[level-membership-for-pediatrics-category]
Chapter 12 Nephrology
Long Cases
Chronic kidney disease (CKD)
There has been much progress in the last few years in paediatric nephrology. Nomenclature has changed recently, to enable more precision when describing the nature and progression of paediatric renal disease. Chronic renal failure (CRF) has been renamed chronic kidney disease (CKD), and is divided into six stages (CKD 1–5, 5D). The first stage has a normal glomerular filtration rate (GFR); the sixth stage requires renal replacement therapy (RRT) by dialysis and renal transplantation (RTx). The stages in between describe mild, moderate and severe reductions in renal function. RTx is the treatment of choice for end-stage renal disease (ESRD) and many children receive renal allografts without prior dialysis (pre-emptive RTx). Pre-emptive RTx avoids the morbidity and mortality associated with dialysis. Many of the multiple long-term problems of CKD involving growth and neurocognitive development are improved with RTx.
The concept of prenatal programming of renal disease has become established: being born with low birth weight (from IUGR [intrauterine growth retardation], being born SGA [small for gestational age] or from being born prematurely) increases the risk of CKD in adulthood. It is now postulated that an adverse intrauterine environment decreases the final number of nephrons; hence the job for the paediatrician is to identify children at risk, avoiding nephrotoxic drugs (e.g. aminoglycosides, non-steroidal, anti-inflammatory drugs), obesity counselling starting in the neonatal period—other risk factors should be discussed with parents of IUGR/SGA or premature babies to minimise exposure to smoking, encourage healthy diet—and monitoring blood pressure to prevent hypertension, early recognition of proteinuria (and treatment with ACE inhibitors or angiotensin blockers) and obesity.
Background information
Aetiology
2. Malformation (structural) of kidney/urinary tract: 30%.
3. Hereditary nephropathies (e.g. nephronophthisis, cystinosis): 20%.
4. Others (e.g. haemolytic uraemic syndrome [HUS], nephrotoxins): 20%.
Hereditary nephropathies are numerous (around 50 types at present—refer to the standard tomes).
There are two important points to remember:
1. Structural problems tend to cause salt- and water-losing forms of CKD.
2. Glomerular disease tends to cause oliguria, salt retention and hypertension.
Many children have dysplastic disease (structural), which leads to polyuric renal failure, so they may drink large amounts of fluid, and do not have any problems with oedema or hypertension. This is quite different to adults with CKD, most of whom are oliguric or anuric. Irrespective of the original cause of kidney damage, once CKD supervenes, there is relentless progression towards kidney failure, but the rate of this is quite variable. Risk factors for rapid decline include lower levels of kidney function at presentation, higher levels of proteinuria, and hypertension. Increased protein in urine causes injury to tubular cells, which leads to interstitial inflammation and fibrosis; in patients with CKD, blocking the RAS decreases proteinuria and slows the deterioration to ESRD. Systemic hypertension causes intraglomerular hypertension, which leads to glomerular hypertrophy and injury; intensified BP control slows progression of renal failure.
Glomerular filtration rate (GFR) and clinical correlates
• Stage 1: normal or increased GFR (≥ 90 mL/min/1.73 m2), with some evidence of kidney damage.
• Stage 2: mildly decreased GFR (60–89 mL/min/1.73 m2); the emphasis is on prevention of disease progression.
• Stage 3: moderately decreased GFR (30–59 mL/min/1.73 m2); the emphasis is on prevention of disease progression, plus managing complications.
• Stage 4: severely decreased GFR (15–29 mL/min/1.73 m2); the emphasis is on treating complications, and preparing for dialysis or transplant.
• Stage 5: very little GFR left (<15 mL/min/1.73 m2); dialysis imminent.

History
Current symptoms
Essentially, this is an extensive systems review, relating to CKD:
1. General health (e.g. tiredness, coping at school, sports, poor exercise tolerance).
2. Urinary (e.g. polyuria, nocturia, anuria, bed wetting).
3. Gastrointestinal (e.g. anorexia, nausea, vomiting, abdominal pain, diarrhoea).
4. Neurological (e.g. headache, paraesthesiae, seizures, confusion).
5. Cardiovascular (e.g. hypertension, cardiac failure symptoms: dyspnoea, oedema).
6. Growth (e.g. monitoring weight, height, causes attributed to poor growth).
7. Skin (e.g. itching [from microscopic subcutaneous calcium deposits], bruising).
8. Skeletal (e.g. bone pain, muscle cramps, muscular weakness).
Family history
Other members of the immediate family affected (e.g. siblings), renal problems (e.g. nephronophthisis, polycystic kidney disease, cystinosis), deafness (e.g. Alport’s syndrome).
Social history
• Disease impact on child: for example, amount of school missed, limitations on lifestyle, body image, self-esteem, peer reactions, future plans, education, career, educational difficulties related to development and/or time lost from school.
• Impact on family: for example, financial considerations such as the cost of frequent hospitalisations, drugs (especially post-transplant if not eligible for [in Australia] a health care card—a problem for those over 16 years of age), special feeds (low-phosphate, high-calorie, low-protein, designed for renal patients—in Australia, Kindergen is now on the Pharmaceutical Benefits Scheme [PBS] and is useful for infants, but other feeds such as Suplena and Nephro have to be bought from the hospital and can be expensive), pumps and disposables (many children are on nasogastric or feeds via gastrostomy overnight), surgery (renal transplant), treatment for other affected children, time lost from parents’ work, transport, private health insurance.
• Parents, other family members, as potential kidney donors; who will be the donor and what they understand about their own potential morbidity; financial considerations.
• Impact on siblings: for example, sibling as kidney donor, sibling rivalry, sibling neglect.
• Benefits received, social supports (e.g. social worker, extended family, Kidney Health Australia [previously Australian Kidney Foundation], dialysis and renal transplant associations).
• Discussion on transition from paediatric to adult renal services by 18 years of age. (In Australia, dialysis machines and disposables are provided by the government.)
• Compliance with and understanding of medications by child/family.
Management
The main goal is for the child to have as normal a life as possible, free of uraemic symptoms, and able to be involved with the usual activities of daily living. The candidate should avoid getting ‘bogged down’ in the acute management of electrolyte problems and should discuss issues such as bone disease, growth including use of rhGH, development and psychosocial issues (especially schooling in chronic patients, transition). Discuss schooling in particular, as children on dialysis often miss key parts of their education (especially maths) and have major problems at school later, often with some acting-out behaviour. Ask about schooling during haemodialysis treatment in children on in centre haemodialysis.
The candidate should recognise that CKD and ESRD are not synonymous.
U. Uraemic complications (includes monitoring for development of neuropathy, encephalopathy; measuring serum urea and creatinine)
R. Renal replacement therapy (dialysis, RTx)
E. Electrolytes and fluids: this includes serum potassium; this is very important, but the candidate should not go directly to this unless the child is already on dialysis; it is usually not the most important problem in CKD; also includes salt and water balance (including hypertension).
Electrolytes and fluid: (a) control of serum potassium
Treatment of acute hyperkalaemia
1. Salbutamol, given nebulised (same doses as in asthma) or intravenously, works quickly and lasts a couple of hours. This is the easiest treatment to give in a paediatric service.
2. Calcium gluconate 10%, or calcium chloride 10% (cardioprotective) intravenously over 2–5 minutes, with electrocardiographic monitoring. This modifies myocardial cells’ action potential, and protects for around 30 minutes.
3. Sodium bicarbonate intravenously over 30 minutes. Alkalosis shifts K+ into cells. The effect lasts 2 hours.
4. An intravenous infusion of 50% dextrose with soluble insulin over 30 minutes. This shifts K+ into cells. The effect lasts 2 hours.
5. Sodium polystyrene sulphonate orally in 70% sorbitol (or in water), or rectally in 1% methylcellulose suspension (or 20% sorbitol). This binds K+ with ion exchange resin. This takes 1–2 hours to take effect, and lasts 4–6 hours.
Electrolytes and fluids: (b) control of salt and fluid balance
Hypertension
In children being medically managed, before needing dialysis, salt restriction and antihypertensive drugs can be used. The drugs used ideally will slow progress to renal failure, and block the RAS: ACE inhibitors (e.g. ramipril, lisinopril); angiotensin receptor blockers (ARBs, e.g. irbesartan). Other drugs used have included diuretics, and calcium channel blockers (e.g. amlodipine). Less frequently used drugs include beta blockers (e.g. atenolol, metoprolol, propranolol) and prazosin (alpha1 post-synaptic blocker). Different nephrologists have different preferences for the order in which various drugs are tried. One suggested general plan is as follows:
2. ACE inhibitors (e.g. lisinopril, an easy-to-use tablet that can be dissolved and dose titrated for small children) or ARBs (e.g. irbesartan). They can cause hyperkalaemia, which limits their use in children with severe reductions in GFR or ESRD.
3. Diuretics (e.g. frusemide); these lose effectiveness once 70% of renal function is lost.
4. Calcium channel blockers (e.g. nifedipine, amlodipine, diltiazem).
Acute hypertensive crisis
• Alpha and beta blocker: labetolol (IV)—also available orally.
• ACE inhibitors: ‘prils’—benzapril, captopril, enalapril, fosinopril, lisinopril, quinapril + enalaprilat (IV). Lisinopril is easy to use, as a tablet can be dissolved in water and then the required dose administered as a liquid.
• Angiotensin receptor blockers: ‘sartans’—irbesartan, losartan.
• Beta blockers: ‘olols’—atenolol, bisoprolol, esmolol (IV), metoprolol, propranolol.
• Calcium channel blockers: diltiazem + ‘dipines’—amlopdipine, felodipine, isradipine, nifedipine, nicardipine (IV).
• Central alpha agonist: clonidine.
• Diuretics: hydrochlorothiazide, chlorthalidone, frusemide (loop diuretic), spironolactone, triamterene, amiloride. Note that the latter three may be associated with hyperkalaemia because of their site of action.
• Dopamine 1 receptor agonist: fenoldopam (IV).
• Peripheral alpha agonists: ‘azosins’—doxazosin, prazosin, terazosin.
• Vasodilators: hydralazine (IV, IM), minoxidil, sodium nitroprusside (IV).
Bone disease: CKD-mineral and bone disorder (CKD-MBD)—renal osteodystrophy; renal rickets plus hyperparathyroidism
Control of serum phosphate
This is important, as hyperphosphataemia can lead to a rapid decline in renal function. It is best to correct the hyperphosphataemia before trying to increase the serum calcium level. A low-phosphate diet is recommended (avoiding ice cream, milk, dairy). Calcium carbonate (e.g. Caltrate, or Cal-Sup chewable) or sevelamer hydrochloride (non-calcium-based) can be used as a phosphate binder, but need to be given with food to be effective. Phosphate binders can be increased in dosage until the calcium level is also returning towards normal. Sevelamer hydrochloride, which is much more expensive than calcium carbonate, is indicated where phosphate levels cannot be controlled with calcium carbonate without causing hypercalcaemia.
Stature (growth)
There are many factors that can adversely affect growth in CKD:
1. Deficient caloric intake: a most important factor in infants and young children.
2. Salt wasting: another important factor in infants and young children.
3. Disease onset (e.g. CKD from infancy [congenital renal diseases] leads to an attained height of −3 Height Standard Deviation Score (SDS) at 3 years of age— probably one third of reduction in height occurs in the first 3 months of life, as most children with CKD have normal birth weights and lengths).
4. Disease type (e.g. cystinosis, or nephrotic syndrome requiring high-dose corticosteroids, may lead to particularly poor growth).
5. Abnormalities in insulin-like growth factor-1 (IGF-1) and IGF-binding proteins (IGFBPs, especially IGFBP-3).
6. Growth hormone resistance, secondary to point 3 above.
7. Poor protein synthesis and low protein turnover.
8. CKD-mineral and bone disorder (renal osteodystrophy).
9. Glucose intolerance (on steroids).
As noted above, growth problems are worse if the disease causing CKD dates from (before) birth. These children may have several problems, especially sodium wasting, leading to significant undernutrition in the first two years of life. Growth problems are also significant around puberty.
Intake: nutrition
In some children, volume constraints will limit the amount of nutritional supplementation that can be given. For younger children with structural disease, nutrition can be optimised by supplemental feeding (e.g. overnight by gastrostomy, or nasogastric feeding), as volume overload is not a problem. Should fluid restriction be necessary, high-calorie supplements (e.g. Suplena, Nutrison Energy Plus or Nepro) can be used for overnight feeds. If overnight feeds are not tolerated because of coexistent gastro-oesophageal reflux, fundoplication may be needed.
Recombinant human erythropoietin (r-HuEPO)
There are three forms of r-HuEPO:
• Epoetin alfa (Eprex): this can be given 2–3 times per week as SC injection, or in those on haemodialysis, given IV on dialysis days, although the IV route is less effective than the SC route.
• Darbepoetin alfa (Aranesp): this can be given SC or IV, has a longer half-life, and so needs to be given only weekly or less frequently (fortnightly or even monthly). The main problem with this is pain at the injection site.
• Epoetin beta (Neorecormon): this can be given once weekly, but can be required more frequently. Its advantage is being less painful SC than darbepoetin.
Renal support and replacement
Dialysis
APD involves use of an automated cycler for overnight instillation and drainage of dialysate fluid. APD has the advantages of only one connection and disconnection between the cycler and the peritoneal catheter per day, and hence less risk of infection, and decreased time demands on the family. It does interfere with the child’s social life, especially for teenagers. APD in the form of continuous cycling peritoneal dialysis (CCPD) is performed nightly and then fluid remains in the peritoneal cavity during the day. Most children need an additional bag change in the late afternoon for optimum dialysis.
Renal transplantation (RTx)
Pre-emptive transplantation (PET) is where children have a transplant as their initial renal replacement therapy, if there is a living related donor. PET is performed before reaching ESRD and dialysis, and has become increasingly popular, with around 30% of transplants in the USA being PETs. Recipients of PETs have improved patient and allograft survival.
1. Very large kidneys: one or both are removed to provide adequate space for a new transplant.
2. Very significant polyuria (e.g. obstructive uropathies, dysplastic kidneys). Polyuria can persist in the native kidney, causing dehydration and exacerbating nephrotoxic effects of post-RTx immunosuppressive regimens.
3. Significant electrolyte disturbance.
4. Persistent proteinuria (e.g. FSGS, SLE, MPGN, congenital nephrotic syndrome). Oedema, poor nutritional status, hyperlipidaemia and hypercoagulability can all increase the risk of allograft thrombosis.
5. Hypertension (many glomerulonephritides). Pretransplant native nephrectomy can lead to improved blood pressure control, less reliance on antihypertensives, and improved surgical and long-term graft survival. In general, however, native kidneys are removed post-transplant if hypertension is difficult to control.
6. Recurrent pyelonephritis (can occur particularly with bladder dysfunction, obstructive uropathy and reflux nephropathy).
Immunosuppressive therapy
A major problem with transplantation is the need for long-term immunosuppressive therapy, with the associated risk of opportunistic infection and an increased risk of malignancy later in life. Corticosteroids are still used. The most common standard treatment used is prednisone, mycophenolate mofetil (MMF) and tacrolimus; cyclosporine A (CSA) is significantly less effective in preventing rejection compared with tacrolimus in a systematic review.
Antiproliferatives
1. Azathioprine (AZA) is a prodrug of 6-mercaptopurine, which inhibits growth of immune cells. It is being used less in RTx, replaced by MMF. Side effects are bone marrow suppression, susceptibility to viruses, pancreatitis, alopecia, malignancy and gastrointestinal upset.
2. Mycophenolate mofetil (MMF) is an antimetabolic agent that interferes with purine metabolism in B- and T-lymphocytes. It is metabolised to mycophenolic acid, which blocks conversion of inosine monophosphate (IMP) to guanosine IMP. It is more effective than AZA in preventing acute rejection in the first year post-transplant, and has similar toxicities to AZA.
3. Sirolimus is a product of Streptomyces hygroscopicus. It forms a complex with FK binding proteins in lymphocytes, which interrupts second messenger signalling, leading to blockage of cytokine-mediated proliferation of T- and B-cells. With the combination of sirolimus and CSA, there is a synergistic effect that allows a lower dose of CSA to be given. Sirolimus is not nephrotoxic, and has no adverse effect on blood pressure. There is a competitive drug interaction with CSA and sirolimus, such that sirolimus should not be given within 4 hours of CSA. Sirolimus is an alternative to CSA or tacrolimus, as it has less nephrotoxicity, but it is also less potent than either of these; thus there is a slightly higher rate of rejection early. Side effects include mouth ulcers, hypercholesterolaemia, pneumonia and rash.
Biological agents
Calcineurin inhibitors
1. Tacrolimus (TAC) is a fungus-derived macrolide, like CSA. It inhibits lymphokines derived from T-cells, including IL-2, IL-3, IL-4 and gamma interferon, and clonal expansion of helper and cytotoxic T-cells. Tacrolimus is effective in inducing graft tolerance. Its side effects are similar to those of CSA, including nephrotoxicity, neurotoxicity and infection, plus lymphoproliferative disease (more common than with CSA), but it does not cause the cosmetic side effects of hypertrichosis or gingival hyperplasia. It may induce diabetes mellitus. TAC was used in 6% of RTx in the USA in 1996; by 2006–07, it was used in 74%.
2. Cyclosporine A (CSA) is a fungus-derived cyclic peptide that blocks T-cell response. It binds to cellular proteins (cyclophilins), blocks IL-2 production and inhibits T-cell proliferation and differentiation. Side effects are nephrotoxicity, hypertension, hyperkalaemia, anaemia, hypertrichosis, gingival hyperplasia, susceptibility to infection and increased risk of malignancy. The newer microemulsion form of CSA has improved, more consistent absorption. If used during induction, it can be given intravenously by continuous infusion or 8-hourly. CSA has more rapid metabolism in children than adults, such that children have higher doses. The dose of CSA is lower when used with sirolimus, as the two drugs have a synergistic immunosuppressive effect. CSA previously used to be used in 82% of RTx in the USA in 1996, but its use declined to 8% by 2006–07, in the USA.
Management
The above agents can be divided into two groups:
• Induction therapy agents: biological (ATG, basiliximab, daclizumab).
• Maintenance therapy agents: steroids, calcineurin inhibitors (CSA, tacrolimus) and antiproliferative agents (AZA, MMF, sirolimus).
The main issues post-transplant can be divided into short term (at time of transplant and next few months), which comprise problems of rejection and infection, and long term, comprising decrease in renal function (50% renal survival at 15 years), malignancies (particularly lymphoma and skin cancer) and cardiovascular disease (long-term effects of left-ventricular hypertrophy, hypertension and hypercholesterolaemia lead to cardiovascular diseases [e.g. stroke, ischaemic heart disease] being more common).
Allograft loss
1. Chronic rejection (or chronic allograft nephropathy) is the most common cause: incomplete understanding of mechanism; presumed combination of immunological and non-immunological mechanisms, non-compliance, drug toxicity and recurrent disease; risk increased by multiple acute rejection episodes; late acute rejection. No effective therapies yet available.
2. Acute rejection (second most common cause). Complete rejection reversal can be achieved in around two thirds of patients, but reversal is less likely with an increased number of rejections, increased age of the recipient and late (over 12 months post-RTx rejection) rejection episodes.
3. Vascular thrombosis (third most common cause). The risks are decreased by use of calcineurin inhibitors or antilymphocyte antibody on day 0–1, and increased by peritoneal dialysis pre-RTx, cadaver donors under 5 years of age, recipients under 2 years of age or repeat RTx recipients.
4. Recurrent disease. See the list below. FSGS is a particular problem, and the most important, as most lose their grafts quickly. Children with FSGS are twice as likely as all other diagnostic groups (below) to have primary acute tubular necrosis (ATN) requiring dialysis post-RTx. If it does recur once, there is an 80% chance it will recur in any subsequent RTx.
Recurrence rates in transplants (histological recurrence)
1. Membranoproliferative glomerulonephritis (MPGN) type II: 100%.
2. Membranoproliferative glomerulonephritis (MPGN) type I: 70%.
3. Henoch–Schönlein purpura (HSP): 55–85%.
5. Focal segmental glomerular sclerosis (FSGS): 25–30%. FSGS is the most important, as most lose their grafts quickly, though some patients may respond to plasma exchange with or without rituximab.
6. Haemolytic uraemic syndrome (HUS): 12–25%. Most children with recurrent HUS associated with Factor H mutations lose their grafts with recurrent disease.
Social problems
Compliance with medications is a real problem. A significant proportion (around 25%) of grafts lost from rejection are associated with non-compliance.
Minor issues
Other issues
Monitoring
1. Routine check of growth, blood pressure, oedema.
2. Electrolytes, bicarbonate, creatinine, urea, calcium, phosphate, alkaline phosphatase.
3. Haemoglobin, white cell count, platelet count.
4. Cholesterol, triglycerides, uric acid.
5. Bone X-rays—hips, knees, ankles, hands.
8. Follow-up by dietician, social worker, occupational therapist, especially for adolescent issues, and in younger children, development.
Nephrotic syndrome
There have been several advances in the understanding of the pathophysiology of nephrotic syndrome in recent years, following on from the discovery in 2006 that the cause of congenital nephrotic syndrome of the Finnish type (CNF1) is a mutation of a protein named nephrin, coded for by the gene NPHS. Nephrin is located within the visceral glomerular epithelium, at the slit diaphragms, which are membranes that bridge filtration pores between neighbouring podocytes. The slit diaphragm is attached to the cell cytoskeleton by adaptor proteins, including podocin and CD2AP. Podocytes are known to regulate integrity and survival of glomerular epithelial cells. It is now recognised that several diseases, both acquired and inherited, are due to defects in the slit diaphragm, which is essentially a multi-protein signalling complex. The phenotype focal segmental glomerulosclerosis (FSGS) can be caused by defects in the gene/proteins: NPHS2/podocin, located at the slit diaphragm; CD2AP/CD2AP, located near the slit diaphragm; TRPC6/TRPC6, located at the podocyte; ACTIN4/alpha actinin 4, located at the podocyte; and other genes located at the podocyte (WT1 [Wilms’ tumour suppressor gene], tRNA (leu), COQ2). Autosomal recessive nephrotic syndrome is an inherited form of FSGS due to mutations in the gene NPHS2 situated on chromosome 1q25–q31, which codes for podocin; it has early onset, minimal changes on early biopsy, FSGS on later biopsy, rapid progression to ESRD, but rare recurrence after RTx; it can present as familial FSGS with later onset, adolescent or adult. Podocin mutations occur in some 10–30% of sporadic steroid responsive nephrotic syndrome in some populations.
Background information
Definition
Nephrotic syndrome (NS) is characterised by four components:
Aetiology
Other important causes of NS are as follows:
1. Focal segmental glomerulosclerosis (FSGS). This is the most common progressive glomerular disease in children, and the second most common cause of ESRD (the most common cause being congenital renal anomalies). Several different genetic forms are known, some autosomal recessive (putative genes NPHS2, and WT1) and some autosomal dominant (putative genes TRP6, and ACTIN4). FSGS can be idiopathic (somes genes not discovered yet) or secondary to postinfectious glomerulonephritis, obstructive uropathy, reflux nephropathy or systemic diseases (e.g. sickle cell disease [SCD], systemic lupus erythematosis [SLE]).
2. Mesangial proliferative glomerulonephritis (MesPGN). This is usually idiopathic, but can be due to chronic infection (bacterial or viral), SCD, SLE, renal transplant or bone marrow transplant. Idiopathic forms of MesPGN may be associated with IgM or C1Q deposition on immunofluoresence, and are then referred to as IgM or C1Q nephropathy. It remains unclear whether MesPGN is a forerunner of FSGS in some patients.
3. Membranous glomerulonephritis. This can be idiopathic, or due to SCD, SLE, drugs (e.g. non-steroidal anti-inflammatory drugs [NSAIDs], captopril), toxins (heavy metals) or infections (e.g. hepatitis B or C).
4. SLE. This can cause FSGS, MesPGN or membranous glomerulonephritis, but the typical histology is of a diffuse proliferative GN with typical immunofluorescent findings. Clinically, SLE is more likely to present with haematuria and acute nephritis, rather than a nephrotic syndrome.
Definitions used in INS
• Remission: urinary protein excretion less than 4 mg/m2/hour, or no trace of protein on urine dipstick, or protein/creatinine ratio below 0.02 g/mmol for three consecutive days.
• Relapse: recurrence of proteinuria as defined in definition of INS, or 2+ or more on dipstick for three consecutive days.
• Frequent relapsers: children with two or more relapses within the first 6 months of initial response, or more than four relapses in 12 months.
• Corticosteroid dependence: two consecutive relapses while on prednisone or within 2 weeks of ceasing prednisone.
• Corticosteroid resistance: failed response after 8 weeks of 2 mg/kg/day prednisone.
• Cyclosporine (CSA) dependent: relapses when CSA is tapered or stopped; it refers to the group of steroid-dependent children who achieve remission with CSA.
Complications of NS
Oedema
For years, the theory has been that children with rapid onset of INS develop loss of oncotic pressure: fluid moves from the intravascular space to the interstitium, resulting in hypovolaemia. In these patients, infusion of albumin with administration of a loop diuretic (e.g. frusemide) has been given, leading to increased excretion of salt and water loss. However, plasma volume is decreased in children during the initial phase of a relapse. Children with chronic nephrotic syndrome, however, may have increased or normal plasma volume. In these patients, diuretics alone can be given (e.g. frusemide, together with spironolactone). Angiotensin-converting enzyme (ACE) inhibitors (e.g. enalapril) have been used in children with CRINS to reduce proteinuria, raise serum albumin and reduce oedema. It is now believed that rather than being due to the oncotic pressure theory, oedema is due to a primary defect in sodium excretion; also vasopressin excess occurs. Anasarca (generalised, massive oedema) can lead to the following: difficulty walking, from severe scrotal or vulval oedema; respiratory distress, from pleural effusions and/or severe ascites splinting the diaphragm; and tissue breakdown with cellulitis. This can be treated successfully with infusing salt-poor albumin followed by frusemide (see the later section on management).
Negative nitrogen balance
Loss of protein in urine, plus poor appetite, and nausea contribute to poor intake.
Management
Investigations
Any child with NS should probably have the following tests.
Urine
Urinalysis, including looking for cellular casts, which do not tend to occur in INS but may well occur in other glomerulonephropathies (note that hyaline or waxy casts are common in INS). Microscopic haematuria is usually transitory in steroid-sensitive NS, but may persist in steroid-resistant NS.
Blood
1. Urea, creatinine and electrolytes (renal function is usually normal in INS; may be abnormal in other glomerulonephropathies).
2. Albumin and total protein levels (to evaluate severity).
3. Serum complements C3 and C4 (low with MesPGN, SLE; normal in INS).
4. Full blood examination. The haemoglobin level is normal in INS. If anaemia is present, this suggests other diagnoses.
5. Hepatitis B and C serology (hepatitis B is associated with membranous nephritis; hepatitis C is associated with MesPGN).
Treatment
1. Corticosteroids
The other use of steroids is for steroid-resistant NS, usually due to FSGS, using IV methylprednisolone. One regimen reported to be effective in observational studies was to give methylprednisolone starting at a dose of 30 mg/kg, three times a week for 2 weeks, then tapering gradually over 80 weeks with concurrent prednisolone on alternate days. While reported to provide remission rates of 60–70% by some researchers, others reported less satisfactory response rates. This regimen is very toxic (side effects: hypertension, delayed growth, cataracts, infections) and has largely been abandoned, though some clinicians may give methylprednisolone for three doses with cyclosporin in steroid-resistant NS.
2(a) Other agents for corticosteroid-sensitive idiopathic nephrotic syndrome (CSINS): additional options including cytotoxic drugs
The main agents that can claim success in NS (mnemonic CLAIM) are as follows:
C. Cyclophosphamide (CPA: 2 mg/kg/day for 8–12 weeks)/Chlorambucil (0.2 mg/kg/day day for 8 weeks)/Cyclosporine A (CSA: 2.5 mg/kg 12-hourly for 12–24 months)
L. Levamisole (2.5 mg/kg alternate daily for 12–24 months)
A. Angiotensin-converting enzyme (ACE) inhibitors
I. Immunisation with pneumococcal vaccine
M. Mycophenolate mofetil (25 mg/kg/day in divided doses for 12–24 months)
• CPA has significant side effects, short term (e.g. bone marrow suppression, risk of viral infections such as varicella, measles) and long term (e.g. gonadal toxicity and risk of carcinogenesis). However, it offers the chance of prolonged remission off therapy, with 36% of children with frequently relapsing CSINS remaining in remission at 5 years.
• CSA can cause nephrotoxicity, hypertension, gingival hyperplasia and hypertrichosis.
• CPA given for 8 weeks and CSA given for 6–9 months are equally effective in maintaining remission in randomised controlled trials while CSA is being given. However, the effect of CSA is not sustained, while that of CPA is.
• Levamisole causes enhanced cellular immune responses in certain conditions with depressed immune function. Levamisole may help to maintain remission in steroid-dependent INS, is well tolerated and has few side effects (neutropenia, rarely vasculitis, liver toxicity, convulsions), and these are all reversible on withdrawing the drug. It decreases the steroid requirement by 50%, and the relapse rate by 50%.
• ACE inhibitors reduce glomerular hyperfiltration. Also, from the ESCAPE trial (see the long case on CKD), it is now known that intensified blood pressure control (keeping blood pressure below the 50th centile) in children aged 3–18 with CKD, using high-dose ACE inhibition with ramipril at a fixed dose, delayed progression of kidney disease. Adverse effects include hypotension and cough.
• Immunisation as per schedule for pneumococcal vaccination of immunocompromised children: heptavalent conjugated pneumococcal vaccine (7vPCV) in children under 5, and polysaccharide pneumococcal vaccine (PPV23) in children 5 or older.
• Mycophenolate mofetil is used for steroid-sensitive NS, largely on the basis of observational data. A single underpowered RCT demonstrated no significant difference in efficacy between MMF and CSA, but there was considerable imprecision in the results. Its side effects include gastrointestinal effects (diarrhoea and abdominal pain), and haematological abnormalities, but its side effect profile is preferable to that of CSA.
Indications for commencing these agents include the following:
1. Failure to respond to steroids (depends on pathology—only CSA and ACE inhibitors have been shown to reduce proteinuria significantly in RCTs).
2. Unacceptable steroid side effects (e.g. reduced height gain).
3. Steroid-dependent NS (only if side effects are unacceptable).
4. Relapses with hypertension or thrombosis (only if frequent relapser or steroid dependent).
6 Hypertension
A minority of children with NS will have hypertension. Agents of choice are ACE inhibitors or ARBs, as these delay deterioration in renal function that can occur with ongoing proteinuria in a minority of children with steroid-sensitive nephrotic syndrome, but in more children with steroid-resistant nephrotic syndrome.
Short Cases
Renal examination
Look at the skin, for sallow complexion (CKD), pallor (anaemia of CKD or haemolytic uraemic syndrome [HUS]), and periorbital or peripheral oedema. Also note any jaundice (hepatorenal syndrome), bruising (CKD), uraemic ‘frost’ (CKD) or scratch marks (pruritis with CKD), although it is unlikely that patients who are that sick would participate in the examination. Note any hirsutism (steroids), hypertrichosis/gum hypertrophy (cyclosporine [CSA]) or Cushingoid features (steroids for transplant or nephrotic syndrome). There may be other peripheral signs, such as nearby bags of peritoneal dialysis fluid (CKD), and a visible peritoneal dialysis catheter (CKD), arteriovenous fistula, subclavian or jugular venous catheter. Look for evidence of previous dialysis access (particularly in renal transplant patients); in the neck for subclavian or internal jugular dialysis catheters, in the arms for previous fistulae and in the abdomen for previous peritoneal dialysis catheters (both the incision and exit sites of the catheter). Also look for scars of previous or current transplants. Most transplants are in the right or left iliac fossae and are easily palpated beneath a ‘hockey stick’ scar. However, in small children there may be a central abdominal incision and the kidney may be palpated bimanually, usually to the left of the scar.
After initial inspection, a systematic examination can be performed, starting with the hands, followed by checking the blood pressure, and then examining in turn the head and neck, chest and abdomen, and finally the gait and lower limbs. This suggested order is outlined in Figure 12.1. Details of findings sought at each step are outlined in Table 12.1.
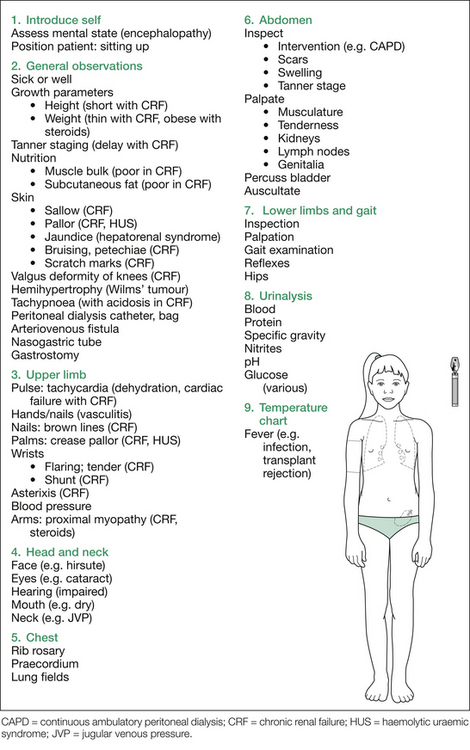
Table 12.1 Additional information: details of possible findings on renal examination
| Head and neck |
| Face |
CAPD = continuous ambulatory peritoneal dialysis; CMV = cytomegalovirus; CKD = chronic kidney disease; CSA = cyclosporine; HUS = haemolytic uraemic syndrome; SLE = systemic lupus erythematosus.
The urinalysis may be requested at any stage (e.g. before laying hands on the patient), but it must not be forgotten.
Hypertension
After inspection, take the blood pressure (BP) reading yourself, in both arms. Make sure you use the right cuff size and the correct technique. The recommended cuff bladder width is 40% of the circumference of the midpoint of the upper limb, midway between the olecranon and the acromion. The cuff bladder must cover at least 80% of the circumference of the arm. The BP should be measured with the cubital fossa at the level of the heart, the arm being supported, with the stethoscope placed over the brachial arterial pulse, medial and proximal to the cubital fossa, inferior to the lower edge of the cuff. Note that current normative BP tables include height percentiles, age and gender, as BP depends on all these variables. A taller child will have a slightly higher BP than a shorter child of the same sex and age. Reference to the appropriate tables is essential. Note also that the fifth Korotkoff sound is used to define diastolic BP.
Note that most children and adolescents with BP levels at or above the 95th centile for their age and sex are overweight. Body size is the most important determinant of BP in childhood and adolescence. If the child has both diastolic and systolic BP above the 95th centile, there will be an underlying cause, usually renal disease. In children under 12 months of age, systolic BP is used to define hypertension.
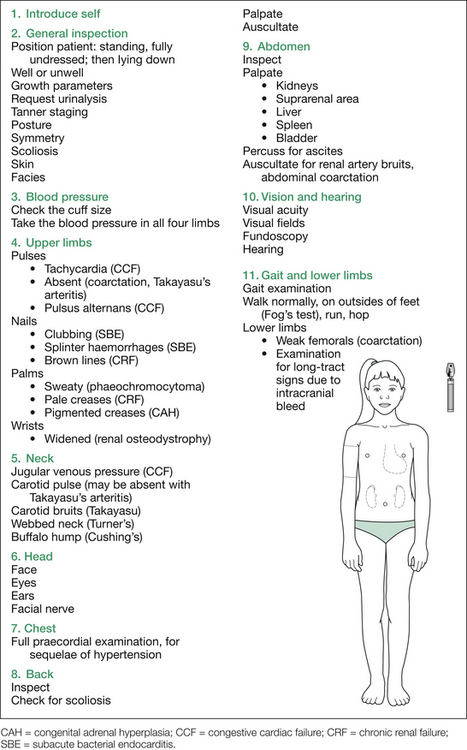
The relevant findings sought at each point are listed in Table 12.2, and the suggested order of approach is outlined in Figure 12.2.
Table 12.2 Additional information: details of procedure and possible findings in the hypertension short case
| Introduction |
| Irritability (encephalopathy) |
| Dysphasia (CVA) |
| Hearing impairment (Alport, congenital rubella, aminoglycoside toxicity) |
| General inspection |
| Parameters |
BPD = bronchopulmonary dysplasia; CAH = congenital adrenal hyperplasia; CCF = congestive cardiac failure; CVA = cerebrovascular accident; HSP = Henoch–Schönlein purpura; HUS = haemolytic uraemic syndrome; ICP = intracranial pressure; LVF = left ventricular failure; NF-1 = neurofibromatosis type 1; SLE = systemic lupus erythematosus; TS = tuberous sclerosis; UAC = umbilical artery catheterisation.
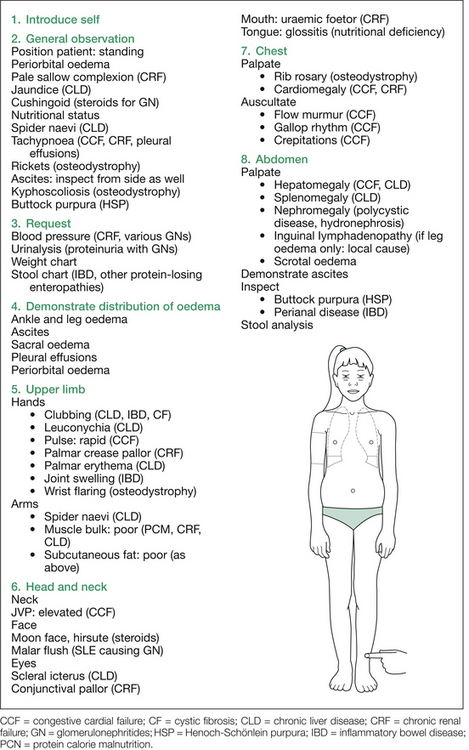
Oedema
1. Renal; for example, nephrotic syndrome or chronic kidney disease (CKD).
2. Liver; for example, chronic liver disease (CLD).
3. Cardiac; for example, congestive cardiac failure (CCF).
4. Bowel; for example, protein-losing enteropathies, including inflammatory bowel disease (IBD) and cystic fibrosis (CF).
5. Nutrition; for example, protein calorie malnutrition (PCM).
6. Local causes; for example, lymphadenopathy causing leg oedema.
Each of these conditions deserves initial consideration.
Begin by asking the child to stand up (in underwear only) and inspect for periorbital oedema, findings of CKD (e.g. sallow complexion, pallor, skeletal changes of renal osteodystrophy), CLD (e.g. jaundice, spider naevi) and CCF (e.g. raised jugular venous pressure), Cushingoid features of steroid-treated glomerulonephritis (especially idiopathic nephrotic syndrome [INS]), and nutrition and abdominal swelling from ascites. Also inspect from the side for ascites and kyphoscoliosis (osteodystrophy) and from the back for scoliosis (osteodystrophy) and buttock rash (Henoch–Schönlein purpura).
1. Ankle oedema: press over the anterior tibiae for one minute (remember: one finger, one spot, one minute).
2. Ascites: shifting dullness, fluid thrill.
3. Sacral oedema: press over sacrum for one minute.
After demonstrating the extent of the oedema, proceed to examine for the aetiology. A suggested order is as follows: hands, blood pressure (if not yet requested), face, chest, abdomen and urinalysis (again, if not already requested). Figure 12.3 outlines the findings sought at each stage of the examination procedure.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 12 Nephrology
Long Cases
Chronic kidney disease (CKD)
There has been much progress in the last few years in paediatric nephrology. Nomenclature has changed recently, to enable more precision when describing the nature and progression of paediatric renal disease. Chronic renal failure (CRF) has been renamed chronic kidney disease (CKD), and is divided into six stages (CKD 1–5, 5D). The first stage has a normal glomerular filtration rate (GFR); the sixth stage requires renal replacement therapy (RRT) by dialysis and renal transplantation (RTx). The stages in between describe mild, moderate and severe reductions in renal function. RTx is the treatment of choice for end-stage renal disease (ESRD) and many children receive renal allografts without prior dialysis (pre-emptive RTx). Pre-emptive RTx avoids the morbidity and mortality associated with dialysis. Many of the multiple long-term problems of CKD involving growth and neurocognitive development are improved with RTx.
The concept of prenatal programming of renal disease has become established: being born with low birth weight (from IUGR [intrauterine growth retardation], being born SGA [small for gestational age] or from being born prematurely) increases the risk of CKD in adulthood. It is now postulated that an adverse intrauterine environment decreases the final number of nephrons; hence the job for the paediatrician is to identify children at risk, avoiding nephrotoxic drugs (e.g. aminoglycosides, non-steroidal, anti-inflammatory drugs), obesity counselling starting in the neonatal period—other risk factors should be discussed with parents of IUGR/SGA or premature babies to minimise exposure to smoking, encourage healthy diet—and monitoring blood pressure to prevent hypertension, early recognition of proteinuria (and treatment with ACE inhibitors or angiotensin blockers) and obesity.
Background information
Aetiology
2. Malformation (structural) of kidney/urinary tract: 30%.
3. Hereditary nephropathies (e.g. nephronophthisis, cystinosis): 20%.
4. Others (e.g. haemolytic uraemic syndrome [HUS], nephrotoxins): 20%.
Hereditary nephropathies are numerous (around 50 types at present—refer to the standard tomes).
There are two important points to remember:
1. Structural problems tend to cause salt- and water-losing forms of CKD.
2. Glomerular disease tends to cause oliguria, salt retention and hypertension.
Many children have dysplastic disease (structural), which leads to polyuric renal failure, so they may drink large amounts of fluid, and do not have any problems with oedema or hypertension. This is quite different to adults with CKD, most of whom are oliguric or anuric. Irrespective of the original cause of kidney damage, once CKD supervenes, there is relentless progression towards kidney failure, but the rate of this is quite variable. Risk factors for rapid decline include lower levels of kidney function at presentation, higher levels of proteinuria, and hypertension. Increased protein in urine causes injury to tubular cells, which leads to interstitial inflammation and fibrosis; in patients with CKD, blocking the RAS decreases proteinuria and slows the deterioration to ESRD. Systemic hypertension causes intraglomerular hypertension, which leads to glomerular hypertrophy and injury; intensified BP control slows progression of renal failure.
Glomerular filtration rate (GFR) and clinical correlates
• Stage 1: normal or increased GFR (≥ 90 mL/min/1.73 m2), with some evidence of kidney damage.
• Stage 2: mildly decreased GFR (60–89 mL/min/1.73 m2); the emphasis is on prevention of disease progression.
• Stage 3: moderately decreased GFR (30–59 mL/min/1.73 m2); the emphasis is on prevention of disease progression, plus managing complications.
• Stage 4: severely decreased GFR (15–29 mL/min/1.73 m2); the emphasis is on treating complications, and preparing for dialysis or transplant.
• Stage 5: very little GFR left (<15 mL/min/1.73 m2); dialysis imminent.
History
Current symptoms
Essentially, this is an extensive systems review, relating to CKD:
1. General health (e.g. tiredness, coping at school, sports, poor exercise tolerance).
2. Urinary (e.g. polyuria, nocturia, anuria, bed wetting).
3. Gastrointestinal (e.g. anorexia, nausea, vomiting, abdominal pain, diarrhoea).
4. Neurological (e.g. headache, paraesthesiae, seizures, confusion).
5. Cardiovascular (e.g. hypertension, cardiac failure symptoms: dyspnoea, oedema).
6. Growth (e.g. monitoring weight, height, causes attributed to poor growth).
7. Skin (e.g. itching [from microscopic subcutaneous calcium deposits], bruising).
8. Skeletal (e.g. bone pain, muscle cramps, muscular weakness).
Family history
Other members of the immediate family affected (e.g. siblings), renal problems (e.g. nephronophthisis, polycystic kidney disease, cystinosis), deafness (e.g. Alport’s syndrome).
Social history
• Disease impact on child: for example, amount of school missed, limitations on lifestyle, body image, self-esteem, peer reactions, future plans, education, career, educational difficulties related to development and/or time lost from school.
• Impact on family: for example, financial considerations such as the cost of frequent hospitalisations, drugs (especially post-transplant if not eligible for [in Australia] a health care card—a problem for those over 16 years of age), special feeds (low-phosphate, high-calorie, low-protein, designed for renal patients—in Australia, Kindergen is now on the Pharmaceutical Benefits Scheme [PBS] and is useful for infants, but other feeds such as Suplena and Nephro have to be bought from the hospital and can be expensive), pumps and disposables (many children are on nasogastric or feeds via gastrostomy overnight), surgery (renal transplant), treatment for other affected children, time lost from parents’ work, transport, private health insurance.
• Parents, other family members, as potential kidney donors; who will be the donor and what they understand about their own potential morbidity; financial considerations.
• Impact on siblings: for example, sibling as kidney donor, sibling rivalry, sibling neglect.
• Benefits received, social supports (e.g. social worker, extended family, Kidney Health Australia [previously Australian Kidney Foundation], dialysis and renal transplant associations).
• Discussion on transition from paediatric to adult renal services by 18 years of age. (In Australia, dialysis machines and disposables are provided by the government.)
• Compliance with and understanding of medications by child/family.
Management
The main goal is for the child to have as normal a life as possible, free of uraemic symptoms, and able to be involved with the usual activities of daily living. The candidate should avoid getting ‘bogged down’ in the acute management of electrolyte problems and should discuss issues such as bone disease, growth including use of rhGH, development and psychosocial issues (especially schooling in chronic patients, transition). Discuss schooling in particular, as children on dialysis often miss key parts of their education (especially maths) and have major problems at school later, often with some acting-out behaviour. Ask about schooling during haemodialysis treatment in children on in centre haemodialysis.
The candidate should recognise that CKD and ESRD are not synonymous.
U. Uraemic complications (includes monitoring for development of neuropathy, encephalopathy; measuring serum urea and creatinine)
R. Renal replacement therapy (dialysis, RTx)
E. Electrolytes and fluids: this includes serum potassium; this is very important, but the candidate should not go directly to this unless the child is already on dialysis; it is usually not the most important problem in CKD; also includes salt and water balance (including hypertension).
Electrolytes and fluid: (a) control of serum potassium
Treatment of acute hyperkalaemia
1. Salbutamol, given nebulised (same doses as in asthma) or intravenously, works quickly and lasts a couple of hours. This is the easiest treatment to give in a paediatric service.
2. Calcium gluconate 10%, or calcium chloride 10% (cardioprotective) intravenously over 2–5 minutes, with electrocardiographic monitoring. This modifies myocardial cells’ action potential, and protects for around 30 minutes.
3. Sodium bicarbonate intravenously over 30 minutes. Alkalosis shifts K+ into cells. The effect lasts 2 hours.
4. An intravenous infusion of 50% dextrose with soluble insulin over 30 minutes. This shifts K+ into cells. The effect lasts 2 hours.
5. Sodium polystyrene sulphonate orally in 70% sorbitol (or in water), or rectally in 1% methylcellulose suspension (or 20% sorbitol). This binds K+ with ion exchange resin. This takes 1–2 hours to take effect, and lasts 4–6 hours.
Electrolytes and fluids: (b) control of salt and fluid balance
Hypertension
In children being medically managed, before needing dialysis, salt restriction and antihypertensive drugs can be used. The drugs used ideally will slow progress to renal failure, and block the RAS: ACE inhibitors (e.g. ramipril, lisinopril); angiotensin receptor blockers (ARBs, e.g. irbesartan). Other drugs used have included diuretics, and calcium channel blockers (e.g. amlodipine). Less frequently used drugs include beta blockers (e.g. atenolol, metoprolol, propranolol) and prazosin (alpha1 post-synaptic blocker). Different nephrologists have different preferences for the order in which various drugs are tried. One suggested general plan is as follows:
2. ACE inhibitors (e.g. lisinopril, an easy-to-use tablet that can be dissolved and dose titrated for small children) or ARBs (e.g. irbesartan). They can cause hyperkalaemia, which limits their use in children with severe reductions in GFR or ESRD.
3. Diuretics (e.g. frusemide); these lose effectiveness once 70% of renal function is lost.
4. Calcium channel blockers (e.g. nifedipine, amlodipine, diltiazem).
Acute hypertensive crisis
• Alpha and beta blocker: labetolol (IV)—also available orally.
• ACE inhibitors: ‘prils’—benzapril, captopril, enalapril, fosinopril, lisinopril, quinapril + enalaprilat (IV). Lisinopril is easy to use, as a tablet can be dissolved in water and then the required dose administered as a liquid.
• Angiotensin receptor blockers: ‘sartans’—irbesartan, losartan.
• Beta blockers: ‘olols’—atenolol, bisoprolol, esmolol (IV), metoprolol, propranolol.
• Calcium channel blockers: diltiazem + ‘dipines’—amlopdipine, felodipine, isradipine, nifedipine, nicardipine (IV).
• Central alpha agonist: clonidine.
• Diuretics: hydrochlorothiazide, chlorthalidone, frusemide (loop diuretic), spironolactone, triamterene, amiloride. Note that the latter three may be associated with hyperkalaemia because of their site of action.
• Dopamine 1 receptor agonist: fenoldopam (IV).
• Peripheral alpha agonists: ‘azosins’—doxazosin, prazosin, terazosin.
• Vasodilators: hydralazine (IV, IM), minoxidil, sodium nitroprusside (IV).
Bone disease: CKD-mineral and bone disorder (CKD-MBD)—renal osteodystrophy; renal rickets plus hyperparathyroidism
Control of serum phosphate
This is important, as hyperphosphataemia can lead to a rapid decline in renal function. It is best to correct the hyperphosphataemia before trying to increase the serum calcium level. A low-phosphate diet is recommended (avoiding ice cream, milk, dairy). Calcium carbonate (e.g. Caltrate, or Cal-Sup chewable) or sevelamer hydrochloride (non-calcium-based) can be used as a phosphate binder, but need to be given with food to be effective. Phosphate binders can be increased in dosage until the calcium level is also returning towards normal. Sevelamer hydrochloride, which is much more expensive than calcium carbonate, is indicated where phosphate levels cannot be controlled with calcium carbonate without causing hypercalcaemia.
Stature (growth)
There are many factors that can adversely affect growth in CKD:
1. Deficient caloric intake: a most important factor in infants and young children.
2. Salt wasting: another important factor in infants and young children.
3. Disease onset (e.g. CKD from infancy [congenital renal diseases] leads to an attained height of −3 Height Standard Deviation Score (SDS) at 3 years of age— probably one third of reduction in height occurs in the first 3 months of life, as most children with CKD have normal birth weights and lengths).
4. Disease type (e.g. cystinosis, or nephrotic syndrome requiring high-dose corticosteroids, may lead to particularly poor growth).
5. Abnormalities in insulin-like growth factor-1 (IGF-1) and IGF-binding proteins (IGFBPs, especially IGFBP-3).
6. Growth hormone resistance, secondary to point 3 above.
7. Poor protein synthesis and low protein turnover.
8. CKD-mineral and bone disorder (renal osteodystrophy).
9. Glucose intolerance (on steroids).
As noted above, growth problems are worse if the disease causing CKD dates from (before) birth. These children may have several problems, especially sodium wasting, leading to significant undernutrition in the first two years of life. Growth problems are also significant around puberty.
Intake: nutrition
In some children, volume constraints will limit the amount of nutritional supplementation that can be given. For younger children with structural disease, nutrition can be optimised by supplemental feeding (e.g. overnight by gastrostomy, or nasogastric feeding), as volume overload is not a problem. Should fluid restriction be necessary, high-calorie supplements (e.g. Suplena, Nutrison Energy Plus or Nepro) can be used for overnight feeds. If overnight feeds are not tolerated because of coexistent gastro-oesophageal reflux, fundoplication may be needed.
