[level-membership-for-pediatrics-category]
Chapter 521 Nephrotic Syndrome
Etiology
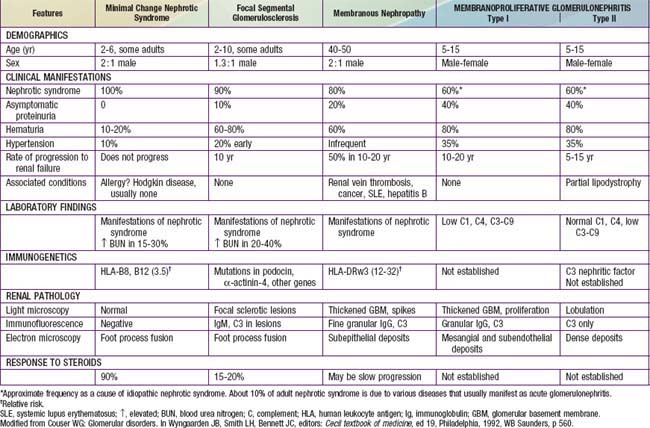
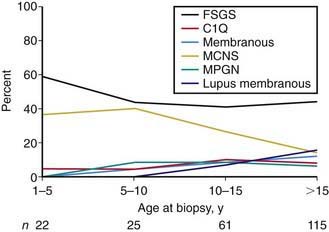
Most children with nephrotic syndrome have a form of primary or idiopathic nephrotic syndrome (Table 521-1). Glomerular lesions associated with idiopathic nephrotic syndrome include minimal change disease (the most common), focal segmental glomerulosclerosis, membranoproliferative glomerulonephritis, membranous nephropathy and diffuse mesangial proliferation (Table 521-2). These etiologies have different age distributions (Fig. 521-1).
Table 521-1 CAUSES OF CHILDHOOD NEPHROTIC SYNDROME
GENETIC DISORDERS
Nephrotic Syndrome (Typical)
Proteinuria with or Without Nephrotic Syndrome
Multisystem Syndromes with or Without Nephrotic Syndrome
Metabolic Disorders with or Without Nephrotic Syndrome
IDIOPATHIC NEPHROTIC SYNDROME
SECONDARY CAUSES
Infections
Drugs
Immunologic or Allergic Disorders
Associated with Malignant Disease
Glomerular Hyperfiltration
Note: Childhood nephrotic syndrome can also be consequence of inflammatory glomerular disorders, normally associated with features of nephritis, e.g., vasculitis, lupus nephritis, membranoproliferative glomerulonephritis, IgA nephropathy.
From Eddy AA, Symons JM: Nephrotic syndrome in childhood, Lancet 362:629–638, 2003.
Nephrotic syndrome may also be secondary to systemic diseases such as systemic lupus erythematosus, Henoch-Schönlein purpura, malignancy (lymphoma and leukemia), and infections (hepatitis, HIV, and malaria) (see Table 521-1).
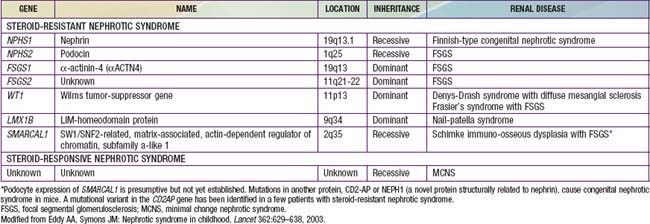
A number of hereditary proteinuria syndromes are caused by mutations in genes that encode critical protein components of the glomerular filtration apparatus (Table 521-3).
Pathophysiology
The underlying abnormality in nephrotic syndrome is an increased permeability of the glomerular capillary wall, which leads to massive proteinuria and hypoalbuminemia. On biopsy, the extensive effacement of podocyte foot processes (the hallmark of idiopathic nephrotic syndrome) suggests a pivotal role for the podocyte. Idiopathic nephrotic syndrome is associated with complex disturbances in the immune system, especially T cell–mediated immunity. In focal segmental glomerulosclerosis, a plasma factor, probably produced by a subset of activated lymphocytes, may be responsible for the increase in capillary wall permeability. Alternatively, mutations in podocyte proteins (podocin, α-actinin 4) and MYH9 (podocyte gene) are associated with focal segmental glomerulosclerosis (see Table 521-3). Steroid-resistant nephrotic syndrome can be associated with mutations in NPHS2 (podocin) and WT1 genes, as well as other components of the glomerular filtration apparatus, such as the slit pore, and include nephrin, NEPH1, and CD-2 associated protein.
Del Rio M, Kaskel F. Evaluation and management of steroid-unresponsive nephrotic syndrome. Curr Opin Pediatr. 2008;20:163-170.
Hodson EM, Alexander SM. Evaluation and management of steroid-sensitive nephrotic syndrome. Curr Opin Pediatr. 2008;20:145-150.
Liapis H. Molecular pathology of nephrotic syndrome in childhood: a contemporary approach to diagnosis. Pediatr Dev Pathol. 2008;11:154-163.
1978 Nephrotic syndrome in children: Prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int. 1978;13:159.
Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387-1399.
Van de Walle JGJ, Donckerwolcke RA. Pathogenesis of edema formation in the nephrotic syndrome. Pediatr Nephrol. 2001;16:283.
521.1 Idiopathic Nephrotic Syndrome
Pathology
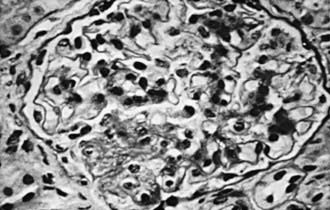
In focal segmental glomerulosclerosis (FSGS), glomeruli show lesions that are both focal (present only in a proportion of glomeruli) and segmental (localized to ≥1 intraglomerular tufts). The lesions consist of mesangial cell proliferation segmental scarring on light microscopy (Fig. 521-2 and see Table 521-2). Immunofluorescence microscopy is positive for IgM and C3 staining in the areas of segmental sclerosis. Electron microscopy demonstrates segmental scarring of the glomerular tuft with obliteration of the glomerular capillary lumen. Similar lesions may be seen secondary to HIV infection, vesicoureteral reflux, and intravenous use of heroin and other drugs of abuse. Only 20% of patients with FSGS respond to prednisone. The disease is often progressive, ultimately involving all glomeruli, and ultimately leads to end-stage renal disease in most patients.
Clinical Manifestations
The idiopathic nephrotic syndrome is more common in boys than in girls (2 : 1) and most commonly appears between the ages of 2 and 6 yr (see Fig. 521-1). However, it has been reported as early as 6 mo of age and throughout adulthood. MCNS is present in 85-90% of patients <6 yr of age. In contrast, only 20-30% of adolescents who present for the first time with nephrotic syndrome have MCNS. The more common cause of idiopathic nephrotic syndrome in this older age group is FSGS. The incidence of FSGS may be increasing; it may be more common in African-American, Hispanic, and Asian patients.
Abeyagunawardena AS, Trompeter RS. Increasing the dose of prednisolone during viral infections reduced the risk of relapse in nephrotic syndrome: a randomized controlled trial. Arch Dis Child. 2008;93:226-228.
Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003;362:629-639.
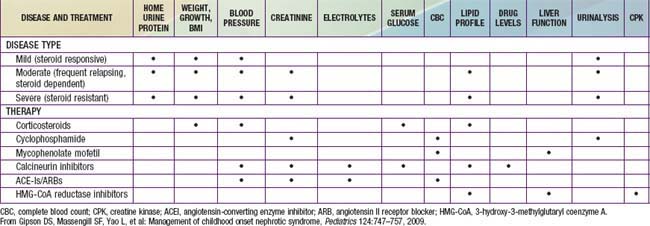
Gipson DS, Massengill SF, Yao L, et al. Management of childhood onset nephrotic syndrome. Pediatrics. 2009;124:747-757.
Hodson EM, Craig JC, Willis NS. Evidence based management of steroid sensitive nephrotic syndrome. Pediatr Nephrol. 2005;20:1523-1530.
Kapur G, Valentini RP, Imam AA, et al. Treatment of severe edema in children with nephrotic syndrome with diuretics alone—a prospective study. Clin J Am Soc Nephrol. 2009;4:907-913.
Kerlin BA, Blatt NB, Fuh B, et al. Epidemiology and risk factors for thromboembolic complications of childhood nephrotic syndrome: a Midwest Pediatric Nephrology Consortium (MWPNC) study. J Pediatr. 2009;155:105-110.
Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40:1175-1184.
Leonard MB, Feldman HI, Shults J, et al. Long-term, high-dose glucocorticoids and bone mineral content in childhood glucocorticoid-sensitive nephrotic syndrome. N Engl J Med. 2004;351:868-875.
Loeffler K, Gowrishankar M, Yiu V. Tacrolimus therapy in pediatric patients with treatment-resistant nephrotic syndrome. Pediatr Nephrol. 2004;19:281-287.
Machuca E, Esquivel EL, Antignac C. Idiopathic nephrotic syndrome: genetic aspects. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:643-666.
Mao J, Zhang Y, Du L, et al. NPHS1 and NPHS2 gene mutations in chinese children with sporadic nephrotic syndrome. Pediatr Res. 2007;61:117-122.
Niaudet P, Boyer O. Idiopathic nephrotic syndrome in children: clinical aspects. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:667-702.
Focal Segmental Glomerulosclerosis Clinical Trial (FSGS-CT). sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). https://clinicaltrials.gov.
521.2 Secondary Nephrotic Syndrome
Nephrotic syndrome can occur as a secondary feature of many forms of glomerular disease. Membranous nephropathy, membranoproliferative glomerulonephritis, postinfectious glomerulonephritis, lupus nephritis, and Henoch-Schönlein purpura nephritis can all have a nephrotic component (see Tables 521-1 and 521-2). Secondary nephrotic syndrome should be suspected in patients >8 yr and those with hypertension, hematuria, renal dysfunction, extrarenal symptoms (rash, arthralgias, fever), or depressed serum complement levels.
Barsoum RS. Schistosomal glomerulopathies. Kidney Int. 1993;44:1-12.
Herman ES, Klotman PE. HIV-associated nephropathy: epidemiology, pathogenesis, and treatment. Semin Nephrol. 2003;23:200-208.
Ronco PM. Paraneoplastic glomerulopathies: new insights into an old entity. Kidney Int. 1999;56:355-377.
Sitprija V. Nephropathy in falciparum malaria. Kidney Int. 1998;34:867.
521.3 Congenital Nephrotic Syndrome
Primary congenital nephrotic syndrome is due to a variety of syndromes inherited as autosomal recessive disorders (see Table 521-3). A number of structural and functional abnormalities of the glomerular filtration barrier causing congenital nephrotic syndrome have been elucidated. The glomerular filtration barrier, which is both size and charge selective, is composed of 3 layers: the fenestrated endothelium, the glomerular basement membrane, and the podocyte foot processes. The foot processes are interconnected by bridging structures, the slit diaphragms, which act as a size selective filter, whereas the glomerular basement membrane restricts molecules based on their ionic charge.
Secondary congenital nephrotic syndrome can resolve with treatment of the underlying cause, such as syphilis (Table 521-5). The management of primary congenital nephrotic syndrome includes intensive supportive care with intravenous albumin and diuretics, regular administration of intravenous gamma-globulin, and aggressive nutritional support (often parenteral), while attempting to pharmacologically decrease urinary protein loss with angiotensin-converting enzyme inhibitors, angiotensin II receptor inhibitors, and prostaglandin synthesis inhibitors or even unilateral nephrectomy. If conservative management fails, and patients suffer from persistent anasarca or repeated severe infections, bilateral nephrectomies are performed and chronic dialysis is initiated. Renal transplantation is the definitive treatment of congenital nephrotic syndrome, though recurrence of the nephrotic syndrome has been reported to occur after transplantation.
Table 521-5 CAUSES OF NEPHROTIC SYNDROME IN INFANTS YOUNGER THAN 1 YEAR
SECONDARY CAUSES
PRIMARY CAUSES
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Saunders, p 418.
Hinkes BG, Mucha B, Vlangos CN, et al. Nephrotic syndrome in the first year of life. Pediatrics. 2007;119:e907-e919.
Jackson LW. Congenital nephrotic syndrome. Neonatal Netw. 2007;26:47-55.
Jalanko H, Holmberg C. Congenital nephrotic syndrome. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:601-620.
Liapis H. Molecular pathology of nephrotic syndrome in childhood. Pediatr Dev Pathol. 2008;11:154-163.
Papez KE, Smoyer WE. Recent advances in congenital nephrotic syndrome. Curr Opin Pediatr. 2004;16:165-170.
VanDeVoorde R, Witte D, Kogan J, et al. Pierson syndrome: a novel cause of congenital nephrotic syndrome. Pediatrics. 2006;118:e501-e505.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 521 Nephrotic Syndrome
Etiology
Most children with nephrotic syndrome have a form of primary or idiopathic nephrotic syndrome (Table 521-1). Glomerular lesions associated with idiopathic nephrotic syndrome include minimal change disease (the most common), focal segmental glomerulosclerosis, membranoproliferative glomerulonephritis, membranous nephropathy and diffuse mesangial proliferation (Table 521-2). These etiologies have different age distributions (Fig. 521-1).
Table 521-1 CAUSES OF CHILDHOOD NEPHROTIC SYNDROME
GENETIC DISORDERS
Nephrotic Syndrome (Typical)
Proteinuria with or Without Nephrotic Syndrome
Multisystem Syndromes with or Without Nephrotic Syndrome
Metabolic Disorders with or Without Nephrotic Syndrome
IDIOPATHIC NEPHROTIC SYNDROME
SECONDARY CAUSES
Infections
Drugs
Immunologic or Allergic Disorders
Associated with Malignant Disease
Glomerular Hyperfiltration
Note: Childhood nephrotic syndrome can also be consequence of inflammatory glomerular disorders, normally associated with features of nephritis, e.g., vasculitis, lupus nephritis, membranoproliferative glomerulonephritis, IgA nephropathy.
From Eddy AA, Symons JM: Nephrotic syndrome in childhood, Lancet 362:629–638, 2003.
Nephrotic syndrome may also be secondary to systemic diseases such as systemic lupus erythematosus, Henoch-Schönlein purpura, malignancy (lymphoma and leukemia), and infections (hepatitis, HIV, and malaria) (see Table 521-1).
A number of hereditary proteinuria syndromes are caused by mutations in genes that encode critical protein components of the glomerular filtration apparatus (Table 521-3).
Pathophysiology
The underlying abnormality in nephrotic syndrome is an increased permeability of the glomerular capillary wall, which leads to massive proteinuria and hypoalbuminemia. On biopsy, the extensive effacement of podocyte foot processes (the hallmark of idiopathic nephrotic syndrome) suggests a pivotal role for the podocyte. Idiopathic nephrotic syndrome is associated with complex disturbances in the immune system, especially T cell–mediated immunity. In focal segmental glomerulosclerosis, a plasma factor, probably produced by a subset of activated lymphocytes, may be responsible for the increase in capillary wall permeability. Alternatively, mutations in podocyte proteins (podocin, α-actinin 4) and MYH9 (podocyte gene) are associated with focal segmental glomerulosclerosis (see Table 521-3). Steroid-resistant nephrotic syndrome can be associated with mutations in NPHS2 (podocin) and WT1 genes, as well as other components of the glomerular filtration apparatus, such as the slit pore, and include nephrin, NEPH1, and CD-2 associated protein.
Del Rio M, Kaskel F. Evaluation and management of steroid-unresponsive nephrotic syndrome. Curr Opin Pediatr. 2008;20:163-170.
Hodson EM, Alexander SM. Evaluation and management of steroid-sensitive nephrotic syndrome. Curr Opin Pediatr. 2008;20:145-150.
Liapis H. Molecular pathology of nephrotic syndrome in childhood: a contemporary approach to diagnosis. Pediatr Dev Pathol. 2008;11:154-163.
1978 Nephrotic syndrome in children: Prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int. 1978;13:159.
Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387-1399.
Van de Walle JGJ, Donckerwolcke RA. Pathogenesis of edema formation in the nephrotic syndrome. Pediatr Nephrol. 2001;16:283.
521.1 Idiopathic Nephrotic Syndrome
Pathology
In focal segmental glomerulosclerosis (FSGS), glomeruli show lesions that are both focal (present only in a proportion of glomeruli) and segmental (localized to ≥1 intraglomerular tufts). The lesions consist of mesangial cell proliferation segmental scarring on light microscopy (Fig. 521-2 and see Table 521-2
[/not-level-membership-for-pediatrics-category]
