Chapter 495 Neoplasms of Bone
495.1 Malignant Tumors of Bone
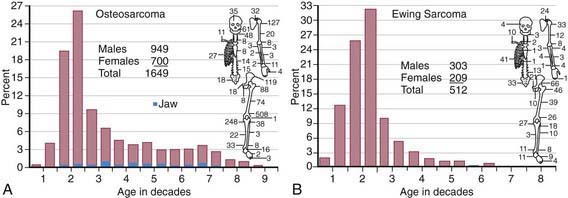
The annual incidence of malignant bone tumors in the USA is approximately 7 cases/million white children <14 yr of age, with a slightly lower incidence in African-American children. Osteosarcoma is the most common primary malignant bone tumor in children and adolescents, followed by Ewing sarcoma (Table 495-1; Fig. 495-1). In children <10 yr of age, Ewing sarcoma is more common than osteosarcoma. Both tumor types are most likely to occur in the second decade of life.
Table 495-1 COMPARISON OF FEATURES OF OSTEOSARCOMA AND THE EWING FAMILY OF TUMORS
| FEATURE | OSTEOSARCOMA | EWING FAMILY OF TUMORS |
|---|---|---|
| Age | Second decade | Second decade |
| Race | All races | Primarily whites |
| Sex (M : F) | 1.5 : 1 | 1.5 : 1 |
| Cell | Spindle cell–producing osteoid | Undifferentiated small round cell, probably of neural origin |
| Predisposition | Retinoblastoma, Li-Fraumeni syndrome, Paget disease, radiotherapy | None known |
| Site | Metaphyses of long bones | Diaphyses of long bones, flat bones |
| Presentation | Local pain and swelling; often, history of injury | Local pain and swelling; fever |
| Radiographic findings | Sclerotic destruction (less commonly lytic); sunburst pattern | Primarily lytic, multilaminar periosteal reaction (“onion-skinning”) |
| Differential diagnosis | Ewing sarcoma, osteomyelitis | Osteomyelitis, eosinophilic granuloma, lymphoma, neuroblastoma, rhabdomyosarcoma |
| Metastasis | Lungs, bones | Lungs, bones |
| Treatment | Chemotherapy | Chemotherapy |
| Ablative surgery of primary tumor | Radiotherapy and/or surgery of primary tumor | |
| Outcome | Without metastases, 70% cured; with metastases at diagnosis, ≤20% survival | Without metastases, 60% cured; with metastases at diagnosis, 20-30% survival |
Osteosarcoma
Diagnosis
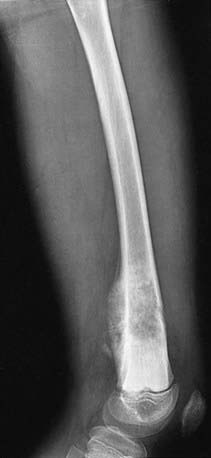
Bone tumor should be suspected in a patient who presents with deep bone pain often causing nighttime awakening in whom there is a palpable mass, and radiographs demonstrate a lesion. The lesion may be mixed lytic and blastic in appearance, but new bone formation is usually visible. The classic radiographic appearance of osteosarcoma is the sunburst pattern (Fig. 495-2). When osteosarcoma is suspected, the patient should be referred to a center with experience in managing bone tumors. The biopsy and the surgery should be performed by the same surgeon so that the incisional biopsy site can be placed in a manner that will not compromise the ultimate limb salvage procedure. Tissue usually is obtained for molecular and biologic studies at the time of the initial biopsy. Before biopsy, MRI of the primary lesion and the entire bone should be performed to evaluate the tumor for its proximity to nerves and blood vessels, soft tissue and joint extension, and skip lesions. The metastatic work-up, which should be performed before biopsy, includes CT of the chest and radionuclide bone scanning to evaluate for lung and bone metastases, respectively. The differential diagnosis of a lytic bone lesion includes histiocytosis, Ewing sarcoma, lymphoma, and bone cyst.
Ewing Sarcoma
Diagnosis
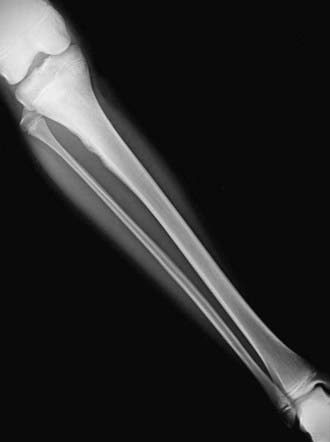
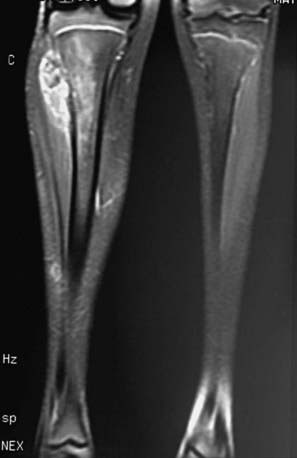
The diagnosis of Ewing sarcoma should be suspected in a patient who presents with pain and swelling, with or without systemic symptoms, and with a radiographic appearance of a primarily lytic bone lesion with periosteal reaction, the characteristic onion-skinning (Fig. 495-3). A large associated soft tissue mass often is visualized on MRI or CT (Fig. 495-4). The differential diagnosis includes osteosarcoma, osteomyelitis, Langerhans cell histiocytosis, primary lymphoma of bone, metastatic neuroblastoma, or rhabdomyosarcoma in the case of a pure soft tissue lesion. Patients should be referred to a center with experience in managing bone tumors for evaluation and biopsy. Thorough evaluation for metastatic disease includes CT of the chest, radionuclide bone scan, and bone marrow aspiration and biopsy specimens from at least two sites. MRI of the tumor and the entire length of involved bone should be performed to determine the exact extension of the soft tissue and bony mass and the proximity of tumor to neurovascular structures. To avoid compromising an ultimate potential for limb salvage by a poorly planned biopsy incision, the same surgeon should perform the biopsy and the surgical procedure. CT-guided biopsy of the lesion often provides diagnostic tissue. It is important to obtain adequate tissue for special stains, cytogenetics, and molecular studies.
495.2 Benign Tumors and Tumor-like Processes of Bone
Eosinophilic granuloma is a monostotic or polyostotic disease with no extraskeletal involvement. This latter finding distinguishes eosinophilic granuloma from the other forms of Langerhans’ cell histiocytosis (Hand-Schüller-Christian or Letterer-Siwe variants), which can have a less favorable prognosis (Chapter 501). Eosinophilic granuloma usually occurs during the first 3 decades of life and is most common in boys 5-10 yr of age. The skull is most commonly affected, but any bone may be involved. Patients usually present with local pain and swelling. Marked tenderness and warmth often are present in the area of the involved bone. Spinal lesions can cause pain, stiffness, and occasional neurologic symptoms. The radiographic appearance of the skeletal lesions is similar in all forms of Langerhans’ cell histiocytosis but is variable enough to mimic many other benign and malignant lesions of bone. The radiolucent lesions have well-defined or irregular margins with expansion of the involved bone and periosteal new bone formation. Spine involvement can cause uniform compression or flattening of the vertebral body. A skeletal survey is warranted because polyostotic involvement and the typical skull lesions strongly suggest the diagnosis of eosinophilic granuloma. Biopsy often is necessary to confirm the diagnosis because of the broad radiographic differential diagnosis. Treatment includes curettage and bone grafting, low-dose radiation therapy, or corticosteroid injection. Observation for symptomatic lesions is reasonable because most osseous lesions heal spontaneously and do not recur. Children with bone lesions should be evaluated for visceral involvement because treatment of Hand-Schüller-Christian disease and Letterer-Siwe disease is more complex and often systemic.
Arndt CAS, Crist WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341:342-352.
Balamuth N, Woner R. Ewing’s sarcoma. Lancet Oncol. 2010;11(2):184-192.
Campanacci M, Capanna R, Picci P. Unicameral and aneurysmal bone cysts. Clin Orthop Relat Res. 1986;204:25-36.
Campanacci M, Laus M. Osteofibrous dysplasia of the tibia and fibula. J Bone Joint Surg Am. 1981;63:367-375.
Dahlin DC, Ivins JC. Benign chondroblastoma: a study of 125 cases. Cancer. 1972;30:401-413.
De Alava E, Gerald WL. Molecular biology of the Ewing’s sarcoma/primitive neuroectodermal tumor family. J Clin Oncol. 2000;18:204-213.
Freiberg AA, Loder RT, Heidelberger KT. Aneurysmal bone cysts in young children. J Pediatr Orthop. 1994;14:86-91.
Ginsberg JP, Woo SY, Johnson ME, et al. Ewing’s sarcoma family of tumors. In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. ed 4. Philadelphia: Lippincott Williams & Wilkins; 2009:973-1016.
Gorlick R, Anderson P, Andrulis I, et al. Biology of childhood osteogenic sarcoma and potential targets for therapeutic development: meeting summary. Clin Cancer Res. 2003;9:5442-5453.
Grier H, Krailo M, Tarbell N, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694-701.
Grier RJ. Surgical options for children with osteosarcoma. Lancet Oncol. 2005;6:85-92.
Kneisl JS, Simon MA. Medical management compared with operative treatment for osteoid osteoma. J Bone Joint Surg Am. 1992;74:179-185.
Marina N, Gebhardt M, Teot L, et al. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist. 2004;9:422-441.
Mavrogenis AF, Papagelopoulos PJ, Soucacos PN. Skeletal osteochondromas revisited. Orthopedics. 2008;31:ii.
Meyers P, Schwartz C, Krailo M, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival—a report from the Children’s Oncology Group. J Clin Oncol. 2008;26:633-638.
Rodriguez-Galindo C, Sount SL, Pappo AS. Treatment of Ewing sarcoma family of tumors: current status and outlook for the future. Med Pediatr Oncol. 2003;40:276-287.
Schmale GA, Conrad EUIII, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Am. 1994;76:986-992.



