incoordination
Cerebellar hemisphere lesion: the patient veers towards the side of the lesion; there is ipsilateral limb incoordination
The spastic arm is held adducted at the shoulder with the elbow flexed and the forearm in front of the chest with flexion at the wrist and fingers
Swinging of the affected arm is reduced
The spastic leg is stiff with impaired knee flexion and ankle dorsiflexion, with abduction of the leg to bring the plantarflexed foot round and forward when taking a step (circumduction)
This term has a number of mainly less satisfactory synonyms, including gait apraxia, lower body parkinsonism, atherosclerotic or vascular parkinsonism, marche à petits pas
Patterns of motor and sensory signs
Patterns of motor signs
Upper motor neurone disorders affecting the leg
The lesion may be in the spinal cord or brain. Weakness affects the flexor muscles more than extensor muscles; hence the abnormal findings are of weakness in hip flexion (iliopsoas), knee flexion (hamstrings) and ankle dorsiflexion (tibialis anterior). Spasticity usually predominates in the hip adductors and the extensor muscles of the leg and foot (gluteus maximus, quadriceps and calf muscles). These circumstances account for the characteristics of the hemiparetic and paraparetic gait (see Table 14.11).
Upper motor neurone disorders affecting the arm and hand
The distribution of weakness in upper motor neurone lesions of the limbs is often referred to as ‘pyramidal’, a useful but not completely accurate term. Table 14.12 summarizes where in the CNS lesions need to be to produce the various categories of upper motor neurone lesions.
| Pattern of weakness | Site of lesion |
|---|---|
| Spastic monoparesis |
High cervical spinal cord lesion, e.g. C3/C4 disc
Diffuse bilateral cerebral hemisphere disease affecting motor pathways bilaterally, e.g. small vessel cerebrovascular disease
Brainstem disease affecting the corticospinal tracts bilaterally, e.g. basilar artery territory stroke (which causes locked-in syndrome)
ACA, anterior cerebral artery; MCA, middle cerebral artery.
Patterns of sensory loss (Table 14.13)
The diagnosis of lesions of individual sensory peripheral nerves or the sensory component of mixed nerves requires knowledge of anatomy, in this case the area of cutaneous sensation mediated by the nerve in question (see Table 14.13 and Figs 14.15 and 14.16). In lesions affecting mixed nerves, sensory symptoms with or without signs commonly precede motor symptoms and signs. Thus, a clear description by a patient of sensory loss in ulnar nerve distribution provides compelling evidence for an ulnar neuropathy even if there are no signs. The right combination of motor and sensory signs allows the diagnosis of a mononeuropathy to be made. Nerve conduction studies and electromyography may contribute to the assessment of the nature of the lesion (precise location, extent of conduction block, extent of axonal damage, subclinical involvement of other nerves, presence of an underlying subclinical polyneuropathy) but is not necessary for making the diagnosis if there are clear-cut signs.
| Clinical sensory findings | Site of lesion |
|---|---|
| Hemianaesthesia (all modalities) Incomplete, e.g. face and arm Complete |
Infarction affecting the anterior (ventral) thoracic spinal cord
Involvement of spinothalamic tracts bilaterally
If the infarction extends down to the lumbar spinal cord, the lower limb reflexes are lost
Otherwise they become brisk because of the paraplegia caused by bilateral corticospinal tract involvement
Entrapment neuropathies of mixed nerves may present with sensory features only, or with motor and sensory features, which makes clinical diagnosis easier
For some nerves, you have to rely solely on clinical acumen – the nerve is not amenable to neurophysiological investigation, e.g. lateral cutaneous nerve of the thigh
Familiarity with the areas of cutaneous sensation mediated by nerve roots (dermatomes – see Figs 14.15 and 14.16) is important for two reasons. First, it enables lesions of individual nerve roots to be diagnosed. Second, the level of a spinal cord lesion can be ascertained to some extent (see below).
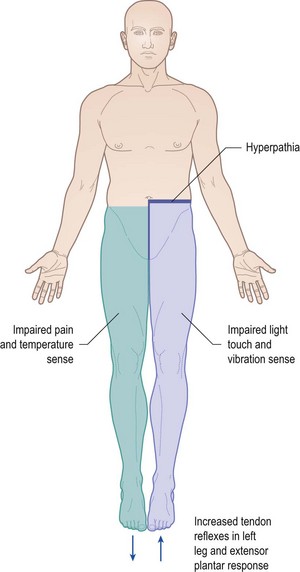
Brown-Séquard syndrome
This syndrome refers to the signs resulting from a focal lesion affecting the right or left half of the spinal cord, such as an intramedullary inflammatory or an extramedullary compressive lesion. Disruption of the descending corticospinal tract on the affected side results in an ipsilateral upper motor neurone disorder below the level of the lesion (commonly in one leg but a high cervical lesion would affect the arm as well). Involvement of the posterior columns on the affected side results in ipsilateral loss of vibration sense and joint position sense in the leg or arm and leg below the lesion. The spinothalamic tracts on the side of the lesion are affected, but sensory input into the spinothalamic tracts comes from pain and temperature afferents on the opposite side which cross over on entering the spinal cord, so loss of pinprick and temperature sensation occurs below the level of the lesion on the opposite side (Fig. 14.19). Some patients with mild lesions comment that while it is one leg which drags and feels weak and stiff, it is the other leg which has impaired sensation.
Cognitive function
The two major syndromes of cognitive impairment, which may be accompanied by no neurological signs, are dementia and delirium. They are covered in Chapter 6. In this section, emphasis is placed on cognitive deficits which, to some extent, have localizing value and which may be seen in patients with focal cerebral lesions as well as those with more generalized neurodegenerative disorders.
Amnesia
Memory is a distributed cognitive function. Dense amnesia requires bilateral hemisphere lesions, but those lesions may be very focal – bilateral thalamic infarction, or damage to the mamillothalamic tracts in Wernicke’s encephalopathy, can cause devastating amnesia – and the anatomy of memory in each hemisphere is localized and well defined. Furthermore, unilateral lesions, such as a posterior cerebral artery occlusion causing temporal lobe infarction, do have discernible effects on memory. Normal episodic memory (memory of events which have been personally experienced) depends on integrity of the hippocampi and entorhinal cortex in the temporal lobes, the fornices, the thalami and the mamillary bodies. Anterior temporal lobes subserve semantic memory (memory for facts, learning and general knowledge). Verbal memory is dependent on the left temporal lobe, visual memory on the right temporal lobe. Some important causes of amnesia are listed in Table 14.14. Amnesia emerges mainly from the history taken from an informant rather than from the patient. Episodic memory may be assessed by testing recall of a name and address that the patient has registered; and by testing the patient’s recall of events from his past, both recent and distant.
| Cause | Comment |
|---|---|
| Acute | |
| Transient global amnesia | The name describes well what happens; the pathology remains obscure; imaging indicates a hippocampal process |
| Transient epileptic amnesia | Here amnesia is the seizure |
| Epilepsy more generally | Temporal lobe seizures may involve amnesia; postictal amnesia follows any generalized convulsion |
| Closed head injury | Concussion involves retrograde and anterograde post-traumatic amnesia |
| Drugs: benzodiazepines, alcohol | Amnesia can occur without significant impairment of alertness |
| Chronic | |
| Hippocampal damage | |
| Herpes simplex encephalitis | Bilateral temporal lobe damage |
| Limbic encephalitis | Bilateral temporal lobe damage |
| Cerebral anoxia | |
| Temporal lobectomy with a lesion in the other temporal lobe | Should not happen nowadays in modern practice because of improvements in preoperative assessment |
| Bilateral posterior cerebral artery territory infarction | |
| Closed head injury | Bilateral temporal lobe damage is common |
| Early Alzheimer’s disease | Later there are other prominent cognitive deficits as well |
| Diencephalic damage | |
| Korsakoff’s syndrome | This is what results from untreated Wernicke’s encephalopathy, due to thiamine deficiency |
| Third ventricular tumours and cysts | Potentially affecting the fornices and thalami bilaterally |
| Bilateral thalamic infarction | Either two separate events or, if the bilateral paramedian thalamosubthalamic arteries of Percheron branch from one common vessel (a not uncommon variant), one stroke |
| Subarachnoid haemorrhage from anterior communicating artery aneurysm | |
The list on the left is adapted from John R. Hodges 1994 Cognitive Assessment for Clinicians, Oxford University Press.
Investigations
Imaging
Headache
 Migraine, tension-type headaches, medication overuse headaches and post-traumatic headaches can be diagnosed with a high degree of accuracy without scans.
Migraine, tension-type headaches, medication overuse headaches and post-traumatic headaches can be diagnosed with a high degree of accuracy without scans.


 Patients with serious intracranial disorders rarely just have headache.
Patients with serious intracranial disorders rarely just have headache.
 Occasionally migraine really is ‘symptomatic’, i.e. caused by a structural intracranial disorder.
Occasionally migraine really is ‘symptomatic’, i.e. caused by a structural intracranial disorder.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
14 Nervous system
The neurological history
The essentials of history taking have been covered elsewhere, but there are some points particularly salient to neurological conditions. The repertoire of neurological symptoms is actually quite limited, though perhaps larger than that of other specialties. Box 14.1 lists the usual ones, and each should be specifically enquired about in taking a neurological history.
Box 14.1 Neurological symptoms
Cognitive symptoms, especially memory impairment
Alteration of perception, including déjà vu
More than double vision (polyopia, palinopsia)
Oscillopsia (a visual sensation of stationary objects swaying back and forth)
Abnormal movements of muscles (including cramp, fasciculations)
Abnormal movements of parts of the body (including tremor, dystonia, myoclonus)
Impairment of control of limbs
The three most common causes of attacks of reduced consciousness or awareness that lead to neurological consultations are: neurocardiogenic syncope, epilepsy and psychogenic non-epileptic attacks. It is far more informative to hear a description from a witness than to request potentially misleading and inappropriate investigations. In the setting of the emergency department, obtaining the witness’s account may involve contact by telephone; this is time well invested. Table 14.1 summarizes some points which help to distinguish syncope from seizures.
Table 14.1 Points which help to distinguish syncope from seizures involving loss of consciousness. Minor injury, incontinence and sleepiness after the event do not distinguish well
The symptoms of focal seizure activity which precede a secondarily generalized attack may or may not be remembered after the attack is over
Vocalization at onset of generalized convulsion (GC)
Postictal amnesia – paramedical staff present when patient ‘comes round’, or patient ‘comes round’ in hospital (although he walked to the ambulance)
The most frequent symptom leading to neurological referral is headache. It is also a common reason for attending an emergency department. The vast majority of headaches are not caused by life-threatening disorders such as aneurysmal subarachnoid haemorrhage, meningitis or brain tumour, but are caused by common primary headache syndromes, particularly migraine. Table 14.2 outlines some of the features which may help to distinguish headaches of different sorts.
Table 14.2 Headaches: points to consider in the history
| Aspect of history | Feature | Diagnosis |
|---|---|---|
| Region/location | Focal, retro-orbital | Cluster headache |
| Migraine | ||
| Retro-orbital lesion | ||
| Focal, frontal | Sinus pathology | |
| Unilateral | Migraine | |
| Chronic paroxysmal hemicrania (CPH) | ||
| Hemicrania continua | ||
| Generalized | Migraine | |
| Tension-type headache | ||
| Temporal aspects | >50% of days | Chronic daily headache (chronic migraine; tension-type headache) |
| Attacks of hours/days | Migraine | |
| Attacks of up to 1 hour | Cluster headache | |
| Many attacks, lasting minutes | CPH | |
| Attacks of seconds | Trigeminal neuralgia | |
| Worse on waking | Raised intracranial pressure | |
| Sleep apnoea | ||
| New, acute/subacute onset | Meningitis | |
| Abscess | ||
| Encephalitis | ||
| Explosive onset, severe | Subarachnoid haemorrhage (SAH) | |
| Character and severity | Tight band around head, bland, featureless, not very severe | Tension-type headache |
| Throbbing, moderately severe | Migraine | |
| Extremely severe, constant | Cluster headache | |
| Severe, stabbing, lancinating | Trigeminal neuralgia | |
| Provoking/relieving factors | Provoked by alcohol | Migraine |
| Cluster headache | ||
| Occur at night, start in sleep | Migraine | |
| Cluster headache | ||
| Hypnic headache | ||
| Relieved by sleep | Migraine | |
| Triggered by touching or moving the face | Trigeminal neuralgia | |
| Caused by cough | Chiari malformation | |
| Idiopathic cough headache | ||
| Exertion | Exacerbates migraine | |
| Benign exertional headache | ||
| Orgasm | Benign coital headache (SAH has to be excluded in a severe single attack) | |
| Associated symptoms | Nausea, vomiting, photophobia, phonophobia | Migraine |
| Migraine aura (visual, sensory, etc.) | Migraine | |
| Neck stiffness, photophobia, vomiting, symptoms of fever | Meningitis | |
| Tear production ipsilateral to a unilateral headache | Migraine | |
| Cluster headache | ||
| SUNCT* | ||
| Ipsilateral conjunctival injection | Cluster headache | |
| SUNCT* | ||
| Visual obscurations | Raised intracranial pressure with papilloedema | |
| Persistent focal neurological symptoms | Intracranial lesion | |
| General health | Indications of systemic neoplasia | Metastasis |
| Polymyalgia and weight loss | Giant cell arteritis |
* SUNCT, short-lasting, unilateral, neuralgiform headache attacks with conjunctival injection and tearing (a rare disorder).
Most organic neurological disorders which give rise to sensory symptoms involve structural or functional damage to nerves somewhere, whether it be in peripheral nerves, nerve roots, spinal cord or brain. Hypersensitivity (hyperaesthesia) to all modalities of sensation is therefore improbable or impossible. Patients who appear to have very sensitive skin as a result of a lesion (e.g. herpes zoster radiculitis) have combinations of paraesthesia, hypoaesthesia, dysaesthesia, allodynia, hyperalgesia and hyperpathia. Some of these terms are not without their ambiguities. Table 14.3 provides a definition for each.
Table 14.3 Nomenclature of cutaneous sensory symptoms
| Hypoaesthesia | Reduced cutaneous sensation of any modality |
| Paraesthesia | Spontaneous abnormal sensation including, tingling, pins and needles and pain |
| Neuralgia | Pain in the distribution of a nerve or nerve root |
| Dysaesthesia | An abnormal perception of a sensory stimulus, e.g. touch causes tingling or pain |
| Allodynia | Pain caused by a stimulus that does not normally cause pain |
| Hyperalgesia | An abnormally intense perception of a mildly painful stimulus |
| Hyperpathia | Perseveration, augmentation and, on occasion, spread of pain |
| The pain threshold is normal or sometimes high | |
| In this, the threshold for perceiving pain may be raised and there may be delay in perceiving a painful stimulus, but once perceived, the pain is severe and prolonged and may spread | |
| Hyperaesthesia | An ambiguous term, best avoided |
Cranial nerve examination
The optic (II) nerves
The optic nerve runs from the back of the globe of the eye to the apex of the orbit and into the skull through the optic canal to the optic chiasm, where it is joined by the optic nerve from the other eye. Directly above the optic chiasm is the hypothalamus. Directly below is the pituitary gland. The pituitary stalk runs from the hypothalamus to the pituitary gland just behind the optic chiasm, between the optic tracts. Sensory afferents from all points of the retina run in the nerve-fibre layer on the inner surface of the retina to enter the optic nerve at the optic disc. Fibres from the temporal retina (nasal visual half-field) are placed laterally while those from the nasal retina (temporal visual half-field) are medial. Fibres from the upper half of the retina run in the upper half of the optic nerve. At the optic chiasm, fibres from the nasal half of the retina (temporal visual half-field) cross (decussate) to the contralateral optic tract, while the fibres from the temporal half of the retina do not cross but proceed posteriorly into the ipsilateral optic tract (Fig. 14.1). Starting within the chiasm and continuing further posteriorly within the optic tract, fibres which convey matching information from each eye (i.e. homonymous fibres, representing equivalent parts in the temporal retina for one eye, nasal retina for the other eye) become aligned with each other. Thus, each optic nerve conveys information from its respective eye, but every part of the sensory visual system behind the optic chiasm on each side deals with vision for the contralateral binocular visual half-field.
2: Optic chiasm lesion (bitemporal hemianopia).
3a or 3b: Uniocular nasal hemianopia (rare).
3a plus 3b: Binasal hemianopia (very rare).
4: Optic tract lesion (incongruous homonymous hemianopia).
5: Visual radiation (homonymous quadrantanopia or hemianopia).
6a: Occipital cortex lesion sparing the occipital pole (homonymous hemianopia with macular sparing).
6b: Occipital pole lesion (homonymous paracentral hemiscotoma).
7: A bilateral occipital cortex lesion (homonymous altitudinal hemianopia).
The pupils
Other common pupillary abnormalities of neurological relevance
The oculomotor (III), trochlear (IV) and abducens (VI) nerves – eye movements
The nucleus for the third cranial nerve is in the midbrain (Fig. 14.2) and emerges ventrally (anteriorly), medial to the cerebral peduncle, passing forward through the cavernous sinus to the superior orbital fissure. In the orbit, the superior ramus supplies superior rectus and levator palpebrae superioris. The inferior ramus supplies inferior rectus, inferior oblique and medial rectus and parasympathetic fibres from the inferior ramus pass to the ciliary ganglion and thence to the ciliary muscle and the pupil sphincter.
The sixth nerve nucleus is beneath the floor of the fourth ventricle in the pons (Fig. 14.3). Nerve fibres run forward (ventrally) through the pons emerging at its lower border, then up the skull base and forward through the cavernous sinus to the superior orbital fissure and into the orbit to supply the lateral rectus muscle. The nerve is long, thin and very susceptible to dysfunction, most notably in the setting of raised intracranial pressure of any aetiology, which may give rise to either unilateral or bilateral sixth nerve lesions. This is referred to as a ‘false localizing sign’, since a focal mass lesion causing raised intracranial pressure may be remote from the sixth nerves and their nuclei or there may be no focal cause of the raised pressure at all (e.g. idiopathic intracranial hypertension).
Table 14.4 and Figure 14.4 outline the actions of each eye muscle.
Examination of eye movements
Diplopia testing
If a patient complains of double vision, first establish that it is true diplopia and not monocular diplopia. In patients who have obvious, easily visible paresis of movement of one or both eyes, the reason for diplopia is self-evident (Fig. 14.5).
Diplopia develops with even very subtle misalignment of the eyes, which cannot be seen on simple inspection. In this situation, if the ophthalmoparesis affects just one eye, it is possible to work out which eye muscles are underactive by diplopia testing. The true image is that generated by the eye with normal movements. The false image is that generated by the eye with the paretic muscle or muscles. For example, if a patient develops double vision on looking to the right, with horizontal separation of the images, the false image will be the one further out to the right. This is true whether it is the right eye which does not abduct adequately (right lateral rectus weakness) or if it is the left eye which does not adduct adequately (left medial rectus weakness). If this does not seem immediately clear, consider the extreme case: one eye moves, the other does not. An image (an examiner’s finger or a white pinhead) moves to the patient’s right. The image remains in the middle of the field of the eye which moves, but moves progressively to the right of the field of the eye which does not. The same rule applies in all directions of gaze. Diplopia is always maximal in the direction in which the weak muscle has its purest action (see Table 14.4).
The severity of the diplopia should be assessed in eight positions: looking to left and right, up and down, and obliquely up and down to the left and obliquely up and down to the right. To work out which muscle is underactive, where the diplopia is maximal, cover each eye in turn and get the patient to tell you which of the two images disappears. Inconsistent answers are common, however, and the assistance of an ophthalmologist or optometrist is frequently desirable. The features of lesions of the third, fourth and sixth cranial nerves are summarized in Table 14.5.
Table 14.5 The effects of lesions of the oculomotor (III), trochlear (IV) and abducens (VI) nerves
| Affected nerve | Signs | Comment |
|---|---|---|
| Oculomotor | Paresis of adduction (medial rectus) | The eye becomes abducted because of unopposed action of lateral rectus, and slightly depressed because of action of superior oblique |
| Paresis of elevation (superior rectus and inferior oblique) | The pure depressor action of superior oblique cannot be tested because the eye cannot be adducted | |
| Intorsion of the eye on attempted down gaze indicates intact trochlear nerve and superior oblique function | ||
| Paresis of depression (inferior rectus) | ||
| Ptosis due to paresis of levator palpebrae superioris | With complete ptosis, there is of course no diplopia | |
| Dilated, unreactive pupil | This feature is not present in pupil-sparing lesions (microvascular lesions of nucleus or nerve) | |
| Trochlear | Paresis of superior oblique | Extorsion of the eye due to unopposed action of inferior oblique leads to diplopia such that a vertical line looks V-shaped |
| The patient compensates with a head tilt to the side opposite the lesion, intact intorsion on that side tending to correct the diplopia | ||
| Abducens | Paresis of lateral rectus | Horizontal diplopia |
In assessing patients who have double vision, it is best first to establish which muscles appear to be weak, and then try to decide what the nature of the problem is likely to be, taking into consideration all the physical signs. Thus, impairment of eye movements in one eye in combination with proptosis of that eye may occur because of mechanical restriction of eye movements by an intraorbital lesion. Weakness of muscles in both eyes with different patterns of involvement of the muscles in the two eyes is likely to be due to a disorder of the muscles themselves (orbital myositis, thyroid eye disease) or due to ocular or generalized myasthenia. The pupils will not be involved. Bilateral, asymmetrical combinations of cranial nerve lesions are relatively uncommon (neoplastic infiltration, cranial polyneuritis). Bilateral sixth cranial nerve lesions are common, usually but not exclusively as a feature of raised intracranial pressure. Multiple oculomotor neuropathies in one eye direct attention to the superior orbital fissure and the cavernous sinus (see Box 14.2).
Box 14.2 Cranial nerve involvement in lesions of the cavernous sinus and superior orbital fissure
1 Lesion of the cavernous sinus (e.g. internal carotid aneurysm; internal carotid artery dissection; meningioma):
2 Lesion of the superior orbital fissure (e.g. Tolosa Hunt syndrome):
Horizontal gaze paresis; internuclear ophthalmoparesis
Neural control of voluntary lateral gaze to the right starts in the left cerebral hemisphere, such that a large left cerebral hemisphere lesion may be associated with failure of right gaze and a tendency for the eyes to deviate to the left (the side of the lesion). Output runs to the right paramedian pontine reticular formation (PPRF); hence,a right-sided pontine lesion may involve a right gaze paresis. Output from the right PPRF goes to the right sixth nerve nucleus, resulting in right eye abduction, and across, via the left medial longitudinal fasciculus (MLF), to the left third nerve nucleus, resulting in simultaneous left eye adduction. Attempted right gaze in the setting of a left MLF lesion results in abduction of the right eye, but failure of adduction of the left eye – an internuclear ophthalmoparesis (INO) (see Fig. 14.6). Bilateral MLF lesions give rise to bilateral INO, in which case, with lateral gaze in either direction, only the abducting eye moves normally. Commonly, nystagmus is seen in the abducting eye. The pathway for adducting both eyes for near vision is separate, and sometimes in bilateral INO preservation of adduction of the eyes for near vision can be demonstrated, proving that the problem is not bilateral medial rectus weakness.
Non-paralytic strabismus
Neurologists need to be able to recognize non-paralytic strabismus. Decompensation of a longstanding squint may be a cause of acquired diplopia, but this is an ophthalmological problem and it is covered in Chapter 19.
The trigeminal (V) nerve
Sensory component of the trigeminal nerve
The primary sensory neurones are in the trigeminal ganglion, just behind the cavernous sinus at the apex of the petrous bone. Central projections run in the trigeminal nerve into the pons. Figure 14.7 shows the cutaneous distribution of the three divisions of the trigeminal nerve: ophthalmic (V1), maxillary (V2) and mandibular (V3). These nerves also mediate general sensation inside the mouth and nose and proprioception. The ophthalmic nerve passes through the cavernous sinus and superior orbital fissure. The maxillary nerve also passes through the cavernous sinus, but leaves the inside of the skull through the foramen rotundum. The mandibular nerve passes through the foramen ovale.
[/not-level-membership-for-internal-medicine-category]
