Neoplasia
Neoplasia
Pulmonary neoplasm is much less common in children than in adults. Affected children may present clinically with respiratory symptoms or pulmonary neoplasm may be detected incidentally on a chest radiograph in an asymptomatic child. In a recent series of 204 pediatric lung tumors, the ratio of primary benign to primary malignant to secondary malignant pulmonary neoplasms (i.e., metastases) is 1.4 : 1 : 11.6.1 Primary lung tumors represent only 0.19% of all pediatric neoplasms. Metastatic lung tumors are approximately 12 times more common than primary lung tumors in children. A list of benign and malignant lung neoplasms is summarized in Table 55-1.
Primary Benign Pulmonary Neoplasms
Primary benign pulmonary neoplasms in children are far less common malignant pulmonary neoplasms, including both primary and secondary lesions. The imaging characteristics of benign lung neoplasms are summarized in Table 55-2.
Table 55-2
Imaging Characteristics of Benign Lung Neoplasm (Nonlymphoproliferative)
| Neoplasm | Imaging Characteristics |
| Hamartoma | Smooth or slightly lobulated, sharply defined mass, occasionally calcified (“popcorn”) Fat and calcification in solitary pulmonary mass on computed tomography is diagnostic |
| Chondroma | Solitary or multiple nodules; commonly (45%) calcified; associated with the Carney triad |
| Respiratory papillomatosis | Rarely (<1%) intrapulmonary; Bilateral, multiple subpleural solid or cystic nodules; may be associated with bronchiectasis or atelectasis |
| Lymphatic malformation | Rarely intrapulmonary; well-marginated, nonenhancing cystic mass; may simulate congenital pulmonary airway malformation or diaphragmatic hernia in neonates; or solid, low-attenuation mediastinal or pulmonary mass in older children |
Hamartoma
Etiology: Pulmonary hamartoma, originally thought to represent a congenital lesion, is now considered a true benign mesenchymal neoplasm. It contains predominantly cartilage, fat, and fibrous tissue. Occasionally, pulmonary hamartoma may also have smooth muscle, bone, and entrapped respiratory epithelium, which grow slowly.2 Pulmonary hamartoma is rare in children (the peak incidence is in the fourth to sixth decades); however, it is the most common primary benign pulmonary neoplasm accounting for 7% to 14% of all solitary pulmonary nodules in children.3 Ninety percent of pulmonary hamartomas are parenchymal in location. They usually present as incidental findings, although a large pulmonary hamartoma could cause respiratory distress.
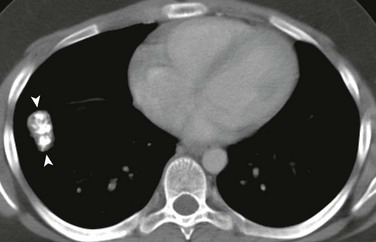
Imaging: The typical radiographic appearance of pulmonary hamartoma is a smooth or slightly lobulated, solitary pulmonary nodule or mass, usually located in the peripheral portion of the lungs. Characteristic punctuate or popcorn-like calcifications may be seen in approximately 10%. On computed tomography (CT), it is typically a well-circumscribed pulmonary nodule that is less than 2.5 cm in diameter. Pulmonary hamartoma often contains fat or calcification, which, in a solitary pulmonary nodule, is considered diagnostic (Fig. 55-1).4
Treatment and Follow-up: Pediatric patients with pulmonary hamartoma require no further treatment unless it grows rapidly or the patient becomes symptomatic because of recurrent pneumonia or atelectasis caused by the mass effect from the tumor.5 In such situations, surgical resection is usually curative.
Chondroma
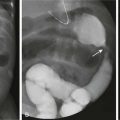
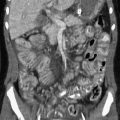
Etiology: Pulmonary chondroma is a benign tumor composed of a well-differentiated benign cartilage with lack of bronchial epithelium.6 Pulmonary chondroma tends to occur before 30 years of age (82%), mainly in young women (85%). Affected persons are usually asymptomatic. Pulmonary chondroma is associated with the Carney triad.7 This syndrome commonly affects three organs: (1) stomach (gastrointestinal stromal tumor, 75%) (Fig. 55-2, B), (2) lung (pulmonary chondroma, 15%), and (3) paraganglionic system (paraganglioma, 10%). However, tumors arising from the adrenal gland (adrenal adenoma or pheochromocytoma, 20%) and the esophagus (leiomyoma, 10%) have also been reported in association with the Carney triad. The majority of patients present with two of three neoplasms. Seventy-five percent of patients with the Carney triad have pulmonary chondroma(s).
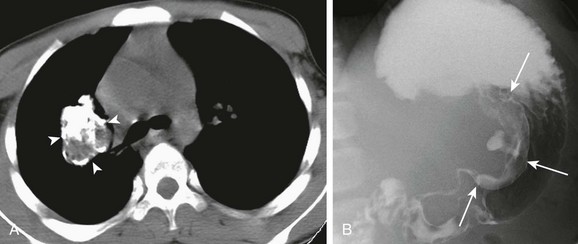
Figure 55-2 Pulmonary chondroma.
A, A 14-year-old boy in the setting of the Carney triad. Axial unenhanced computed tomography image shows a large right parahilar mass (arrowheads) with extensive calcifications. B, Upper gastrointestinal study of the same patient demonstrates a large ulcerated exophytic soft tissue mass (arrows) in the stomach consistent with gastric leiomyosarcoma.
Imaging: Pulmonary chondroma may be single (40%), multiple unilateral (25%), or bilateral (15%) without predilection for specific lobe or side of lung.7 Chest radiography usually shows well-demarcated, often multiple, lung masses with central or popcorn-like calcification (Fig. 55-2, A). When calcified (45%), the appearance of chondroma is indistinguishable from that of pulmonary hamartoma on imaging studies.
Recurrent Respiratory Papillomatosis
Etiology: Recurrent respiratory papillomatosis (RRP) is characterized by mucosal papillomas, which are ingrowths of squamous cell–lined fibrovascular core in the lumen of central airways. It is the most common neoplasm that occurs in the larynx of children. RRP is caused by the human papilloma virus (HPV) types 6 and 11, which is typically acquired during vaginal birth. The primary lesions commonly affect the larynx but may spread to the lung parenchyma (1.8%) especially if surgical or laser therapy had been provided earlier.8 The ultimate prognosis is poor when pulmonary involvement occurs. Malignant transformation into squamous cell carcinoma may occur.9
Imaging: On chest radiography, RRP typically presents as bilateral, multiple nodular and cystic lesions of varying size containing air or debris. Postobstructive atelectasis, bronchiectasis, or secondary infection presenting as consolidation may be also seen.10 On CT, usually, scattered nodules are seen in the lungs (Fig. 55-3), and these nodules may enlarge, become air-filled cysts, or form large cavities with thin or thick walls.11 CT virtual bronchoscopy may be useful to evaluate mural nodules within the central airways.
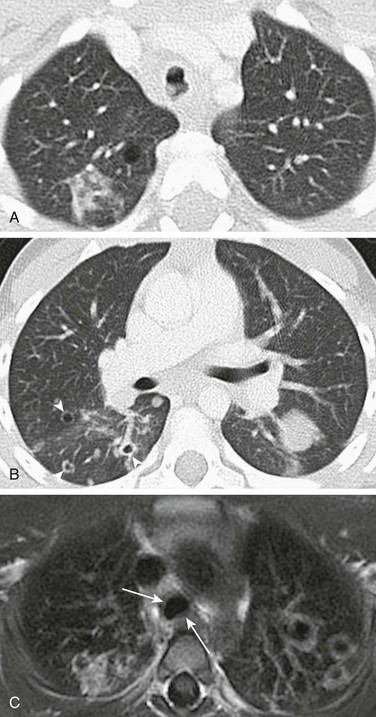
Figure 55-3 Recurrent respiratory papillomatosis.
A, Axial lung window computed tomography image obtained in a 5-year-old girl with recurrent respiratory papillomatosis shows multiple intratracheal soft tissue nodules. B, Multiple parenchymal papillomas are also present, some of which show cavitation (arrowheads). C, An axial T1-weighted magnetic resonance image shows cavitating lesions in both lungs and mildly thickened tracheal walls (arrows).
Lymphatic Malformation
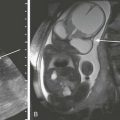
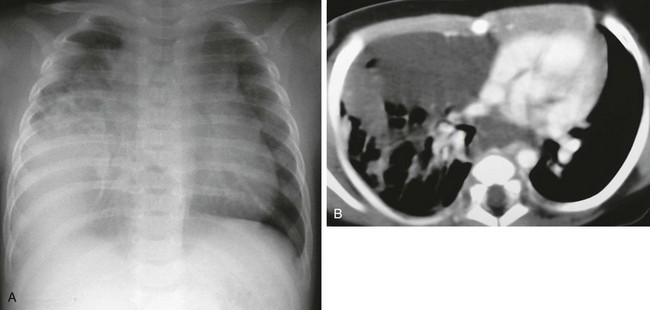
Etiology: Lymphatic malformation, previously referred to as lymphangioma, is characterized by a benign proliferation of nonfunctional lymphatic tissue that may involve nearly every organ of the body. Only 1% of lymphatic malformations remain confined to the chest. Primary pulmonary lymphatic malformation is even less common.14 Affected persons are often asymptomatic. However, some may present with compressive symptoms such as cough, dyspnea, stridor, or even pneumothorax. Younger patients tend to have lesions that affect the lung.15
Imaging: On chest radiography, lymphatic malformation typically presents as a masslike opacity. The most common CT finding is a smooth, well-marginated, nonenhancing, cystic mass. In neonates and infants, this may simulate congenital pulmonary airway malformation (CPAM) or diaphragmatic hernia. Older children may have a solid, low-attenuation, masslike lesion that may mimic pneumonia or tumors (e-Fig. 55-4).
Lymphoproliferation
In recent years, the incidence of lymphoproliferative disorders has increased because of increase in pediatric organ transplantation, prevalence of human immunodeficiency virus (HIV) infection, and development of more potent immunosuppressive therapies. The most common lymphoproliferative disorders affecting lungs in children are plasma cell granuloma, mucosa-associated and bronchus-associated lymphoid tissue, lymphocytic interstitial pneumonitis, lymphomatoid granulomatosis, and posttransplantation lymphoproliferative disorder. The imaging characteristics of lymphoproliferative disorders involving lungs are listed in Table 55-3.
Table 55-3
Lymphoproliferative Disorders of the Lungs
| Disorder | Imaging Characteristics |
| Plasma cell granuloma or inflammatory myofibroblastic tumor | Solitary or multiple; sharply circumscribed mass; may be locally invasive; 15% to 25% have calcification; may be composed of both solid and cystic components |
| Mucosa-associated lymphoid tissue proliferation (pseudolymphoma) | 2- to 5-cm discrete lesions with air bronchograms; may evolve into malignant lymphoma; pleural effusion common |
| Bronchus-associated lymphoid tissue proliferation | Diffuse reticulonodular pattern on chest radiography Small centrilobular nodules and ground glass opacity on computed tomography |
| Lymphoid interstitial pneumonia | Seen in patients with acquired or congenital immunodeficiency as reticular or reticulonodular opacities on chest radiography; diffuse centrilobular and subpleural nodules on high-resolution computed tomography; associated with ground-glass opacity; bronchovascular and interstitial thickening |
| Lymphomatoid granulomatosis | Seen in immunocompromised patients Multiple nodules or confluent masses that frequently cavitate; basal predominance |
| Posttransplantation lymphoproliferative disorder | Solitary or multiple lung masses or consolidation noted months to years after solid organ or bone marrow transplantation; may have associated mediastinal and extrathoracic adenopathy; large lesions may cavitate |
Plasma Cell Granuloma or Inflammatory Myofibroblastic Tumor
Etiology: Plasma cell granuloma is the most common tumorlike abnormality in the lungs of children. It arises in the lung parenchyma but may also involve the mediastinum or pleura. Due to the complexity and variable histologic characteristics, it is known by several different terms, including inflammatory or postinflammatory pseudotumor, fibroxanthoma, myofibroblastic tumor, fibrous histiocytoma, xanthogranuloma, or histiocytoma.17,18 Recently, it has been termed inflammatory myofibroblastic tumor (IMT) because myofibroblasts, fibroblasts, and histiocytes are the main constituents of this tumor (Fig. 55-5, B).19 A substantial proportion of tumors have ALK1 gene mutations.20 The World Health Organization (WHO) currently recognizes IMT as a low-grade mesenchymal malignancy. The majority of children with IMT are older than 5 years, although it has been reported in younger children and even infants. Approximately 60% of affected children are symptomatic, typically presenting with fever, cough, chest pain, dyspnea, wheezing, or hemoptysis.
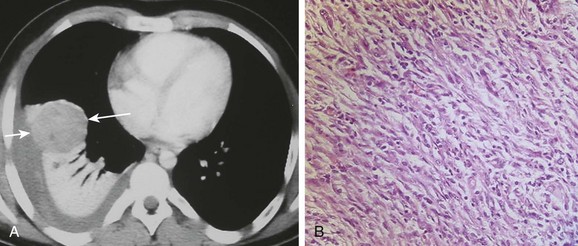
Figure 55-5 Inflammatory myofibroblastic tumor.
An afebrile 13-year-old boy who presented with increasing dyspnea and right-sided pleuritic chest pain. A, An axial contrast-enhanced computed tomography of the chest shows a rounded heterogeneously enhancing lesion (arrows) located adjacent to an area of atelectatic lung. Pleural fluid at the same level demonstrates increased attenuation consistent with a hemothorax. B, A photomicrograph (hematoxylin-eosin stain) of the surgical specimen reveals the presence of both spindle-shaped myofibroblasts and inflammatory cells consistent with an inflammatory myofibroblastic tumor of the lung.
Imaging: Radiographically, IMTs may be seen as solitary (95%) or multiple (5%). IMTs are usually sharply circumscribed, lobulated mass(es) of varying sizes, typically located in the peripheral portion of the lungs. IMT may occasionally be endobronchial. On CT, it usually presents as a soft tissue mass with either homogeneous or heterogeneous attenuation (see Fig. 55-5, A). Although IMT does not enhance substantially, a thick enhancing rim has been reported. Less commonly, it may present with both solid and cystic components. IMT may also have an infiltrative pattern simulating an aggressive malignancy.21 If the mediastinum is involved or the mass contains calcifications (15% to 25%), IMT may mimic a germ cell tumor, neuroblastoma, or metastatic osteosarcoma in pediatric patients.
Mucosa-Associated Lymphoid Tissue and Bronchus-Associated Lymphoid Tissue
Etiology: Mucosa-associated lymphoid tissue (MALT, or pseudolymphoma) is rare in children. Affected persons generally are not severely ill but may have nonspecific respiratory symptoms. Lymphoma has been reported to develop in some cases of MALT. It may be quite difficult to clearly differentiate pseudolymphoma from a true lymphoproliferative condition, even by using modern immunofluorescence techniques. Experts currently disagree about whether MALT should be considered a premalignant or a postinflammatory condition. Bronchus-associated lymphoid tissue (BALT) is a subcategory of the more widely distributed MALT. BALT is a lymphoid aggregate located in the submucosal area of bronchioles, which may become hyperplastic because of chronic antigen stimuli.22 It has been suggested that BALT is related to a hypersensitivity response to unidentified antigens. BALT has two forms: (1) lymphoid interstitial pneumonia and (2) follicular bronchitis or bronchiolitis. Lymphoid interstitial pneumonia type BALT is commonly seen in pediatric patients with acquired immunodeficiency syndrome (AIDS) or other immune compromise. Follicular bronchitis or bronchiolitis type of BALT typically occurs in children with chronic infection, connective tissue disorders, or immunodeficiency disorders and as a hypersensitivity reaction.
Imaging: On chest radiography, MALT typically presents as discrete, often multiple lesions, usually with air bronchograms ranging from 2 to 5 cm in diameter. A pleural effusion is commonly seen. A diffuse reticulonodular opacity, often associated with hyperinflation, is the common radiographic finding of BALT (e-Fig. 55-6, A). On CT, the usual imaging appearance of BALT consists of small centrilobular nodules (foci of lymphoid proliferation) and a ground-glass opacity predominantly involving the lower lobes (see e-Fig. 55-6, B). In patients with advanced BALT, bronchiectasis and peribronchovascular consolidation caused by recurrent infection and chronic airway obstruction may present.
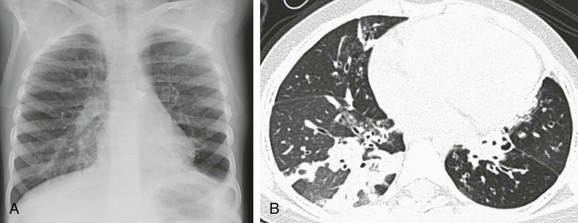
e-Figure 55-6 Bronchus-associated lymphoid tissue.
A 5-year-old girl who presented with worsening shortness of breath and decreased oxygen saturation. A, A chest radiograph shows hyperinflation with increased streaky interstitial opacities in both lungs. B, An axial lung window computed tomography image demonstrates air trappings, mosaic perfusion, and diffuse opacity with cystic bronchiectasis.
Treatment and Follow-up: Aggressive management (e.g., complete surgical excision, radiotherapy, chemotherapy, or a combination of all of these) is the current treatment of choice for MALT. Because of the wide extent of disease often associated with BALT, surgical excision may be considered only for a minority of patients. Chemotherapy is the treatment of choice for the majority of patients with BALT.
Lymphocytic Interstitial Pneumonitis
Etiology: Lymphocytic interstitial pneumonitis (LIP) is characterized by diffuse proliferation of polyclonal lymphocytes and plasma cells in the pulmonary parenchymal interstitium, which expands to the alveolar septa. It is considered an AIDS-defining illness in children younger than 13 years old who are infected with HIV because LIP is rarely idiopathic.
Imaging: On chest radiography, LIP typically presents with reticular or reticulonodular opacities predominantly in the bilateral lower lung zones.23 On high-resolution CT, diffuse centrilobular and subpleural nodules (representing local proliferation of lymphoid germinal centers), areas of ground-glass opacity, and bronchovascular and interstitial thickening in both lungs are often seen (Fig. 55-7). Thin-walled cysts and bronchiectasis may be also present.24
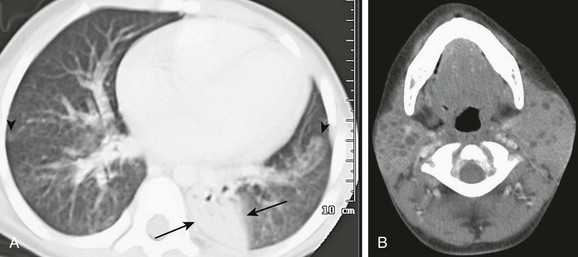
Figure 55-7 Lymphocystic interstitial pneumonitis.
An 8-year-old girl with human immunodeficiency virus (HIV) infection with increasing shortness of breath. A, An axial computed tomography (CT) image shows bilateral patchy areas of ground-glass attenuation, right greater than left, poorly defined nodules (arrowheads) and an area of consolidation in the left lower lobe (arrows). B, An axial, enhanced CT image of neck demonstrates multiple, bilateral parotid lymphoepithelial cysts consistent with HIV parotitis.
Treatment and Follow-up: Steroid and other immunosuppressive agents are used for treating LIP with variable results. Prognosis of children with LIP depends on the associated underlying disease. Progressive honeycomb fibrosis or infectious complications may lead to increased mortality. Low grade B-cell lymphoma may develop in rare cases.25
Lymphomatoid Granulomatosis
Etiology: Lymphomatoid granulomatosis, also known as pseudolymphoma, angiocentric lymphoma, or angiocentric immunoproliferative lesion, is characterized pathologically by an angiocentric and angiodestructive lymphocytic infiltration. It is considered an aggressive multisystem disease with a poor prognosis. The lung is often the primary site of involvement. This disease is associated with Epstein-Barr virus (EBV) infection in immunocompromised individuals. Progression of lymphomatoid granulomatosis to lymphoma occurs in 12% to 47% of patients, with the mortality rate over 50%.26
Imaging: On chest radiography, bilateral, poorly defined, nodular and confluent lesions with a basilar predominance are usually seen (e-Fig. 55-8). Characteristic CT findings of lymphomatoid granulomatosis include peribronchovascular distribution of nodules (which reflects the tendency of lymphomononuclear cells to infiltrate the subintimal region of medium-sized arteries and veins), small thin-walled cysts, and conglomerate small nodules.24,27 Lesions may cavitate, mimicking Wegener granulomatosis.
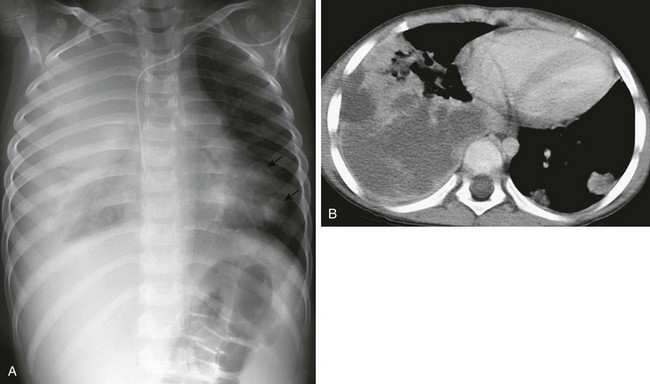
e-Figure 55-8 Lymphomatoid granulomatosis type III (form of B cell lymphoma).
A 5-year-old boy with severe combined immunodeficiency who presented with cough and shortness of breath. A, A chest radiograph demonstrates marked opacity of the right lung and cardiomediastinal shift to the left, with multiple left-sided nodules (arrows). B, An axial enhanced computed tomography image shows multiple left-sided cavitating nodules in addition to a large heterogeneous, predominantly low-attenuation, and confluent density within the right lung.
Posttransplantation Lymphoproliferative Disorder
Etiology: Posttransplantation lymphoproliferative disorder (PTLD) is a consequence of chronic immunosuppression following solid organ transplantation or, less often, bone marrow transplantation.29–32 It is believed to be induced by exposure to EBV. The lesions consist of uncontrolled proliferation of B lymphocytes ranging from benign lymphoid hyperplasia to invasive malignant lymphoma. The incidence of PTLD (1% to 18%) varies with the type of organ transplanted. It occurs most frequently in lung or heart-lung transplantation patients, likely because of the higher levels of immunosuppression required for these organ transplantations.29,33–35 It is also more common in pediatric patients than in adult transplantation patients, possibly because of the lack of prior exposure to EBV. Improved surveillance, earlier diagnosis, and close monitoring of immunosuppression have led to a decreased incidence of PTLD in recent years, as well as a more favorable outcome. The most common sites for PTLD are the tonsils, cervical nodes, gastrointestinal tract, and the chest.36,37 Intrathoracic PTLD tends to present earlier compared with extrathoracic PTLD. PTLD tends to occur within the allograft organ itself, as well as in adjacent anatomic regions.38 Heart transplantation is the sole exception to this predilection.39 Clinical symptoms of PTLD are often vague and include lethargy, fever, and weight loss. Biopsy is typically required to confirm the diagnosis.
Imaging: The imaging appearance of PTLD is not specific and overlaps with that of many opportunistic infections. On chest radiography, the most common finding in PTLD is the presence of multiple well-defined pulmonary nodules with or without mediastinal adenopathy (Fig. 55-9).39 These are best visualized on CT. Large pulmonary nodules and mediastinal adenopathy tend to show central low attenuation, likely representing necrosis. Other less frequent patterns of thoracic involvement include air space consolidation, pleural or chest wall masses, pleural or pericardial effusions, and thymic enlargement.40–42 For a more confident diagnosis, it is helpful to search for extrathoracic PTLD such as thickening of bowel loops, enlarged abdominal lymph nodes, cervical adenopathy, or enlarged oropharyngeal lymphatic tissues.39,43 Fludeoxyglucose positron emission tomography or CT may also increase sensitivity and specificity for the diagnosis.44
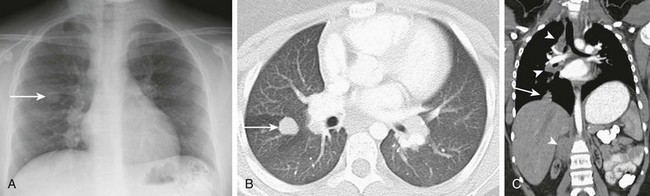
Figure 55-9 Posttransplantation lymphoproliferative disorder (PTLD).
An 11-year-old girl who presented with PTLD following renal transplantation. A, A chest radiograph shows a nodular opacity (arrow) in the right upper lobe and bilateral hilar lymphadenopathy. B, An axial lung window computed tomography (CT) image demonstrates a right upper lobe lung nodule (arrow) corresponding to the opacity noted on chest radiograph. C, A coronal reformatted CT image shows mediastinal, hilar, and retroperitoneal lymphadenopathy (arrowheads) and an additional nodule at the right lung base (arrow). Note the atrophic native bilateral kidneys.
Treatment and Follow-up: Reduction in immunosuppressive therapy remains a primary component of treatment for EBV-positive PTLD, which may lead to resolution of disease in the majority of cases.45 Chemotherapy is used when reduced immunosuppression fails to control disease progression. Newer treatments of PTLD include B-lymphocyte–depleting antibodies, adoptive T-cell immunotherapy using allogeneic, or autologous EBV-specific cytotoxic T-lymphocytes.46 EBV vaccination is advocated to be effective prophylaxis against PTLD.
Primary Malignant Pulmonary Neoplasms
Primary malignant pulmonary neoplasms are rare in children and are histologically diverse. The current WHO classification system of primary malignant pulmonary neoplasms differs substantially from the prior classification system, particularly with regard to new tumors (e.g., pleuropulmonary blastoma) and reclassification of benign and malignant tumors (e.g., IMT).47 A series published in 2008 showed that the most common primary lung malignancies in children are pleuropulmonary blastoma and carcinoid tumor.1 Because of the rarity of these malignant neoplasms and the nonspecific clinical symptoms, they are often not considered in the differential diagnosis in children presenting with persistent pneumonitis, cough, and atelectasis. This often leads to delayed definitive treatment and generally a worse prognosis. The imaging characteristics of primary malignant lung tumors are listed in Table 55-4.
Table 55-4
Imaging Characteristics of Primary Malignant Lung Tumors
| Neoplasm | Imaging Characteristics |
| Carcinoid or salivary gland tumor | Centrally located lesion: intraluminal soft tissue mass with distal atelectasis or obstructive pneumonitis Peripherally located lesion: oval or lobulated intraluminal or exophytic mass and occasionally calcify |
| Bronchogenic carcinoma | Central mass lesions with bronchial obstruction or, less commonly, small peripheral lesions |
| Pleuropulmonary blastoma | Cystic or mixed cystic and solid lesions adjacent to pleura; usually very large, with mediastinal displacement |
| Epithelioid hemangioendothelioma | Multiple well- or ill-defined nodular opacities up to 3 cm in diameter; very rare in childhood |
Bronchial Adenoma (Carcinoid Tumor or Salivary Gland Tumor)
Etiology: The term bronchial adenoma, which implies a benign disease process, was recently recategorized as either carcinoid or salivary gland tumors by the WHO.47 Historically, bronchial adenomas encompassed bronchial carcinoid tumor, mucoepidermoid tumor, and adenoid cystic carcinoma. Carcinoid tumor accounts for approximately 80% of previously classified pediatric bronchial adenomas, which are low-grade neuroendocrine carcinoma arising in lobar bronchi (75%), main stem bronchi (10%), or the lung parenchyma (15%).48–50 Mucoepidermoid carcinoma (e-Fig. 55-10) and adenoid cystic carcinoma (cylindromas) (Fig. 55-11) arise from the salivary-type mucous cells of the submucosa along the tracheobronchial tree. Adenoid cystic carcinoma is extremely rare in children.51 Patients with carcinoid tumor and mucoepidermoid carcinoma frequently present with wheezing, cough, hemoptysis, or pneumonia.48,52,53 Association with the carcinoid syndrome in children with carcinoid tumor is exceedingly rare. The differential diagnosis of these rare tumors includes foreign body aspiration, granulomatous infection, and asthma with mucus plugging.
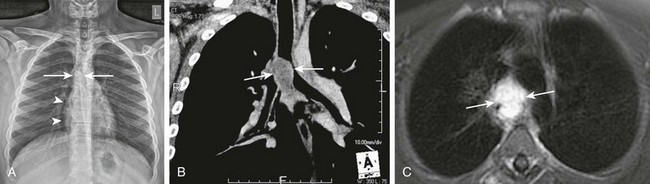
Figure 55-11 Adenoid cystic carcinoma.
A 14-year-old boy with progressively worsening chronic cough and respiratory difficulty for 1 year who presented with hoarseness and crepitations over the neck. A, A chest radiograph shows pneumomediastinum (arrowheads) and an apparent soft tissue density projecting over the carina (arrows). B, A coronal enhanced computed tomography image shows a lobulated soft tissue mass (arrows) centered near the carina, which results in narrowing of the adjacent airway. C, An axial contrast-enhanced T1-weighted magnetic resonance image demonstrates avid enhancement of the mass (arrows).
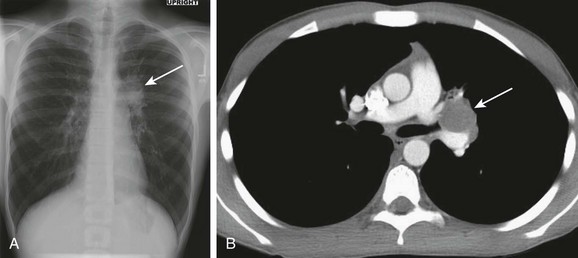
e-Figure 55-10 Endobronchial mucoepidermoid carcinoma.
A 14-year-old boy presented with cough and fever that had lasted for 1 month. A, A chest radiograph shows a right hilar mass (arrow). B, An enhanced axial computed tomography image demonstrates a well-circumscribed, heterogeneously enhancing mass within the left main stem bronchus (arrow). Surgical pathology was consistent with endobronchial mucoepidermoid carcinoma.
Imaging: The radiographic presentation depends on the size and location of tumor within the airway or lungs. A centrally located tumor is typically seen as an intraluminal soft tissue attenuation mass with associated distal atelectasis or obstructive pneumonitis. A peripherally located tumor is commonly seen as a sharply marginated, oval, or lobulated intraluminal or exophytic mass.54–58 Associated stippled calcification is seen in up to 30% of carcinoid tumors. CT is particularly helpful for detecting and defining extrabronchial portion of the tumor and associated adenopathy.
Treatment and Follow-up: The current treatment of choice is surgical resection. Chemotherapy and radiotherapy are reserved for tumors with incomplete surgical resection. Overall survival rate for carcinoid and mucoepidermoid carcinoma is approximately 90%, whereas adenoid cystic carcinoma has a poorer survival rate of 55% because of higher likelihood of distant metastases.59
Bronchogenic Carcinoma
Etiology: Traditionally, the term bronchogenic carcinoma has been used to include both small cell and non–small cell lung cancers (including squamous cell carcinoma, large cell carcinoma, and adenocarcinoma). These tumors are very rare in childhood. In the most recent WHO classification guideline, the term bronchogenic carcinoma is not used and each tumor type is listed individually.47 Among these tumors, adenocarcinoma is the most common in children.51 Most children with adenocarcinoma present with advanced disease, which results in high mortality. Small cell carcinoma, squamous cell carcinoma, and large cell carcinoma are very rare in children.60,61
Imaging: On chest radiography, these tumors often present as a solitary pulmonary nodule or central mass, often associated with postobstructive atelectasis or consolidation (e-Fig. 55-12). On CT, the attenuation and enhancement of masses vary substantially among different types of tumors. Concomitant mediastinal or hilar adenopathy as well as malignant pleural effusion may also present. Aggressive tumors may also invade adjacent mediastinal or osseous structures.
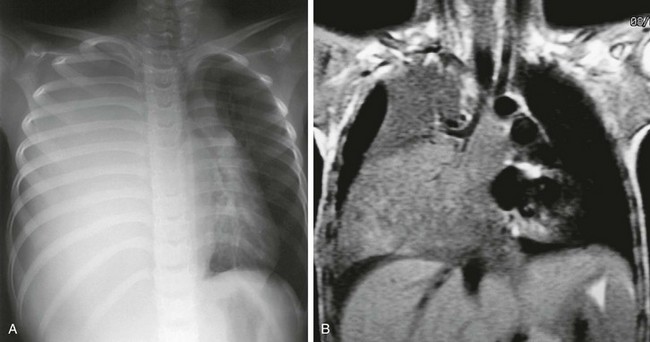
e-Figure 55-12 Bronchogenic carcinoma.
A 7-year-old girl with cough. A, A chest radiograph demonstrates a large mass occupying the right hemithorax, with contralateral mediastinal shift. B, A coronal T1-weighted magnetic resonance image shows a large right-sided mass encasing the bronchus, abutting the thymus, and extending into the mediastinum. Pathology identified the mass as squamous cell carcinoma. (From Fudge TL, Ochsner JL, Mills NL. Clinical spectrum of pulmonary hamartomas. Ann Thorac Surg. 1980;30(1):36-39.)
Pleuropulmonary Blastoma
Etiology: Pleuropulmonary blastoma (PPB) is an embryonal tumor of the lung, and it only occurs in young children. Over 90% of affected patients are below 6 years old at diagnosis. It recapitulates the morphogenesis of the fetal lung and may be regarded as a dysontogenetic analog to Wilms tumor, neuroblastoma, and hepatoblastoma. PPB contains primitive mesenchyma and varying degrees of more mature cartilage, skeletal and smooth muscle, and fibrous tissue. PPB is associated with hereditary tumor predisposition syndrome. Of the affected children, 25% to 30% have family members at risk of other dysplastic and neoplastic conditions.62 The three PPB types are (1) type I (purely cystic with primitive mesenchymal cells beneath an intact epithelium), (2) type II (cystic and solid, mesenchymal cells overgrow the septa), and (3) type III (purely solid, complex sarcomatous neoplasm). Type I lesions occur earlier at a median age of 9 months and have a more favorable prognosis, with 85% to 90% overall survival rate.63 Type II and III lesions occur at a median age of 36 and 42 months, with an overall survival of 60% and 45%, respectively.64 It has been suggested that PPB is probably the same malignant tumor that has been reported in previous studies as mesenchymal sarcoma, malignant mesenchymoma, embryonal sarcoma, primary pulmonary rhabdomyosarcoma, embryonal sarcoma, primary pulmonary rhabdomyosarcoma arising in congenital lung cysts, or pulmonary blastoma in children.65,66 Current data support the assertion that congenital lung cysts do not degenerate to become PPB but that cystic type I PPB may progress to more aggressive type II or type III PPB.67–71 PPB may metastasize to the central nervous system, bone, and liver.
Imaging: Imaging appearances of PPB depend on the type. PPBs present as solid, cystic, or mixed lesions. On chest radiography, the tumor may appear as a nodule or small mass that rapidly grows or as a large mass occupying the hemithorax. A large PPB may often result in mass effect on mediastinal structures. On CT, type 1 PPB is typically a cystic lesion often associated with multiple septations (Fig. 55-13), whereas type 3 PPB is a heterogeneously enhancing solid mass. Type 2 PPB has a combination of both cystic and solid components (Fig. 55-14). Type 1 PPB is often indistinguishable from CPAM on the basis of radiologic evaluation.72 Approximately, 25% of patients with PPB have other embryonal tumors, most commonly renal cystic nephroma.73,74 Other tumors less commonly seen in patients with PPB include medulloblastoma, thyroid dysplasia, germ cell neoplasms, and ovarian teratoma.75
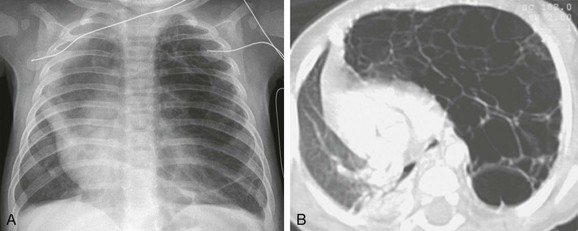
Figure 55-13 Cystic pleuropulmonary blastoma.
A 4-month-old boy with worsening respiratory distress. A, A chest radiograph demonstrates a large radiolucent lesion with fine septations and multiple cysts within the left lung resulting in mediastinal shift. B, An axial lung window computed tomography image shows multiple cysts of varying sizes with mass effect on the mediastinum and contralateral right lung.
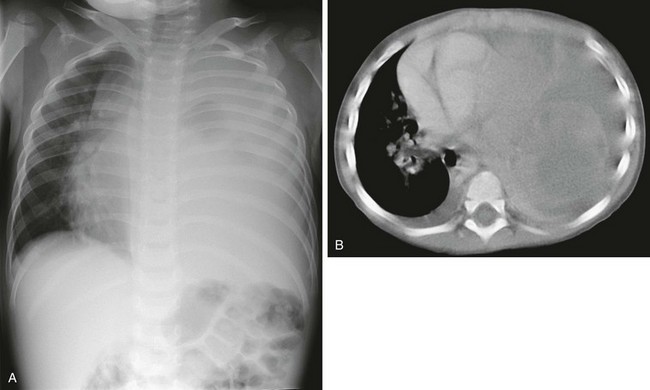
Figure 55-14 Mixed cystic and solid pleuropulmonary blastoma in a 2-year-old boy.
A, A chest radiograph shows a completely opacified left hemithorax with mass effect producing a rightward cardiomediastinal shift. B, An axial enhanced computed tomography image demonstrates bilateral pleural effusion and a large, left-sided predominantly solid, pulmonary and mediastinal heterogeneously enhancing mass.
Treatment and Follow-up: The current treatment of choice for PPB is lobectomy or pneumonectomy.76 Chemotherapy and local radiotherapy are adjuvant therapies if residual disease is present. Many congenital cystic lung lesions are removed surgically because of the risk of recurrent infection and the possibility of underlying neoplasm. Lesions that are not removed should be monitored closely. If the cystic lesion develops a solid component or if a family history of pediatric neoplasm exists, surgical resection is recommended.
Epithelioid Hemangioma
Etiology: Pulmonary epithelioid hemnagioendothelioma (PEH) is a rare tumor arising from endothelium with borderline malignant potential. PEH was first described by Dail and Liebow in 1975 as an intravascular bronchoalveolar tumor that may affect bone, soft tissue, liver, and lung. Later studies revealed that intravascular bronchoalveolar tumor and PEH are different manifestations of the same disease.77,78 Although most of the affected patients are adult women, it may also occur in children.79 The majority of patients are asymptomatic, and lesions are typically detected incidentally.80
Imaging: On chest radiography, PEH typically presents as bilateral multiple pulmonary nodules ranging from 5 mm to 2 cm in diameter (e-Fig. 55-15, A). Some patients may present with a solitary lesion. Concomitant hilar adenopathy and pleural effusion are seen in less than 10% of cases.81 On CT, multiple well-defined or ill-defined perivascular nodules located near medium-sized vessels and bronchi are usually seen (see e-Fig. 55-15, B). Some pulmonary nodules may show calcification.82,83
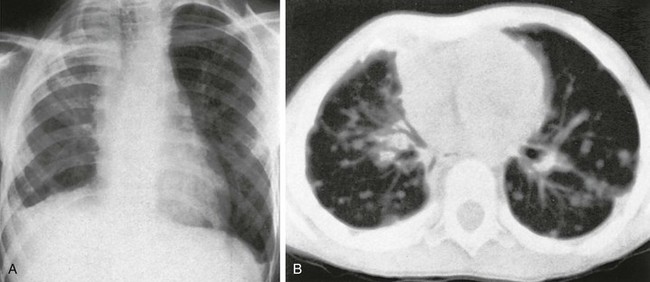
e-Figure 55-15 Epithelial hemangioendothelioma in an 8-year-old girl.
A, A chest radiograph shows mild scoliosis, a smaller right hemithorax, and extensive pleural thickening. Nodular parenchymal opacities are present in both lungs. B, An axial lung window computed tomography (CT) image shows multiple noncalcified parenchymal nodules, greater in number on the right, associated hilar adenopathy, and right-sided volume loss. Other CT sections revealed bilateral pleural thickening, greater on the right than on the left, and extensive periosteal reaction involving several ribs on the right. (Courtesy Dr. Robert Kaufmann, Memphis, TN.)
Treatment and Follow-up: Surgical resection is preferred in patients with solitary or a limited number of pulmonary lesions localized within the one lobe. Chemotherapy, radiotherapy, and interferon are other treatment choices that are associated with variable results.84 Close follow-up with no active therapy is usually used for asymptomatic patients with multiple lesions.80 Recently, vascular endothelium growth factor was proposed as a potential treatment for PEH.
Secondary Malignant Conditions
Secondary malignant conditions of the lungs may be caused by metastatic disease or from systemic diseases such as leukemia or lymphoma. Metastatic disease is, by far, the most common cause of pulmonary malignancy in childhood. Metastatic tumors accounts for approximately 80% of all lung tumors in children.1 Osteogenic sarcoma and Wilms tumor are most common tumors with pulmonary metastasis in children. Secondary pulmonary involvement in systemic diseases such as leukemia or lymphoma is rare but could occur.
Metastases
Etiology: Pulmonary metastatic disease most often occurs hematogenously via the pulmonary arterial system. However, it may also occur via lymphatics, airways, or direct invasion. Pediatric tumors that have a propensity to metastasize to the lung are listed in the order of frequency by anatomic system in Table 55-5.
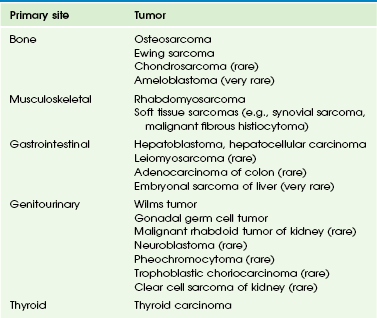
Imaging: On imaging studies, most metastatic lesions are seen as round and sharply marginated and of homogeneous soft tissue attenuation. They tend to be located subpleurally in the outer two thirds of the lung. Metastatic lesions often appear to be directly contiguous with a pulmonary artery branch, reflecting their hematogenous origin. A greater number of metastatic lesions are located at the lung bases rather than in the upper lobes, likely because of gravity-dependent increased basilar blood flow. Lymphangitic tumors usually present as reticular or reticulonodular opacities on chest radiography. Thickening of interlobular or interlobar septa and bronchovascular bundles are often seen on CT of children with lymphangitic tumor spread.
CT is sensitive but not specific in detecting pulmonary metastasis. No specific CT features (e.g., location, attenuation, size, margin characteristics) can reliably distinguish benign lesions from malignant pulmonary lesions. However, in general, solitary pulmonary lesion larger than 5 mm in diameter with sharp margins, especially when multiple, are usually malignant.85 Pulmonary nodules that decrease in size during antineoplastic therapy are usually assumed to be malignant, whereas those that decrease in size without therapy or remain stable during 12 months of follow-up are likely benign.
Although the imaging appearances of the majority of metastastic pulmonary nodules are nonspecific, some primary tumors may have a characteristic appearance of pulmonary metastasis. Metastatic osteosarcoma may ossify (Fig. 55-16), cavitate, or present with acute pneumothorax. Lymphangitic spread is most commonly seen in children with rhabdomyosarcoma, neuroblastoma, and lymphoma.
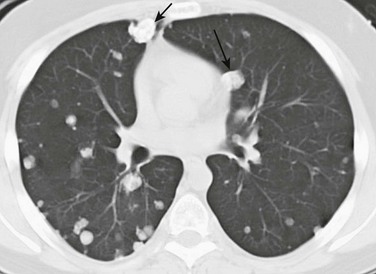
Treatment and Follow-up: Pulmonary metastases have been reported in 10% to 20% of patients with osteogenic sarcoma at initial diagnosis. Approximately 40% to 55% of patients with nonmetastatic osteosarcoma develop lung metastases in the later stages of the disease.86–90 Number, distribution, and timing but not the size of lung metastases are of prognostic value for survival.91,92 The best treatment for this group of patients is a combination of metastatectomy and adjuvant chemotherapy.93,94
The lung is the most common site of metastatic disease in children with Wilms tumor. Traditionally, treatment strategy for metastatic Wilms tumor has been based on lung lesions detectable with chest radiography. Currently, controversy exists over the optimal way for managing small pulmonary lesions that are detected only with CT and are not apparent on chest radiography because of the potential lung toxicity associated with aggressive therapy.95 Previous studies96,97 suggested that majority of these pulmonary lesions represent metastases and patients with these lesions probably require more potent chemotherapy than those children without metastatic disease.98
Leukemia
Etiology: Approximately 20% to 60% of patients with leukemia have histologic evidence of lung involvement at autopsy; but fewer than 5% of these patients present with lung abnormality suggesting leukemic infiltrates on chest radiography.99 Pulmonary involvement of leukemia is most often seen in patients with acute monocytic and myelogenous leukemia.100
Imaging: On chest radiography, leukemic infiltrates usually show a diffuse reticular pattern of opacities (Fig. 55-17). Pulmonary nodules and focal homogeneous opacities are also reported in patients with leukemic involvement of lungs. On CT, interstitial thickening in peribronchial distribution, small pulmonary nodules in centrilobular or peribronchovascular distribution, and focal areas of consolidation are common imaging findings.101,102
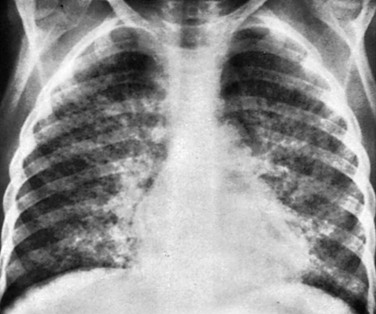
Figure 55-17 Pulmonary leukemia.
A 7-year-old boy with biopsy-proven leukemia of the lungs. A chest radiograph shows a reticulonodular pattern opacity with central confluence. Relative sparing of the apices and the lateral segments of both lungs is evident. No radiographic evidence for adenopathy is present.
Lymphoma
Etiology: The lung is not commonly involved in lymphoma; involvement is 12% with Hodgkin disease (HD) and 10% with non-Hodgkin lymphoma (NHL) in pediatric patients. Lung involvement is almost always seen at initial presentation rather than at disease relapse.104 In both HD and NHL, the underlying mechanisms of lung involvement from lymphoma are hematogenous spread, lymphangitic dissemination, and, less frequently, direct invasion.
Imaging: Lung involvement of lymphoma in pediatric patients typically presents as one of three patterns: (1) presence of single or multiple pulmonary nodules with irregular borders and sometimes central cavitation, which is the most common radiographic pattern (Figs. 55-18 and 55-19); (2) reticular interstitial opacities, which result from venous or lymphatic obstruction caused by hilar or mediastinal adenoapthy, or from interstitial tumor deposition; and (3) lobar or segmental consolidation, which may mimic an infectious process.105–108 Occasionally, multiple tiny pulmonary nodules in a miliary pattern in children with HD may simulate miliary tuberculosis.109,110 Pleural effusion is found in less than 5% of children with lymphoma.
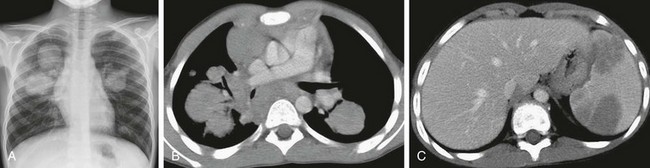
Figure 55-18 Hodgkin lymphoma.
A 13-year-old boy who presented with a cough that had lasted for 6 months. A, A chest radiograph shows several pulmonary masses in both lungs of varying sizes predominantly in central location. B, An axial enhanced computed tomography (CT) image demonstrates several, bilateral pulmonary masses of soft tissue density and varying sizes in addition to anterior and posterior mediastinal lymphadenopathy. C, An axial enhanced CT image of the upper abdomen shows several hypodense lesions in the spleen likely representing splenic involvement of lymphoma.
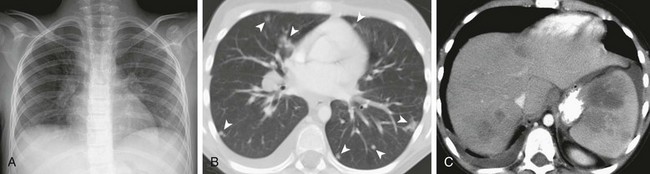
Figure 55-19 Hodgkin lymphoma.
A 12-year-old boy with a history of ataxia telangiectasia, who presented with fevers and intermittent dry cough. He had daily fevers with a maximum temperature of 104°F, worse at night, and night sweats. A, A chest radiograph shows mediastinal and hilar lymphadenopathy, which prompted computed tomography (CT) evaluation. B, An axial lung window CT image demonstrates multiple, small, bilateral ill-defined pulmonary nodules (arrowheads), a small right pleural effusion in addition to lymphadenopathy. C, An axial enhanced CT image shows numerous low attenuation lesions in the liver and spleen. Biopsy samples from his liver and pleural fluid confirmed the diagnosis of Hodgkin lymphoma.
Although the imaging appearance of lung involvement in children with HD and NHL is similar on imaging studies, lung involvement is usually concomitantly present with mediastinal or hilar lymphadenopathy in children with HD, whereas lung involvement may occur without associated lymphadenopathy in NHL.105,111
Treatment and Follow-up: Chemotherapy is the current treatment of choice. Although infection is more common in pediatric lymphoma patients, especially those undergoing treatment, development of new pulmonary lesions with a poor response to antibiotics may represent lymphoma lesions and therefore should promptly be biopsied for a definitive diagnosis. Pulmonary lesions from lymphoma usually decrease in size, disappear, or leave a parenchymal scar after chemotherapy treatment.112
 What the Clinician Needs to Know
What the Clinician Needs to Know
1. Pulmonary mass: size, number, site (parenchymal, intraluminal, extraluminal)
2. Characteristics of mass: fat, calcification, cyst, cavitation, enhancement
3. Reticulonodular interstitial pattern: distribution (centrilobular, intralobular, peribronchial, perivascular, subpleural)
4. Associated parenchymal changes: consolidation, atelectasis, ground glass opacification
5. Associated features: mediastinal adenopathy, pleural effusion, extrathoracic manifestation, associated tumoral lesions (syndrome)
Dishop, MK, Kuruvilla, S. Primary and metastatic lung tumors in the pediatric population: a review and 25-year experience at a large children’s hospital. Arch Pathol Lab Med. 2008;132(7):1079–1103.
Do, KH, Lee, JS, et al. Pulmonary parenchymal involvement of low-grade lymphoproliferative disorders. J Comput Assist Tomogr. 2005;29(6):825–830.
McCahon, E. Lung tumours in children. Paediatr Respir Rev. 2006;7(3):191–196.
Pickhardt, PJ, Siegel, MJ, et al. Posttransplantation lymphoproliferative disorder in children: clinical, histopathologic, and imaging features. Radiology. 2000;217(1):16–25.
Yikilmaz, A, Lee, EY. CT imaging of mass-like nonvascular pulmonary lesions in children. Pediatr Radiol. 2007;37(12):1253–1263.
References
1. Dishop, MK, Kuruvilla, S. Primary and metastatic lung tumors in the pediatric population: a review and 25-year experience at a large children’s hospital. Arch Pathol & Lab Med. 2008;132(7):1079–1103.
2. Gjevre, JA, Myers, JL, Prakash, UB. Pulmonary hamartomas. Mayo Clin Proc. 1996;71(1):14–20.
3. Nicholson, A, Tomashefski, J, Popper, H. Tumours of the lung. In: Travis W, Brambilla E, Muler-Hermelink K, et al, eds. World Health Organization classification of tumours, pathology & genetics, tumors of the lung, pleura, thymus and heart. Lyon: IARC; 2004:13–14.
4. Siegelman, SS, Khouri, NF, Scott, WW, Jr., et al. Pulmonary hamartoma: CT findings. Radiology. 1986;160(2):313–317.
5. Fudge, TL, Ochsner, JL, Mills, NL. Clinical spectrum of pulmonary hamartomas. Ann Thorac Surg. 1980;30(1):36–39.
6. Rodriguez, FJ, Aubry, MC, Tazelaar, HD, et al. Pulmonary chondroma: a tumor associated with Carney triad and different from pulmonary hamartoma. Am J Surg Pathol. 2007;31(12):1844–1853.
7. Carney, JA. Carney triad: a syndrome featuring paraganglionic, adrenocortical, and possibly other endocrine tumors. J Clin Endocrinol Metab. 2009;94(10):3656–3662.
8. Soldatski, IL, Onufrieva, EK, Steklov, AM, et al. Tracheal, bronchial, and pulmonary papillomatosis in children. Laryngoscope. 2005;115(10):1848–1854.
9. Szyszko, T, Gnanasegaran, G, Barwick, T, et al. Respiratory papillomatosis of lung and F-18 FDG PET-CT. Clin Nucl Med. 2009;34(8):521–522.
10. Kramer, SS, Wehunt, WD, Stocker, JT, et al. Pulmonary manifestations of juvenile laryngotracheal papillomatosis. AJR. 1985;144(4):687–694.
11. McKay, SP, Shinhar, SY, Belenky, WM. Pulmonary involvement in a case of juvenile-onset recurrent respiratory papillomatosis. Ear Nose Throat J. 2003;82(6):447–449.
12. Scott, KJ, Greinwald, JH, Jr., Darrow, D, et al. Endobronchial tumors in children: an uncommon clinical entity. Ann Otol Rhinol Laryngol. 2001;110(1):63–69.
13. Gelinas, JF, Manoukian, J, Cote, A. Lung involvement in juvenile onset recurrent respiratory papillomatosis: a systematic review of the literature. Int J Pediatr Otorhinolaryngol. 2008;72(4):433–452.
14. Faul, JL, Berry, GJ, Colby, TV, et al. Thoracic lymphangiomas, lymphangiectasis, lymphangiomatosis, and lymphatic dysplasia syndrome. Am J Respir Crit Care Med. 2000;161(3 Pt 1):1037–1046.
15. Minato, H, Kaji, S, Kinoshita, E, et al. Solitary intrapulmonary cystic lymphangioma in an infant: a case report with literature review. Pathol Res Pract. 2010;206(12):851–856.
16. Arbell, D, Koplewitz, BZ, Udassin, R. Anterior mediastinal cystic lesion: excise rather than delay. Isr Med Assoc J. 2007;9(8):622–623.
17. Cerfolio, RJ, Allen, MS, Nascimento, AG, et al. Inflammatory pseudotumors of the lung. Ann Thorac Surg. 1999;67(4):933–936.
18. Narla, LD, Newman, B, Spottswood, SS, et al. Inflammatory pseudotumor. Radiographics. 2003;23(3):719–729.
19. Coffin, CM, Watterson, J, Priest, JR, et al. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19(8):859–872.
20. Coffin, CM, Hornick, JL, Fletcher, CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 1995;31(4):509–520.
21. Rasalkar, DD, Chu, WC, To, KF, et al. Radiological appearance of inflammatory myofibroblastic tumour. Pediat Blood Cancer. 2010;54(7):1029–1031.
22. Varoczy, L, Gergely, L, Illes, A. Diagnostics and treatment of pulmonary BALT lymphoma: a report on four cases. Ann Hematol. 2003;82(6):363–366.
23. Swigris, JJ, Berry, GJ, Raffin, TA, et al. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122(6):2150–2164.
24. Do, KH, Lee, JS, Seo, JB, et al. Pulmonary parenchymal involvement of low-grade lymphoproliferative disorders. J Comp Assist Tomogr. 2005;29(6):825–830.
25. Teruya-Feldstein, J, Kingma, DW, Weiss, A, et al. Chemokine gene expression and clonal analysis of B cells in tissues involved by lymphoid interstitial pneumonitis from HIV-infected pediatric patients. Mod Pathol. 2001;14(10):929–936.
26. Fauci, AS, Haynes, BF, Costa, J, et al. Lymphomatoid Granulomatosis. Prospective clinical and therapeutic experience over 10 years. NEJM. 1982;306(2):68–74.
27. Lee, JS, Tuder, R, Lynch, DA. Lymphomatoid granulomatosis: radiologic features and pathologic correlations. AJR. 2000;175(5):1335–1339.
28. Jung, KH, Sung, HJ, Lee, JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55(5):386–390.
29. Basgoz, N, Preiksaitis, JK. Post-transplant lymphoproliferative disorder. Infect Dis Clin North Am. 1995;9(4):901–923.
30. Newell, KA, Alonso, EM, Whitington, PF, et al. Posttransplant lymphoproliferative disease in pediatric liver transplantation. Interplay between primary Epstein-Barr virus infection and immunosuppression. Transplantation. 1996;62(3):370–375.
31. Shroff, R, Rees, L. The post-transplant lymphoproliferative disorder-a literature review. Pediatr Nephrol. 2004;19(4):369–377. [(Berlin, Germany)].
32. Allen, U, Hebert, D, Moore, D, et al. Epstein-Barr virus-related post-transplant lymphoproliferative disease in solid organ transplant recipients, 1988-97: a Canadian multi-centre experience. Pediatr Transplant. 2001;5(3):198–203.
33. Pickhardt, PJ, Siegel, MJ, Hayashi, RJ, et al. Posttransplantation lymphoproliferative disorder in children: clinical, histopathologic, and imaging features. Radiology. 2000;217(1):16–25.
34. Montone, KT, Litzky, LA, Wurster, A, et al. Analysis of Epstein-Barr virus-associated posttransplantation lymphoproliferative disorder after lung transplantation. Surgery. 1996;119(5):544–551.
35. Nalesnik, MA, Makowka, L, Starzl, TE. The diagnosis and treatment of posttransplant lymphoproliferative disorders. Curr Prob Surg. 1988;25(6):367–472.
36. Ohta, H, Fukushima, N, Ozono, K. Pediatric post-transplant lymphoproliferative disorder after cardiac transplantation. Int J Hematol. 2009;90(2):127–136.
37. Harris, KM, Schwartz, ML, Slasky, BS, et al. Posttransplantation cyclosporine-induced lymphoproliferative disorders: clinical and radiologic manifestations. Radiology. 1987;162(3):697–700.
38. Donnelly, LF, Frush, DP, Marshall, KW, et al. Lymphoproliferative disorders: CT findings in immunocompromised children. AJR. 1998;171(3):725–731.
39. Lim, GY, Newman, B, Kurland, G, et al. Posttransplantation lymphoproliferative disorder: manifestations in pediatric thoracic organ recipients. Radiology. 2002;222(3):699–708.
40. Siegel, MJ, Lee, EY, Sweet, SC, et al. CT of posttransplantation lymphoproliferative disorder in pediatric recipients of lung allograft. AJR. 2003;181(4):1125–1131.
41. Carignan, S, Staples, CA, Muller, NL. Intrathoracic lymphoproliferative disorders in the immunocompromised patient: CT findings. Radiology. 1995;197(1):53–58.
42. Scarsbrook, AF, Warakaulle, DR, Dattani, M, et al. Post-transplantation lymphoproliferative disorder: the spectrum of imaging appearances. Clin Radiol. 2005;60(1):47–55.
43. Borhani, AA, Hosseinzadeh, K, Almusa, O, et al. Imaging of posttransplantation lymphoproliferative disorder after solid organ transplantation. Radiographics. 2009;29(4):981–1000. [discussion 1002].
44. McCormack, L, Hany, TI, Hubner, M, et al. How useful is PET/CT imaging in the management of post-transplant lymphoproliferative disease after liver transplantation? Am J Transplant. 2006;6(7):1731–1736.
45. Dierickx, D, Tousseyn, T, De Wolf-Peeters, C, et al. Management of posttransplant lymphoproliferative disorders following solid organ transplant: an update. Leukemia Lymphoma. 2011;52(6):950–951.
46. Taylor, AL, Marcus, R, Bradley, JA. Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit Rev Oncol Hematol. 2005;56(1):155–167.
47. Travis, W. Pathology and genetics of tumours of the lung, pleura, thymus and heart. In: World Health Organization, International Agency for Research on Cancer. Lyon, Oxford: IARC Press, Oxford University Press; 2004.
48. McDougall, JC, Gorenstein, A, Unni, K, et al. Carcinoid and mucoepidermoid carcinoma of bronchus in children. Ann Otol Rhinol Laryngol. 1980;89(5 Pt 1):425–427.
49. Al-Qahtani, AR, Di Lorenzo, M, Yazbeck, S. Endobronchial tumors in children: Institutional experience and literature review. J Ped Surg. 2003;38(5):733–736.
50. McCahon, E. Lung tumours in children. Pediatr Respir Rev. 2006;7(3):191–196.
51. Hartman, GE, Shochat, SJ. Primary pulmonary neoplasms of childhood: a review. Ann Thorac Surg. 1983;36(1):108–119.
52. Broaddus, RR, Herzog, CE, Hicks, MJ. Neuroendocrine tumors (carcinoid and neuroendocrine carcinoma) presenting at extra-appendiceal sites in childhood and adolescence. Arch Pathol Lab Med. 2003;127(9):1200–1203.
53. Hause, DW, Harvey, JC. Endobronchial carcinoid and mucoepidermoid carcinoma in children. J Surg Oncol. 1991;46(4):270–272.
54. Yikilmaz, A, Lee, EY. CT imaging of mass-like nonvascular pulmonary lesions in children. Pediatr Radiol. 2007;37(12):1253–1263.
55. Granata, C, Battistini, E, Toma, P, et al. Mucoepidermoid carcinoma of the bronchus: case report and review of the literature. Pediatr Pulmonol. 1997;23(3):226–232.
56. Kim, TS, Lee, KS, Han, J, et al. Mucoepidermoid carcinoma of the tracheobronchial tree: radiographic and CT findings in 12 patients. Radiology. 1999;212(3):643–648.
57. Chong, S, Lee, KS, Chung, MJ, et al. Neuroendocrine tumors of the lung: clinical, pathologic, and imaging findings. Radiographics. 2006;26(1):41–57. [discussion 58].
58. Connor, GF, Fishman, EK. Endobronchial carcinoid in a child: depiction with three-dimensional volume rendering. Pediatr Radiol. 2004;34(12):1008–1011.
59. Molina, JR, Aubry, MC, Lewis, JE, et al. Primary salivary gland-type lung cancer: spectrum of clinical presentation, histopathologic and prognostic factors. Cancer. 2007;110(10):2253–2259.
60. Travis, WD, Travis, LB, Devesa, SS. Lung cancer. Cancer. 1995;75(1 suppl):191–202.
61. Yu, DC, Grabowski, MJ, Kozakewich, HP, et al. Primary lung tumors in children and adolescents: a 90-year experience. J Pediatr Surg. 2010;45(6):1090–1095.
62. Priest, JR, Watterson, J, Strong, L, et al. Pleuropulmonary blastoma: a marker for familial disease. J Pediatr. 1996;128(2):220–224.
63. Priest, JR, Hill, DA, Williams, GM, et al. Type I pleuropulmonary blastoma: a report from the International Pleuropulmonary Blastoma Registry. J Clin Oncol. 2006;24(27):4492–4498.
64. Priest, JR, McDermott, MB, Bhatia, S, et al. Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer. 1997;80(1):147–161.
65. Pai, S, Eng, HL, Lee, SY, et al. Rhabdomyosarcoma arising within congenital cystic adenomatoid malformation. Pediatr Blood Cancer. 2005;45(6):841–845.
66. Pai, S, Eng, HL, Lee, SY, et al. Correction: Pleuropulmonary blastoma, not rhabdomyosarcoma in a congenital lung cyst. Pediatr Blood Cancer. 2007;48(3):370–371.
67. Berbel, O, Pellicer, C, Lopez-Andreu, JA, et al. Cystic adenomatoid malformation of the lung: clinical evolution and management. Eur J Pediatr. 2000;159(8):621–622.
68. Laberge, JM, Puligandla, P, Flageole, H. Asymptomatic congenital lung malformations. Semin Pediatr Surg. 2005;14(1):16–33.
69. Langston, C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12(1):17–37.
70. Wright, JR, Jr. Pleuropulmonary blastoma: A case report documenting transition from type I (cystic) to type III (solid). Cancer. 2000;88(12):2853–2858.
71. Miniati, DN, Chintagumpala, M, Langston, C, et al. Prenatal presentation and outcome of children with pleuropulmonary blastoma. J Pediatr Surg. 2006;41(1):66–71.
72. Orazi, C, Inserra, A, Schingo, PM, et al. Pleuropulmonary blastoma, a distinctive neoplasm of childhood: report of three cases. Pediatr Radiol. 2007;37(4):337–344.
73. Ishida, Y, Kato, K, Kigasawa, H, et al. Synchronous occurrence of pleuropulmonary blastoma and cystic nephroma: possible genetic link in cystic lesions of the lung and the kidney. Med Pediatr Oncol. 2000;35(1):85–87.
74. Boman, F, Hill, DA, Williams, GM, et al. Familial association of pleuropulmonary blastoma with cystic nephroma and other renal tumors: a report from the International Pleuropulmonary Blastoma Registry. J Pediatr. 2006;149(6):850–854.
75. Lallier, M, Bouchard, S, Di Lorenzo, M, et al. Pleuropulmonary blastoma: a rare pathology with an even rarer presentation. J Pediatr Surg. 1999;34(7):1057–1059.
76. Indolfi, P, Casale, F, Carli, M, et al. Pleuropulmonary blastoma: management and prognosis of 11 cases. Cancer. 2000;89(6):1396–1401.
77. Dail, DH, Liebow, AA, Gmelich, JT, et al. Intravascular, bronchiolar, and alveolar tumor of the lung (IVBAT). An analysis of twenty cases of a peculiar sclerosing endothelial tumor. Cancer. 1983;51(3):452–464.
78. Weiss, SW, Ishak, KG, Dail, DH, et al. Epithelioid hemangioendothelioma and related lesions. Semin Diagn Pathol. 1986;3(4):259–287.
79. Madhusudhan, KS, Srivastava, DN, Gamanagatti, S. Multifocal epithelioid hemangioendothelioma presenting with hemoptysis. Indian J Pediatr. 2010;77(6):699–700.
80. Kitaichi, M, Nagai, S, Nishimura, K, et al. Pulmonary epithelioid haemangioendothelioma in 21 patients, including three with partial spontaneous regression. Eur Respir J. 1998;12(1):89–96.
81. Ross, GJ, Violi, L, Friedman, AC, et al. Intravascular bronchioloalveolar tumor: CT and pathologic correlation. J Comp Assis Tomogr. 1989;13(2):240–243.
82. Luburich, P, Ayuso, MC, Picado, C, et al. CT of pulmonary epithelioid hemangioendothelioma. J Comp Assis Tomogr. 1994;18(4):562–565.
83. Mukundan, G, Urban, BA, Askin, FB, et al. Pulmonary epithelioid hemangioendothelioma: atypical radiologic findings of a rare tumor with pathologic correlation. J Comp Assist Tomogr. 2000;24(5):719–720.
84. Mehrabi, A, Kashfi, A, Fonouni, H, et al. Primary malignant hepatic epithelioid hemangioendothelioma: a comprehensive review of the literature with emphasis on the surgical therapy. Cancer. 2006;107(9):2108–2121.
85. Grampp, S, Bankier, AA, Zoubek, A, et al. Spiral CT of the lung in children with malignant extra-thoracic tumors: distribution of benign vs malignant pulmonary nodules. Eur Radiol. 2000;10(8):1318–1322.
86. Link, MP, Gebhardt, MC, Meyers, PA. Osteosarcoma. In: Pizzo PA, Poplack DG, eds. Principles and practice of pediatric oncology. Baltimore, MD: Lippincott Williams and Wilkins; 2002:1051–1089.
87. Ward, WG, Mikaelian, K, Dorey, F, et al. Pulmonary metastases of stage IIB extremity osteosarcoma and subsequent pulmonary metastases. J Clin Oncol. 1994;12(9):1849–1858.
88. Saeter, G, Hoie, J, Stenwig, AE, et al. Systemic relapse of patients with osteogenic sarcoma. Prognostic factors for long term survival. Cancer. 1995;75(5):1084–1093.
89. Kaste, SC, Pratt, CB, Cain, AM, et al. Metastases detected at the time of diagnosis of primary pediatric extremity osteosarcoma at diagnosis: imaging features. Cancer. 1999;86(8):1602–1608.
90. Kager, L, Zoubek, A, Potschger, U, et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2003;21(10):2011–2018.
91. Rasalkar, DD, Chu, WC, Lee, V, et al. Pulmonary metastases in children with osteosarcoma: characteristics and impact on patient survival. Pediatr Radiol. 2011;41(2):227–236.
92. Chen, F, Miyahara, R, Bando, T, et al. Prognostic factors of pulmonary metastasectomy for osteosarcomas of the extremities. Eur J Cardiothorac Surg. 2008;34(6):1235–1239.
93. Harting, MT, Blakely, ML, Jaffe, N, et al. Long-term survival after aggressive resection of pulmonary metastases among children and adolescents with osteosarcoma. J Pediatr Surg. 2006;41(1):194–199.
94. Kayton, ML. Pulmonary metastasectomy in pediatric patients. Thorac Surg Clin. 2006;16(2):167–183. [vi].
95. Meisel, JA, Guthrie, KA, Breslow, NE, et al. Significance and management of computed tomography detected pulmonary nodules: a report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys. 1999;44(3):579–585.
96. Green, DM. Use of chest computed tomography for staging and treatment of Wilms’ tumor in children. J Clin Oncol. 2002;20(12):2763–2764.
97. Green, DM, Fernbach, DJ, Norkool, P, et al. The treatment of Wilms’ tumor patients with pulmonary metastases detected only with computed tomography: a report from the National Wilms’ Tumor Study. J Clin Oncol. 1991;9(10):1776–1781.
98. Brisse, HJ, Smets, AM, Kaste, SC, et al. Imaging in unilateral Wilms tumour. Pediatr Radiol. 2008;38(1):18–29.
99. Maile, CW, Moore, AV, Ulreich, S, et al. Chest radiographic-pathologic correlation in adult leukemia patients. Investigat Radiol. 1983;18(6):495–499.
100. Azoulay, E, Fieux, F, Moreau, D, et al. Acute monocytic leukemia presenting as acute respiratory failure. Am J Respir Crit Care Med. 2003;167(10):1329–1333.
101. Sueyoshi, E, Uetani, M, Hayashi, K, et al. Adult T-cell leukemia with multiple pulmonary nodules due to leukemic cell infiltration. AJR. 1996;167(2):540–541.
102. Heyneman, LE, Johkoh, T, Ward, S, et al. Pulmonary leukemic infiltrates: high-resolution CT findings in 10 patients. AJR. 2000;174(2):517–521.
103. Darmon, M, Thiery, G, Ciroldi, M, et al. Intensive care in patients with newly diagnosed malignancies and a need for cancer chemotherapy. Crit Care Med. 2005;33(11):2488–2493.
104. Maturen, KE, Blane, CE, Strouse, PJ, et al. Pulmonary involvement in pediatric lymphoma. Pediatr Radiol. 2004;34(2):120–124.
105. Au, V, Leung, AN. Radiologic manifestations of lymphoma in the thorax. AJR. 1997;168(1):93–98.
106. Toma, P, Granata, C, Rossi, A, et al. Multimodality imaging of Hodgkin disease and non-Hodgkin lymphomas in children. Radiographics. 2007;27(5):1335–1354.
107. Lewis, ER, Caskey, CI, Fishman, EK. Lymphoma of the lung: CT findings in 31 patients. AJR. 1991;156(4):711–714.
108. Abramson, SJ, Price, AP. Imaging of pediatric lymphomas. Radiol Clin North Am. 2008;46(2):313–338. [ix].
109. Kalra, M, Dinand, V, Sachdeva, A, et al. Pediatric Hodgkin lymphoma presenting with pulmonary nodules and leukemoid reaction. Pediatr Blood Cancer. 2010;55(1):193–195.
110. Miyake, S, Yoshizawa, Y, Ohkouchi, Y, et al. Non-Hodgkin’s lymphoma with pulmonary infiltrates mimicking miliary tuberculosis. Int Med. 1997;36(6):420–423. [(Tokyo, Japan)].
111. Bae, YA, Lee, KS. Cross-sectional evaluation of thoracic lymphoma. Radiol Clin North Am. 2008;46(2):253–264. [viii].
112. White, KS. Thoracic imaging of pediatric lymphomas. J Thorac Imag. 2001;16(4):224–237.