62 Mycosis Fungoides
Epidemiology, Etiology, Genetics, and Cytogenetic Abnormalities
Mycosis fungoides is a rare lymphoma. The estimated annual incidence rate in the United States is only approximately 0.64 per 100,000, or fewer than 1000 new cases diagnosed each year.1 It accounts for only 2% of new cases of non-Hodgkin lymphoma. Although it is uncommon, it is the most common of the primary cutaneous T-cell lymphomas (CTCLs) and is distinguished from other CTCLs by its unique clinical and histologic features. It commonly affects older adults (median age 55 to 60 years); however, it may present in younger individuals with similar clinical findings and course.2 There is a 2 : 1 male predominance, without an established racial predilection.
The causal factors of mycosis fungoides and Sézary syndrome are unknown. Some retrospective studies have suggested an causal role for environmental chemical exposure, as a source of either chronic antigenic stimulation or toxic exposure. However, case-controlled studies failed to reveal any relation between occupational or recreational exposures to chemicals and the development of mycosis fungoides, refuting this hypothesis.3,4
A viral cause for mycosis fungoides was once proposed, based on the isolation of human T-cell leukemia/lymphoma virus 1 (HTLV-1) from the peripheral blood lymphocytes of a patient with a cutaneous lymphoma that resembled mycosis fungoides.5 However, it was later demonstrated that this patient actually had a peripheral T-cell lymphoma with skin involvement. The clinical characteristics of this distinct entity, HTLV-1–associated T-cell lymphoma, have now been described more precisely and are quite different from those of typical mycosis fungoides.
A few studies have demonstrated histocompatibility antigen associations linked to mycosis fungoides and Sézary syndrome, in particular Aw31, Aw32, B8, Bw38, and DR5.6,7 The significance of these immunogenetic findings is unclear, but may suggest a genetic predisposition. Specific chromosomal abnormalities also have been demonstrated in some cases, mostly deletions and translocations in chromosome 1 or 6. In one series, the presence of clonal abnormalities in 11 of 19 patients correlated with advanced-stage disease and a significantly reduced survival.8 In a second study, the detection of a chromosomal clone preceded relapse or progression of the disease.9
Cytokines have been implicated in the pathophysiology of mycosis fungoides and Sézary syndrome.10–12 However, whether cytokine abnormalities are primarily involved or are secondary processes in the pathogenesis is unclear. Studies have reported that soluble interleukin-2 (IL-2) receptor (sIL-2R) values in Sézary syndrome were significantly higher than for other malignant or inflammatory T-cell diseases and that the serum levels correlated with clinical course and Sézary cell count. The highest sIL-2R levels were found in patients with advanced disease.12 Other investigators have shown that peripheral blood mononuclear cells from patients with the Sézary syndrome expressed higher levels of IL-4 and lower levels of IL-2 and interferon (IFN) gamma after phytohemagglutinin stimulation, compared with normal controls.10
A number of studies have suggested that the malignant T cells in Sézary syndrome account for aberrant cytokine production, with increased production of T helper type 2 cytokines (e.g., IL-4, IL-5, IL-10) and decreased production of T helper type 1 cytokines (e.g., IL-2 and IFN gamma).13,14 Moreover, this aberrant cytokine production may cause the immune abnormalities seen occasionally in Sézary syndrome. These immune abnormalities may include decreased T-cell responses to antigens and mitogens, impaired cell-mediated cytotoxicity, including natural killer cell and lymphokine-activated killer cell activities, increased levels of serum immunoglobulin E (IgE) and IgA, and peripheral eosinophilia.
Pathologic Conditions
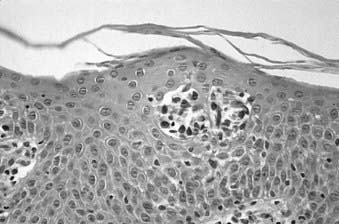
Skin biopsy with routine histologic examination is still considered the most important study to assist the clinician in establishing the diagnosis. The characteristic histopathologic findings of mycosis fungoides demonstrate abnormal cells infiltrating the epidermis (epidermotropism) as single cells or in clusters (Pautrier microabscesses) (Fig. 62-1). Typically, there is also an upper dermal infiltrate that includes cells similar to those seen in the epidermis, as well as variable proportions of histiocytes, eosinophils, and plasma cells. The criteria for a diagnosis of mycosis fungoides vary among pathologists. Based on the severity of the epidermal and dermal involvement, the categories “diagnostic of,” “consistent with,” and “suggestive of” mycosis fungoides have been recommended.15 Treatment programs specific for mycosis fungoides should be considered only in patients who have a biopsy that is diagnostic of or consistent with, not merely suggestive of, mycosis fungoides.
Immunoperoxidase staining indicates that the majority of cases of mycosis fungoides are associated with the helper T-cell phenotype (CD4+) and are also positive for CD2, CD3, CD45, and CD5. These cells may also be positive for CD25 and the p55 (alpha chain) subunit of the IL-2 receptor.16 Usually, although the neoplastic cells of mycosis fungoides retain the CD4 antigen on their cell surface, they lose other mature T-cell antigens, such as CD26 or Leu-9 (CD7). The loss of these mature T-cell antigens may help in the differential diagnosis of mycosis fungoides from benign dermatoses. Rare cases of mycosis fungoides have been demonstrated to be CD8+ (cytotoxic/suppressor T-cell phenotype).
Evaluation of skin biopsies to detect T-cell receptor (TCR) gene rearrangements (genotyping) can be helpful in the differential diagnosis of early mycosis fungoides. TCR gene rearrangements can be detected by Southern blot analysis17 or by methods using polymerase chain reaction (PCR) amplification.18 PCR amplification methods for frozen or paraffin tissue analysis are widely available and affordable. Genotyping is becoming an important diagnostic procedure whenever the routine histologic examination or immunophenotyping is equivocal in patients whose clinical presentation is strongly suggestive of mycosis fungoides or Sézary syndrome.
The pathologic characteristics of extracutaneous disease pose special problems. In the most common situation, enlarged lymph nodes may be biopsied and demonstrate changes of dermatopathic lymphadenitis, including the presence of sinus histiocytosis and an abundance of pigment-laden macrophages. In addition, there may be a variable number of atypical lymphocytes with cerebriform nuclei. The prognostic relevance of different degrees of infiltration by these abnormal cells led to the development of a lymph node classification system. In this system, lymph nodes are classified as LN0 to LN4. Category I (LN0–2) includes dermatopathic nodes and nodes with clusters of less than six atypical cells, category II (LN3) designates lymph nodes with clusters of 10 or more atypical cells, and category III (LN4) includes nodes that are partially or completely effaced by atypical cells.19 Detection of abnormal cells in the lymph nodes is facilitated by the use of Southern blot or PCR analysis. Potential neoplastic involvement with clonal TCR rearrangement may be demonstrated even in lymph nodes that show only dermatopathic changes on routine evaluation.19,20
Flow cytometry studies of the peripheral blood may show expansion of the CD4+/CD7− population reflective of circulating atypical lymphocytes of Sézary type.21 Patients may have atypical lymphocytes with cerebriform nuclei, the Sézary cells, in their peripheral blood. It is controversial as to how many or what percentage of Sézary cells constitutes a significant level to define the Sézary syndrome, and low levels of Sézary-like cells can be detected in the peripheral blood of patients with benign skin conditions.22 Although the original National Cancer Institute (NCI) classification used the criterion of greater than 5% of lymphocytes for significant blood involvement,23 the current practice by many mycosis fungoides referral centers is to use an absolute Sézary cell count of more than 1000/μL or a CD4+/CD8+ T-cell ratio of more than 10 for defining peripheral blood involvement.
Clinical Presentation
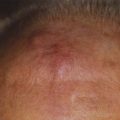
Mycosis fungoides often has a long natural history, and the median duration from the onset of skin symptoms to a diagnosis of mycosis fungoides may be 5 years or longer.24 In many patients, the disease presents initially in a premycotic phase with nonspecific, slightly scaling skin lesions that wax and wane for years. Biopsies are generally nondiagnostic during this phase of disease, and patients may respond to treatment with topical corticosteroids. Some of these patients experience an evolution of their disease and develop more typical patches or infiltrated plaques, from which a definitive diagnostic biopsy may be obtained. Repeated biopsies must be obtained from patients suspected of having mycosis fungoides, even when an initial biopsy is negative.
Infiltrated plaques may evolve into ulcerating or fungating tumors; however, the rapidity of this progression is unpredictable. The majority of patients with initial patch or plaque disease who have been treated at Stanford University have never progressed to have more advanced cutaneous disease.25,26 Cutaneous tumors may become infected, and sepsis may be the cause of death in individuals so affected. Occasional patients present de novo with tumors, so-called tumor d’emblée. Generalized dermal thickening from infiltrative disease may cause the classic but very unusual leonine facies of mycosis fungoides.
Another manifestation of skin involvement in mycosis fungoides is generalized erythroderma. The erythema may be accompanied by either atrophic or lichenified skin, and plaques or tumors may also be present. These patients are almost always intensely symptomatic from pruritus and scaling, and often have lymphadenopathy caused by diffuse and severe skin involvement. Many of these patients also have circulating abnormal cells in the peripheral blood that have the same microscopic appearance, immunophenotyping, and genotyping characteristics as the cells that infiltrate the epidermis. Patients with this complex of findings, generalized erythroderma, lymphadenopathy, and atypical T cells (Sézary cells) in the peripheral blood have Sézary syndrome.27 Patients with Sézary syndrome have a worse prognosis than erythrodermic patients with mycosis fungoides who do not have the other findings of the Sézary syndrome.
Routes of Spread
Many patients with mycosis fungoides have evidence of cutaneous disease only throughout their lifetime. Although molecular studies may reveal extracutaneous disease to be present in a significant proportion of patients, only 15% to 20% of patients with mycosis fungoides develop clinical problems related to extracutaneous disease. The likelihood of developing extracutaneous disease is related to the extent of skin involvement. In a series of patients at Stanford with extracutaneous disease, at that time none had limited plaque disease, 11 had generalized plaque disease, 39 had cutaneous tumors, and 27 had erythroderma.28 The most commonly identified route of extracutaneous spread of mycosis fungoides is to the regional lymphatics, usually in areas that drain significant sites of skin involvement. Visceral disease may be identified subsequently. The most common visceral sites of involvement identified are the lungs, the oral cavity and pharynx, and the central nervous system, but virtually any organ may be involved at autopsy in patients who have died of the disease.29
The risk of disease progression in 525 patients with mycosis fungoides treated at Stanford has been analyzed. Patients were considered to have had disease progression when one of the following events occurred: progression of their mycosis fungoides to a more advanced tumor-node-metastases-blood (TNMB) classification, a more advanced clinical stage, or death resulting from mycosis fungoides. The actuarial risk of disease progression at 10 years was 13% in limited plaque, 32% in generalized plaque, 72% in tumorous, and 57% for patients with erythroderma.30
The actuarial risk for developing extracutaneous disease in our 491 patients who presented with stage I to III disease was analyzed according to their initial T classification. The risk at 10 years was 2% in limited plaque, 9% in generalized plaque, 40% in tumorous, and 10% in patients with erythroderma. However, at the time these patients developed extracutaneous disease, none had limited plaque disease.30
Diagnostic and Staging Studies
The standard staging evaluation for patients with mycosis fungoides includes a comprehensive physical examination with careful examination of the skin (including the scalp, palms, soles, and perineum) and lymph nodes, a complete blood count with Sézary cell studies, screening chemistries (including lactate dehydrogenase), and chest x-ray examination. Based on observations that it is unlikely for patients with T1 or T2 skin involvement to present with extracutaneous disease at diagnosis, additional imaging studies (computed tomography [CT] or magnetic resonance imaging) or nuclear medicine scans (positron emission tomography [PET]) are not recommended unless the patient has clinically significant lymphadenopathy. Because patients with T3 or T4 disease are at greater risk for extracutaneous involvement, further imaging studies should be considered. At Stanford we obtain a CT scan of the chest, abdomen, and pelvis or total-body integrated PET-CT scan in these patients.31 Lymph node biopsies should be obtained if lymphadenopathy is present, because the presence of lymph node involvement affects the stage, prognosis, and management. Suspected sites of visceral involvement must be confirmed by appropriate imaging studies and biopsy when possible. Bone marrow involvement may often be detected in patients who meet the clinical criteria for Sézary syndrome,32 but is extremely uncommon in classical mycosis fungoides. Therefore, a bone marrow biopsy is not routinely used as part of the initial staging procedure for patients with mycosis fungoides.
Staging System
A TNMB staging system that has proved useful for mycosis fungoides was proposed at the Workshop on Mycosis Fungoides held at the NCI in 1978.23 This staging system has been revised to reflect updated prognostic information and be more consistent with current practice.33 Table 62-1 and Table 62-2 summarize the classification and TNMB categories.
Table 62-1 TNMB Classification for Mycosis Fungoides (AJCC 2010)
Rights were not granted to include this table in electronic media. Please refer to the printed book.

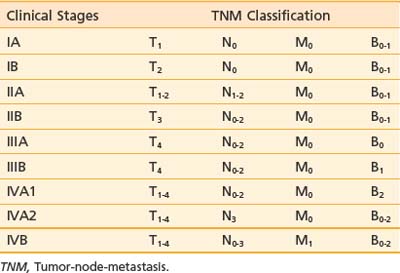
The T classification reflects the extent of skin involvement. The N classification indicates the presence of lymph node involvement. Enlarged lymph nodes should be biopsied (excisional biopsies are recommended), because palpable enlargement is often associated only with changes of dermatopathic lymphadenitis; however, patients with lymph nodes exhibiting rearranged TCR genes have a worse prognosis, regardless of the histologic grade.33 In the M classification, suspected disease should also be documented and treatment programs for visceral disease should be considered only if there is definite proof of extracutaneous disease. In the B classification, the presence of a significant proportion of abnormal, cerebriform (Sézary) cells should be noted; however, low levels of Sézary-like cells can be detected in the peripheral blood of patients with benign skin conditions. The current practice is to use the criterion of an expanded peripheral blood CD4+ population with increased ratio of CD4/CD8 T lymphocytes (greater than 10:1), expanded populations of abnormal T cells with CD4+/CD7− or CD4+/CD26− phenotype, and molecular evidence of a relevant TCR gene rearrangement in the peripheral blood.33
Standard Therapeutic Approaches
Multiple therapeutic options exist for mycosis fungoides and the Sézary syndrome. The National Comprehensive Cancer Network (NCCN) has established consensus guidelines for the therapy of these diseases (www.nccn.org, non-Hodgkin lymphoma mycosis fungoids/Sézary syndrome practice guidelines). Selection of treatment is based primarily on the clinical stage of the disease (Fig. 62-2). However, factors such as access to special treatment approaches, the patient’s age, other social and medical problems, and the cost/benefit ratio should also be taken into consideration. For patients with patch or plaque skin involvement (T1 and T2) without extracutaneous disease, the treatment plan may be limited to topical therapeutic measures, whereas patients with any extracutaneous disease should receive systemic (cytotoxic or biologic) therapy as part of their management. There is no evidence that early aggressive combined modality therapy is more effective than conservative sequential therapies in the management of limited or advanced disease.34 Despite decades of experience in the treatment of mycosis fungoides and Sézary syndrome, well-designed, prospective, controlled clinical studies comparing the efficacy of various therapies are lacking.
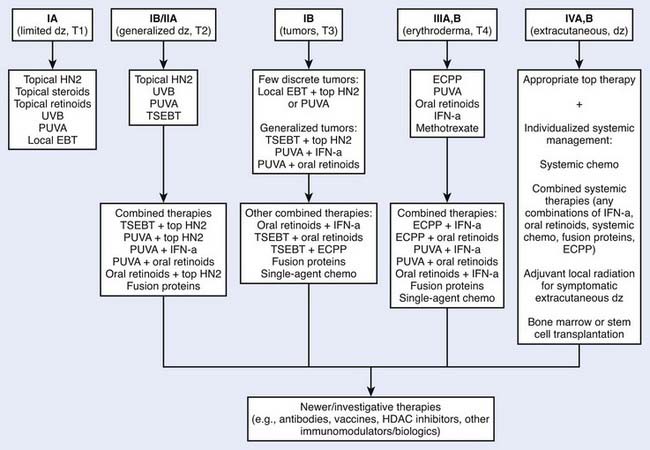
Figure 62-2 • An algorithm summarizing management approaches for patients with all stages of mycosis fungoides.
Topical Chemotherapy
Nearly all patients with mycosis fungoides require treatment directed at their skin. Common therapies include topical corticosteroids, psoralen plus ultraviolet (UV) A (PUVA), UVB, topical chemotherapy, topical retinoids, and irradiation. Topical nitrogen mustard (mechlorethamine, mustargen) is a very effective form of topical chemotherapy for patients with mycosis fungoides.30,35,36 The activity of intravenous nitrogen mustard as an alkylating agent for the management of systemic malignancy is well documented. The mechanism of action when nitrogen mustard is applied topically for the management of mycosis fungoides is less certain and may not be related simply to its alkylating agent properties. Its activity may be mediated by immune mechanisms or by interaction with the epidermal cell–Langerhans cell–T cell axis. It may be mixed in water or in an ointment base.
Because of its efficacy and ease of application, topical nitrogen mustard is employed widely as primary or secondary therapy in the management of patients with mycosis fungoides, especially those who have a limited or generalized patch or plaque phase of skin involvement (see Fig. 62-2). In patients with a discrete number of refractory lesions, treatment may be supplemented with local irradiation.37
Topical Retinoids
Bexarotene (Targretin) 1% gel, a retinoid X receptor–selective synthetic retinoid, may be an effective agent for the topical treatment of mycosis fungoides. Bexarotene gel is applied with a thin application to the patches or plaques as often as twice daily. Because of the irritant effect of the retinoids, it is not suitable for generalized application, but is used only when there are a discrete number of patches or plaques. A phase III trial of bexarotene included 50 patients with refractory or persistent early stage disease (stages IA to IIA).38 Responses were seen in 62% of patients with stage IA and 50% of patients with stage IB disease. Three patients with stage IIA or IIB disease did not respond. The most common toxicity of bexarotene gel is irritation at the sites of application, which occurs in the majority of patients. Because of the erythema from the irritant reaction, it may be necessary to withhold therapy for a few weeks to assess disease activity. Bexarotene gel is approved by the Food and Drug Administration (FDA) for patients with stages IA and IB disease who have refractory or persistent disease after other therapies or who have not tolerated other therapies.
Phototherapy
Phototherapy involves using UV radiation in the form of UVA or UVB wavelengths, which can be used alone, together, or with psoralen, a photosensitizing agent, as PUVA. The long-wave UVA has the advantage over UVB in its greater depth of penetration into the dermal infiltrates of mycosis fungoides. For early limited diseases, UVB alone39 or home UV phototherapy (UVA + UVB)40 has been shown to be effective.
PUVA is the most commonly used form of phototherapy for mycosis fungoides and Sézary syndrome patients. It was first used in the treatment of psoriasis but has proved effective for patients with mycosis fungoides as well.41–43 In the presence of UVA, the psoralen drug intercalates with deoxyribonucleic acid (DNA), forming both monofunctional and bifunctional adducts, which inhibit DNA synthesis.
Indications for PUVA treatment include its use as the primary therapy for patients with limited or generalized patch or plaque disease or as a secondary therapy following the failure of other topical modalities. PUVA may also be effective for patients with erythroderma, provided that very low daily exposures are used to avoid phototoxic reactions. For patients with Sézary syndrome, PUVA may be supplemented by systemic biologic therapies, such as IFN-alpha or systemic retinoids.44,45 The treatment of patients with more advanced cutaneous disease may be facilitated by the addition of localized irradiation to particular refractory plaques or tumors.
The primary acute complications of PUVA therapy include nausea and phototoxic reactions such as erythroderma and blistering, as well as skin dryness. Patients should shield their skin and eyes from the sun for at least 24 hours following psoralen ingestion. The potential long-term complications of PUVA therapy include cataract formation (requiring the use of UVA-opaque goggles during therapy) and secondary cutaneous malignancies. Among patients treated for mycosis fungoides, this risk is greatest for patients who have undergone long-term treatment with multiple topical therapies.46
PUVA may often be used in combination with other therapies, such as IFN or retinoids. For the combined regimen with PUVA, IFN-alpha and PUVA therapy are initiated concurrently, each usually given thrice weekly. When skin clearance is complete, the frequency of PUVA treatment is reduced. Complete and partial response (PR) rates of 79% to 80% and 14% to 20% in patients with generalized patch and plaque (stages IB and IIA) disease treated with the combination of IFN-alpha and PUVA have been reported.44 The clinical response and response duration results may be better with the combined regimen of PUVA and IFN-alpha compared with either treatment alone; however, prospective randomized clinical trials are needed to confirm this impression.47,48
When retinoids are used in combination with PUVA (RePUVA), the response rate is similar to that of PUVA alone; however, the responses may be achieved with fewer PUVA treatments and with a lower cumulative UVA dose.45 The duration of remission may be more prolonged if retinoids are given as maintenance therapy.49
Radiation Therapy
Mycosis fungoides is an exquisitely radiosensitive neoplasm, and irradiation may be exploited in several ways for its management.15 Individual plaques or tumors of mycosis fungoides may be treated with orthovoltage or electron-beam radiation therapy (electron-beam therapy) to total doses of 15 to 25 Gy in 1 to 3 weeks, with a high likelihood of achieving long-term local control. Recently, CRs were reported for 92% of lesions treated with a palliative regimen of 4 Gy × 2, whereas the CR rate to 2 Gy × 2 was only 30%.50 Electrons have an intrinsic advantage over photons in the treatment of mycosis fungoides because the lesions are often superficial, the thickness can be estimated accurately, and their depth of penetration can be controlled by the appropriate selection of electron energy. For indurated plaques, electron energies as low as 6 MeV are generally sufficient. Use of bolus may be indicated because of the relative “skin-sparing” effect of low-energy electrons. For the unusual patient with unilesional or localized mycosis fungoides, local electron-beam therapy achieves the most efficient and complete clearance of the disease.
Several centers have developed expertise in the use of total-skin electron-beam therapy (TSEBT).51,52 Generally, the dose range used has been 30 to 36 Gy and the overall response (OR) rates are nearly 100%, with CR rates ranging from 40% to 98%, depending on the extent of skin involvement. As many as 50% of patients with limited plaque disease and 25% of patients with generalized plaque disease may remain free of disease for more than 5 years after completion of a single course of electron-beam therapy. Although the curative potential of this treatment remains disputed, there is no doubt that it provides an important palliative benefit, especially for patients with extensive disease. Often, when disease recurs, it is in a more limited distribution and may be controlled more readily with localized topical therapies. Because relapse is so common, there have been recent attempts to re-examine the concept of low-dose TSEBT. The rationale is that the long-term benefit of irradiation may be spread out for a longer period with multiple courses, and that the relative morbidity of indivdual low-dose courses would be less. Doses as low as 4 Gy, adminstered over a period of 4 days, achieve only incomplete, nondurable responses.53 However, a recent review of the early Stanford experience suggests that doses in the range of 10 to 20 Gy are associated with a high OR rate and quite a long time to progression, suggesting that low-dose TSEBT (10–20 Gy) should be revisited.54
Some patients may be candidates for a second course of TSEBT.55 Eligibility criteria should include a good response of reasonable duration to the initial course of treatment, failure of other subsequent therapies, and generalized symptomatic skin involvement. These patients develop more pronounced skin changes secondary to treatment, including more prominent telangiectasia, skin dryness, atrophy, and hair loss.
Systemic Chemotherapy
Systemic chemotherapy is the most appropriate choice for patients with extracutaneous disease and has only limited indications for patients who have skin disease alone. Recently, the NCCN published guidelines that recommend stage-based therapy and, using their algorithm, only a minority of patients with mycosis fungoides (10%–20%) require systemic management.56 With combination chemotherapy regimens, overall CR or PR rates can reach 80% to 100%; however, in most cases, the median duration of response is less than a year, and in many patients, less than several months.57,58 Virtually all single-agent and drug combinations that have proved useful in the management of patients with non-Hodgkin lymphomas have been tested in patients with mycosis fungoides. The most effective and commonly used combinations include cyclophosphamide, vincristine, and prednisone (CVP) alone or with doxorubicin.59 Other regimens used include cyclophosphamide, doxorubicin, vincristine, and etoposide, or CVP with methotrexate.34,60,61 Chemotherapy may be used alone or in combination with topical therapy (e.g., radiation) or biologic response modifiers (e.g., IFN-alpha). A randomized trial comparing electron-beam radiation plus chemotherapy with topical therapy in the initial treatment of mycosis fungoides failed to demonstrate an improved survival with the more aggressive management approach.34
Methotrexate, the purine analogs (fludarabine, 2′-deoxycoformycin), gemcitabine, and liposomal doxirubicin (Doxil) are the most commonly used single-agent chemotherapy regimens in mycosis fungoides and Sézary syndrome.57,58 The OR rates are generally 20% to 80% and the median duration of responses reported has ranged from 3 to 22 months. The purine analogs show promise in early clinical trials, with response rates of 20% to 70% for pentostatin,62 Chlorodeoxyadenosine (2-CDA),63 and fludarabine,64 but complications related to immunosuppression are significant.65–67 Recently, single-agent gemcitabine has been reported to achieve response rates as high as 70%, but the complete remission rate is low.68,69 Another active single agent is liposomal doxorubicin. An OR rate of 33% has been reported in refractory patients with stage IVB disease.70
Autologous hematopoietic stem cell transplant (HSCT) has not proved to be useful. Only about 20 cases have been reported and the relapse risk is quite high.71 One of the limitations of an autologous approach, which might account for the short-lived responses, is the potential reinfusion of contaminating malignant T cells. In an attempt to overcome this problem, autotransplantation with T cell depletion has been evaluated; however, these patients also had a high risk of relapse.72
The concept of allogeneic HSCT is provocative, because even in the absence of a CR, a graft-versus-tumor effect may provide an immune mechanism to control the malignant T-cell process. Eligibility and preparatory regimens have varied. In a report by Guitart and associates, three patients received an allograft from human leukocyte antigen–matched siblings.73 Complete and sustained clinical and histologic remissions were achieved in two. The third patient achieved a CR followed by a limited cutaneous recurrence. Molina and associates reported a CR in six patients transplanted for refractory disease.74 Five patients remained in CR at a median follow-up of 17 months (range 3-65 months). Mild acute and chronic graft-versus-host disease (GVHD) developed in all patients and chronic GVHD was ongoing in patients with sustained remissions, suggesting a possible graft-versus-lymphoma (GVL) effect.
Although the results of allogeneic transplant are encouraging and may be durable, it has limited applicability because of associated toxicities, especially in older patients. A nonmyeloablative approach with reduced intensity conditioning is associated with a reduced risk.71,75,76
The largest series included 15 patients with refractory mycosis fungoides who had failed three prior regimens. With a median follow-up of 41 months, 9 were still in a CR with a 5-year progression-free survival of 60%.77
Thus, it appears that allogeneic HSCT (myeloablative or with reduced-intensity conditioning regimens) may result in durable long-term remissions, perhaps related to a GVL effect. Larger prospective studies will be required to identify the optimal timing of transplant, the best conditioning regimen, and resultant efficacy and safety of the therapy.72,78
Extracorporeal Photopheresis
Photopheresis (extracorporeal photopheresis [ECPP] or systemic photochemotherapy) is a method of delivering PUVA systemically via an extracorporeal technique.79 The patient’s white blood cells are collected (leukapheresis), exposed to a photoactivating drug, and then irradiated with UVA. Then the irradiated cells are returned to the patient. The mechanism of action of ECPP remains unclear. It is hypothesized that there may be a dual effect: a direct cytotoxic or antiproliferative effect on the neoplastic cells and an immune-enhancing effect on the competent lymphocytes against the neoplastic cells. Photopheresis is usually administered every 4 weeks, but in patients with severe disease, the frequency can be as often as every 2 to 3 weeks. Once complete clearance is achieved, the frequency can be gradually reduced and then discontinued.
Compared with other systemic therapies, ECPP has minimal adverse effects.79 Some patients may experience nausea, mostly caused by the ingested psoralen, and some have a transient low-grade fever or slight malaise after treatment. There are no reports of any significant organ damage or bone marrow or immune suppression.
In erythrodermic (T4) mycosis fungoides and Sézary syndrome, ECPP may be the initial treatment of choice.80,81 Its effectiveness in T1, T2, and T3 diseases remains unclear. The largest study of erythrodermic patients revealed that the majority experienced some level of response to ECPP, with an OR rate of 83%.82 The CR rate was only 21%, but 41% of patients showed at least a 50% improvement in their skin disease. If the response to ECPP alone is partial or slow, IFN-alpha or systemic retinoidscan be added as a combined-modality regimen.83,84 The dose for IFN-alpha in the combined program is usually low, 1.5 to 5 million units three to five times weekly, not exceeding 10 million units per dose. Topical therapies such as corticosteroids, nitrogen mustard, phototherapy, or TSEBT can be combined with ECPP if additional skin-directed treatment is needed.56,85
Interferon Alpha
IFN-alpha is indicated primarily for the palliative management of refractory or advanced disease. It may be used alone or more often combined with topical or other systemic therapies. The dosage of IFN-alpha for mycosis fungoides is usually initiated at 3 to 5 million units daily or three times a week, and is gradually increased, depending on the clinical response and severity of adverse effects. Reported OR rates when used as monotherapy range from 53% to 74%, with CR rates of 21% to 35%.86,87
The clinical efficacy of combined IFN-alpha and PUVA has been demonstrated in mycosis fungoides and Sézary syndrome.44 Patients in clinical stages IB to IVB have been treated, with an OR rate of 85% to 100% and CR rate of 33% to 80%, depending on disease severity.
Systemic Retinoids
Systemic therapy with retinoids has been beneficial in the management of mycosis fungoides and Sézary syndrome. The reported response rate is approximately 45%, with a 20% CR rate.88 Systemic retinoids are indicated for palliative therapy for refractory or advanced disease, often in combination with other topical or systemic therapies, including PUVA (Re-PUVA), IFN-alpha, or TSEBT.45,89–91 The systemic retinoid most commonly used is bexarotene.92 This agent is administered orally. The initial dose is 300 mg/m2 per day, which can be adjusted according to clinical response and the severity of adverse effects. The most common adverse effects include photosensitivity, xerosis, myalgia, arthralgia, headaches, and impaired night vision. The well-known teratogenic effects of retinoids must be carefully addressed in female patients. Because of its potential hepatotoxic and hyperlipidemic effects, liver function and serum lipid levels should be monitored in each patient during treatment. In addition, central hypothyroidism is often induced, so patients are routinely started on levothyroxine (Synthroid) immediately before or simultaneously with bexarotene.
Recombinant Fusion Proteins
Recombinant fusion protein therapy, such as the IL-2–diphtheria toxin fusion protein (Ontak, denileukin diftitox), involves the use of growth factor–diphtheria toxin fusion proteins designed specifically to kill defined neoplastic cell populations. Ontak has undergone a multicenter, phase III trial in patients with IL-2 receptor (CD25+)-expressing mycosis fungoides.93 Patients with stage IB to III mycosis fungoides who failed to respond to at least four prior therapies, or stage IVA mycosis fungoides who failed to respond to at least one prior therapy, were included in the phase III trial. The OR rate was 30%, with CR and PR rates of 10% and 20%, respectively. The main complication is related to a “capillary leak” syndrome, which may be ameliorated by pretreatment with corticosteroids.
Histone Deacetylase Inhibitors
Histone deacetylase (HDAC) inhibitors are a novel class of agents that can induce growth arrest, differentiation, or apoptosis by affecting gene expression and protein function. Vorinostat (suberoylanilide hydroxamic acid) is an orally available pan-HDAC inhibitor that has activity in patients with mycosis fungoides and Sézary syndrome. This agent has been approved by the FDA for the treatment of patients with refractory disease.94 The OR rate was 30% (n = 74); however, only one patient achieved CR. Vorinostat is available as 100-mg capsules and the recommended starting dose is 400 mg daily.
Combined Modality Therapy
Patients who fail to respond to a single-agent topical regimen can be treated with combined-modality therapy. This may include combination topical therapies (e.g., TSEBT, topical nitrogen mustard, PUVA, bexarotene gel), or combined topical and systemic biologic therapies (e.g., PUVA or TSEBT with IFN-alpha or systemic bexarotene).44,45,89
Most patients with tumor stage IIB mycosis fungoides have generalized involvement with tumor and plaque disease, and the greatest likelihood of a response is with TSEBT. However, in view of the high risk for relapse after irradiation, adjuvant therapy (e.g., topical nitrogen mustard) is generally employed after completion of electron-beam therapy. Topical nitrogen mustard in Aquaphor provides the dual benefit of treatment for any residual disease and emolliation of the skin, which is often chronically dry after completion of electron-beam therapy. Patients with a discrete number of tumors may be treated with topical nitrogen mustard or PUVA combined with localized electron-beam therapy to individual tumors. Although topical (nitrogen mustard or PUVA) or systemic (IFN-alpha or photopheresis) adjuvant therapy after skin clearance with TSEBT may improve the disease-free interval, it does not improve survival.95–97
Patients with recalcitrant disease may require a combination of systemic therapies, either with biologic therapies or a combination of biologic therapies and chemotherapies, with or without topical therapy. IFN-alpha has been used in combination with systemic retinoids with variable results.90,98 Combinations of systemic biologic therapies have been used successfully and with potential synergistic effects.99 For example, when retinoids are used in combination with PUVA, the response rate is similar to that of PUVA alone; however, the responses may be achieved with fewer PUVA treatments and with a lower cumulative UVA dose.45 The duration of remission tended to be prolonged if retinoids were given as maintenance therapy.
Various chemotherapy regimens have been used in combination with TSEBT.34,100 However, there is no evidence that an aggressive combination regimen employing systemic chemotherapy at the outset results in superior survival.34 Focal resistant tumors can be boosted with local electron-beam therapy or orthovoltage radiation.
PUVA can be given in combination with IFN-alpha. In one of the largest studies of this combination, a CR rate of 33% and PR rate of 50% was observed in patients with stage IIB disease.44 Studies using PUVA alone for erythrodermic (stage III) patients have reported CR rates of 33% to 70%.41–43,48,101 Despite these good response rates, the majority of the patients relapse during maintenance therapy. In another study, the CR and PR rates for a combination regimen of PUVA plus IFN-alpha in stage III disease were 62% and 25%, respectively.44 It is thought that the combined regimen may improve clinical response or response duration beyond that observed with PUVA or IFN alone. However, there is no clear evidence that prolongation of response duration leads to improvement in the overall survival.
Techniques of Radiotherapy
Ionizing irradiation is the most effective single agent for the treatment of mycosis fungoides. Reports of dramatic responses to low doses of x-ray therapy (including photo-documentation) appeared within a decade of Roentgen’s first discovery of x-irradiation.102 Clinical dose-time fractionation studies indicate that permanent local control of lesions may be achieved with doses as low as 7 Gy. Reconstruction of data on a semilog plot of cell survival indicates a D0 of 0.90 Gy and n (extrapolation number) less than 2.103
For palliative treatment of individual lesions with 100 to 200 Kv x-rays or 6 to 9 MeV electrons, fractionated doses of as low as 12 Gy (e.g., 3 Gy × 4 or 2 Gy × 6) are often adequate to achieve long-term local control. Doses in excess of 30 Gy are rarely required.104
The ability to irradiate the entire skin depended on the development of electron-beam therapy. By the proper choice of electron energy, the depth dose characteristics of the electron beam make it possible to treat large surfaces of the skin in a single field, concentrating the dose of irradiation in the epidermis and upper dermis, while limiting the dose to the deep dermis and subcutaneous tissue. By using rotational, four-field, six-field, or eight-field techniques, it is possible to irradiate the entire cutaneous surface. The concept of TSEBT for the management of mycosis fungoides was first described by Trump et al.105 using a van de Graaff generator. The more widespread use of TSEBT was facilitated by the development of the modern medical linear accelerator. At Stanford, the original Stanford medical linear accelerator was modified to treat the entire skin, first using a four-field technique, and later a six-field technique.106,107 Modifications of the Stanford technique have been adopted widely for the management of patients with mycosis fungoides.108–113
In general, the dosimetry of total skin electron irradiation improves as the number of fields of treatment increases.114 With four-field treatment, there is significant overlap of adjacent fields, creating “hot spots” that may result in long-term telangiectasia, subcutaneous fibrosis, and even necrosis. These complications may be accentuated by fractionation programs that use larger doses per fraction or fewer fractions per week.
In a typical setup, patients are treated in the standing position at a distance of 3.5 meters from the isocenter (electron source). A 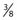 -inch polymethyl methacrylate plate is placed as close as possible to the patient surface to degrade and scatter the electrons. During treatment, the machine is angled upward or downward at an angle of 18 degrees to provide for better homogeneity of dose at the patient’s surface and to minimize photon contamination, which is greatest in the central axis of the beam (Fig. 62-3).
-inch polymethyl methacrylate plate is placed as close as possible to the patient surface to degrade and scatter the electrons. During treatment, the machine is angled upward or downward at an angle of 18 degrees to provide for better homogeneity of dose at the patient’s surface and to minimize photon contamination, which is greatest in the central axis of the beam (Fig. 62-3).
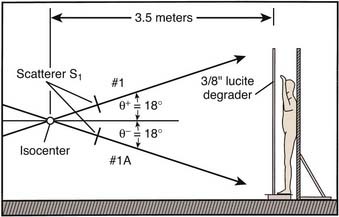
Patients are treated with a six-field technique that includes anterior, posterior, and four opposed oblique fields (Fig. 62-4).115 A full “cycle” of treatment is administered over a 2-day period. On day 1, the anterior and two posterior oblique fields are treated at each of the two accelerator angles. On day 2, the posterior and two anterior oblique fields are treated at each of the two accelerator angles. The total dose administered with each cycle is 1.5 to 2 Gy. Most patients will tolerate 2 Gy per cycle, but lower doses are used for patients with erythroderma, atrophic skin, or a previous course of electron-beam therapy. The prescribed total dose is generally 36 Gy administered over a 10-week period, with a 1-week split after delivering a dose of 18 to 20 Gy to provide relief from the generalized skin erythema that usually accompanies treatment.
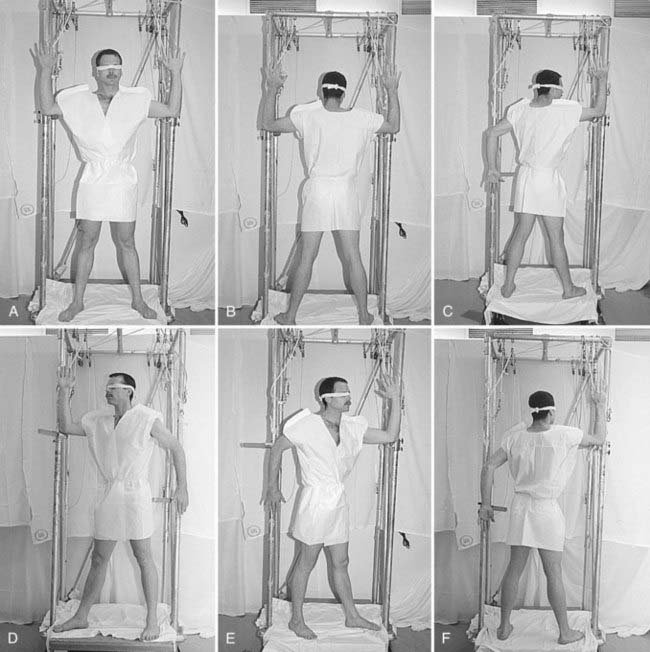
In the standard course of treatment, only the eyes are shielded. Internal lead eye shields with an inner paraffin coating are used whenever disease is present on the face or scalp. The shields are placed under the lids after the eyes have been anesthetized topically. If disease is absent from these areas, external lead eye shields, which are taped over the closed eyes, are used. In addition, in the absence of involvement of the scalp or face, a scalp shield is used after a dose of 25 Gy to facilitate adequate regrowth of scalp hair. Complete scalp shielding is contraindicated and may result in extension of disease to this area.111
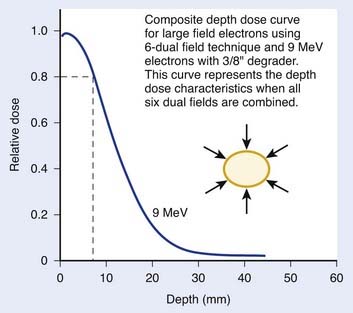
The composite depth-dose distribution curve for TSEBT with 9-MeV electrons using the six–dual field technique is displayed in Fig. 62-5. Using a 9-MeV beam and the six–dual field technique, the 80% dose is located at a depth of 0.7 cm. The 50% depth dose is at 1.25 cm. These depth-dose characteristics are ideal for mycosis fungoides, in which the malignant infiltrates are generally limited to the epidermis and upper portions of the dermis.
 -inch polymethyl methacrylate degrader near the patient surface.
-inch polymethyl methacrylate degrader near the patient surface.Critical Normal Tissues—Radiation Injury
The most common acute complications of this therapy are erythema and dry desquamation.111,113,115 The severity of these problems can be minimized by proper fractionation techniques. In addition, a 1-week split may be given in the middle of the course of treatment to permit skin recovery. Intermediate-term complications include alopecia, which is incomplete and only temporary if the scalp dose is limited to 25 Gy.113 Most patients will also suffer temporary loss of fingernails and toenails 2 to 4 months following completion of therapy. Most patients report the inability to sweat properly for 6 to 12 months following therapy116 and chronically dry skin thereafter, which requires the regular use of emolliation. In long-term follow-up, evidence for chronic radiation dermatitis is uncommon in our experience.117 Occasional patients may display scattered telangiectasia, and rarely these may be evident on casual examination. These skin changes may be more evident with irradiation techniques that employ fewer than six fields or use larger treatment fractions, administered only once weekly. Although secondary malignancies such as squamous cell and basal cell cancers of the skin are increased after the use of TSEBT, the only patients in whom these have become problematic are those who have received repeated treatment with multiple therapies, including irradiation, topical nitrogen mustard, and PUVA.43
Selected Outcome
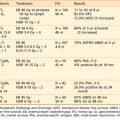
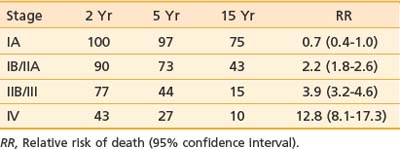
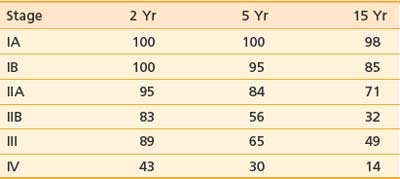
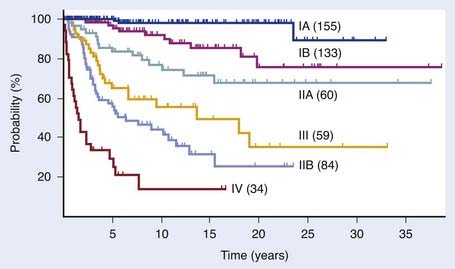
The T classification and presence of extracutaneous disease are the most important predictors of survival in patients with mycosis fungoides.30,118 Table 62-3 and Table 62-4 display the survival and disease-specific survival as a function of disease stage. Fig. 62-6 shows the disease-specific survival for 525 patients treated at Stanford.
Table 62-3 Survival (%) and Relative Risk of Death for 525 Patients With Mycosis Fungoides Treated at Stanford University
Stage IA (Limited Patch or Plaque, T1) Disease
Most patients with limited patch or plaque (T1) involvement have stage IA disease. The primary therapies for patients with stage IA (T1) disease are topical. These include topical chemotherapy (mostly nitrogen mustard), phototherapy (UVB or PUVA), bexarotene gel, or localized radiation therapy (usually electron-beam). The CR rate to nitrogen mustard is 60% to 70%119 and to TSEBT is 85% to 95%.120 However, there is no evidence that any one approach is superior to the others with respect to long-term disease control or survival. Treatment selection is often based on convenience and cost.
Patients with limited patch or plaque (T1, overall stage IA) disease who are treated with conventional therapies have an excellent prognosis with an overall long-term life expectancy that is similar to an age-, gender-, and race-matched control population. In a retrospective study of 122 patients with stage IA disease at Stanford, the median survival was not reached at 33 years.25 Nearly all patients with stage IA disease who die will die from causes other than mycosis fungoides. Furthermore, only 9% of treated patients at this stage will ever progress to a more advanced stage of disease. Early, aggressive therapy with combination chemotherapy does not result in a more favorable survival outcome compared with patients managed more conservatively with sequential topical regimens.34
Stage IB/IIA (Generalized Patch or Plaque, T2) Disease
Topical nitrogen mustard, PUVA, and TSEBT are commonly used for patients with T2 disease. CR rates using topical nitrogen mustard are 50% to 70%,26,36,121 whereas the CR rate for total skin electron-beam therapy is 80% to 90%.26,120 However, in a study at Stanford, patients treated with TSEBT did not have an improved long-term survival compared with those who received topical nitrogen mustard as initial therapy, despite the superior CR rate.26 In patients treated with PUVA, the CR rate ranges from 50% to 80%.38,40,122
Patients who fail to respond to one topical therapy, or who progress after an initial response, may be treated with an alternative topical therapy. There is no evidence that development of resistance to one modality affects subsequent response to an alternative topical therapy.25,26 Combination topical or topical plus biologic therapies may also be used as initial therapy for patients with T2 disease. This may provide for better long-term control of disease, but the ultimate outcome is not usually affected.45,74,76 Patients who have generalized patch or plaque disease without evidence of extracutaneous involvement and who are treated with these modalities have a median survival greater than 11 years and 25% of deaths in this group are attributable to mycosis fungoides.26
Stage IIB (Tumorous) Disease
Patients who have a limited number of cutaneous tumors in the setting of generalized patch or plaque disease may be treated with local radiation to the tumors plus one of the topical therapies noted earlier. However, those who have generalized tumorous disease will be treated most effectively with TSEBT followed by maintenance therapy with nitrogen mustard76 or combinations of PUVA with IFN-alpha45 or PUVA plus oral bexarotene.
Stage III (Erythrodermic, T4) Disease
The long-term outcome for these patients is quite variable and depends on patient age at presentation (younger than versus older than 65 years), overall stage (III versus IV), and peripheral blood involvement (B0 versus B1).123 The median survival varies widely, depending on the number of independent adverse prognostic factors present: three distinct prognostic subgroups may be identified (favorable, intermediate, and unfavorable), with median survivals of 10.2, 3.7, and 1.5 years, for patients with zero, one, or more than one unfavorable prognostic factor, respectively.
Stage IV (Extracutaneous) Disease
In a Stanford series, 77 patients either presented with or later developed extracutaneous (stage IV) disease (56 stage IVA, 21 stage IVB). The median survival of the 77 patients was only 13 months and was similar, regardless of the extent of their skin involvement (T2 versus T3 versus T4, P = 0.69 to 0.88) or the site of extracutaneous disease (IVA versus IVB).30
1 Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973–2002. Arch Dermatol. 2007;143(7):854-859.
2 Crowley JJ, Nikko A, Varghese A, et al. Mycosis fungoides in young patients: clinical characteristics and outcome. J Am Acad Dermatol. 1998;38(5 Pt 1):696-701.
3 Whittemore AS, Holly EA, Lee IM, et al. Mycosis fungoides in relation to environmental exposures and immune response: a case-control study. J Natl Cancer Inst. 1989;81(20):1560-1567.
4 Tuyp E, Burgoyne A, Aitchison T, et al. A case-control study of possible causative factors in mycosis fungoides. Arch Dermatol. 1987;123(2):196-200.
5 Poiesz BJ, Ruscetti FW, Gazdar AF, et al. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A. 1980;77(12):7415-7419.
6 Safai B, Myskowski PL, Dupont B, et al. Association of HLA-DR5 with mycosis fungoides. J Invest Dermatol. 1983;80(5):395-397.
7 Rosen ST, Radvany R, Roenigk HJr, et al. Human leukocyte antigens in cutaneous T cell lymphoma. J Am Acad Dermatol. 1985;12(3):531-534.
8 Thangavelu M, Finn WG, Yelavarthi KK, et al. Recurring structural chromosome abnormalities in peripheral blood lymphocytes of patients with mycosis fungoides/Sézary syndrome. Blood. 1997;89(9):3371-3377.
9 Karenko L, Hyytinen E, Sarna S, et al. Chromosomal abnormalities in cutaneous T-cell lymphoma and in its premalignant conditions as detected by G-banding and interphase cytogenetic methods. J Invest Dermatol. 1997;108(1):22-29.
10 Vowels BR, Cassin M, Vonderheid EC, et al. Aberrant cytokine production by Sézary syndrome patients: cytokine secretion pattern resembles murine Th2 cells. J Invest Dermatol. 1992;99(1):90-94.
11 Bernengo MG, Fierro MT, Novelli M, et al. Soluble interleukin-2 receptor in Sézary syndrome: its origin and clinical application. Br J Dermatol. 1993;128(2):124-129.
12 Wasik MA, Vonderheid EC, Bigler RD, et al. Increased serum concentration of the soluble interleukin-2 receptor in cutaneous T-cell lymphoma, clinical and prognostic implications. Arch Dermatol. 1996;132(1):42-47.
13 Rook AH, Heald P. The immunopathogenesis of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9(5):997-1010.
14 Yoo EK, Cassin M, Lessin SR, et al. Complete molecular remission during biologic response modifier therapy for Sézary syndrome is associated with enhanced helper T type 1 cytokine production and natural killer cell activity. J Am Acad Dermatol. 2001;45(2):208-216.
15 Hoppe RT, Wood GS, Abel EA. Mycosis fungoides and the Sézary syndrome: pathology, staging, and treatment. Curr Probl Cancer. 1990;14(6):293-371.
16 Wood G, Weiss L, Warnke R. The immunopathology of cutaneous lymphomas: immunophenotypic and immunogenotypic characteristics. Semin Dermatol. 1986:334.
17 Weiss LM, Hu E, Wood GS, et al. Clonal rearrangements of T-cell receptor genes in mycosis fungoides and dermatopathic lymphadenopathy. N Engl J Med. 1985;313(9):539-544.
18 Ashton-Key M, Diss TC, Du MQ, et al. The value of the polymerase chain reaction in the diagnosis of cutaneous T-cell infiltrates. Am J Surg Pathol. 1997;21(7):743-747.
19 Ralfkiaer ECL, Sander CA. Mycosis fungoides. Press: IARC; 2008.
20 Kern DE, Kidd PG, Moe R, et al. Analysis of T-cell receptor gene rearrangement in lymph nodes of patients with mycosis fungoides. Prognostic implications. Arch Dermatol. 1998;134(2):158-164.
21 Harmon CB, Witzig TE, Katzmann JA, et al. Detection of circulating T cells with CD4+CD7-immunophenotype in patients with benign and malignant lymphoproliferative dermatoses. J Am Acad Dermatol. 1996;35(3 Pt 1):404-410.
22 Vonderheid EC, Sobel EL, Nowell PC, et al. Diagnostic and prognostic significance of Sézary cells in peripheral blood smears from patients with cutaneous T cell lymphoma. Blood. 1985;66(2):358-366.
23 Bunn PAJr, Lamberg SI. Report of the Committee on Staging and Classification of Cutaneous T-Cell Lymphomas. Cancer Treat Rep. 1979;63(4):725-728.
24 Kim YH, Hoppe RT. Mycosis fungoides and the Sézary syndrome. Semin Oncol. 1999;26(3):276-289.
25 Kim YH, Jensen RA, Watanabe GL, et al. Clinical stage IA (limited patch and plaque) mycosis fungoides. A long-term outcome analysis. Arch Dermatol. 1996;132(11):1309-1313.
26 Kim YH, Chow S, Varghese A, et al. Clinical characteristics and long-term outcome of patients with generalized patch and/or plaque (T2) mycosis fungoides. Arch Dermatol. 1999;135(1):26-32.
27 Ralfkiaer EWR, Whittake S. Mycosis fungoides. IARC Sci Publ. 2008:299.
28 De Coninck EC, Kim YH, Varghese A, et al. Clinical characteristics and outcome of patients with extracutaneous mycosis fungoides. J Clin Oncol. 2001;19(3):779-784.
29 Epstein EHJr, Levin DL, Croft JDJr, et al. Mycosis fungoides. Survival, prognostic features, response to therapy, and autopsy findings. Medicine (Baltimore). 1972;51(1):61-72.
30 Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139(7):857-866.
31 Tsai EY, Taur A, Espinosa L, et al. Staging accuracy in mycosis fungoides and Sézary syndrome using integrated positron emission tomography and computed tomography. Arch Dermatol. 2006;142(5):577-584.
32 Salhany KE, Greer JP, Cousar JB, et al. Marrow involvement in cutaneous T-cell lymphoma. A clinicopathologic study of 60 cases. Am J Clin Pathol. 1989;92(6):747-754.
33 Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110(6):1713-1722.
34 Kaye FJ, Bunn PAJr, Steinberg SM, et al. A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med. 1989;321(26):1784-1790.
35 Ramsay DL, Halperin PS, Zeleniuch-Jacquotte A. Topical mechlorethamine therapy for early stage mycosis fungoides. J Am Acad Dermatol. 1988;19(4):684-691.
36 Vonderheid EC, Tan ET, Kantor AF, et al. Long-term efficacy, curative potential, and carcinogenicity of topical mechlorethamine chemotherapy in cutaneous T cell lymphoma. J Am Acad Dermatol. 1989;20(3):416-428.
37 Zackheim HS, Epstein EHJr, Crain WR. Topical carmustine (BCNU) for cutaneous T cell lymphoma: a 15-year experience in 143 patients. J Am Acad Dermatol. 1990;22(5 Pt 1):802-810.
38 Heald P, Mehlmauer M, Martin AG, et al. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: results of the phase III clinical trial. J Am Acad Dermatol. 2003;49(5):801-815.
39 Ramsay DL, Lish KM, Yalowitz CB, et al. Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol. 1992;128(7):931-933.
40 Resnik KS, Vonderheid EC. Home UV phototherapy of early mycosis fungoides: long-term follow-up observations in thirty-one patients. J Am Acad Dermatol. 1993;29(1):73-77.
41 Honigsmann H, Brenner W, Rauschmeier W, et al. Photochemotherapy for cutaneous T cell lymphoma. A follow-up study. J Am Acad Dermatol. 1984;10(2 Pt 1):238-245.
42 Abel EA, Sendagorta E, Hoppe RT, et al. PUVA treatment of erythrodermic and plaque-type mycosis fungoides. Ten-year follow-up study. Arch Dermatol. 1987;123(7):897-901.
43 Herrmann JJ, Roenigk HHJr, Hurria A, et al. Treatment of mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. J Am Acad Dermatol. 1995;33(2 Pt 1):234-242.
44 Kuzel TM, Roenigk HHJr, Samuelson E, et al. Effectiveness of interferon alfa-2a combined with phototherapy for mycosis fungoides and the Sézary syndrome. J Clin Oncol. 1995;13(1):257-263.
45 Thomsen K, Hammar H, Molin L, et al. Retinoids plus PUVA (RePUVA) and PUVA in mycosis fungoides, plaque stage. A report from the Scandinavian Mycosis Fungoides Group. Acta Derm Venereol. 1989;69(6):536-538.
46 Abel EA, Sendagorta E, Hoppe RT. Cutaneous malignancies and metastatic squamous cell carcinoma following topical therapies for mycosis fungoides. J Am Acad Dermatol. 1986;14(6):1029-1038.
47 Mostow EN, Neckel SL, Oberhelman L, et al. Complete remissions in psoralen and UV-A (PUVA)-refractory mycosis fungoides-type cutaneous T-cell lymphoma with combined interferon alfa and PUVA. Arch Dermatol. 1993;129(6):747-752.
48 Roenigk HHJr, Kuzel TM, Skoutelis AP, et al. Photochemotherapy alone or combined with interferon alpha-2a in the treatment of cutaneous T-cell lymphoma. J Invest Dermatol. 1990;95(6 Suppl):198S-205S.
49 Breneman D, Duvic M, Kuzel T, et al. Phase 1 and 2 trial of bexarotene gel for skin-directed treatment of patients with cutaneous T-cell lymphoma. Arch Dermatol. 2002;138(3):325-332.
50 Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy for cutaneous B- and T-cell lymphomas. Int J Radiat Biol Phys. 2008;74(1):154-158.
51 Hoppe RT. Total skin electron beam therapy in the management of mycosis fungoides. Front Radiat Ther Oncol. 1991;25:80-89.
52 Jones GW, Tadros A, Hodson DI, et al. Prognosis with newly diagnosed mycosis fungoides after total skin electron radiation of 30 or 35 GY. Int J Radiat Oncol Biol Phys. 1994;28(4):839-845.
53 Kamstrup MR, Specht L, Skovgaard GL, et al. A prospective, open-label study of low-dose total skin electron beam therapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 2008;71(4):1204-1207.
54 Harrison C, Young J ND, Navi D, et al: Revisiting low dose total skin electron beam therapy in the management of mycosis fungoides, Int J Radiat Oncol Biol Phys (submitted for publication).
55 Becker M, Hoppe RT, Knox SJ. Multiple courses of high-dose total skin electron beam therapy in the management of mycosis fungoides. Int J Radiat Oncol Biol Phys. 1995;32(5):1445-1449.
56 Horwitz SM, Olsen EA, Duvic M, et al. Review of the treatment of mycosis fungoides and sezary syndrome: a stage-based approach. J Natl Compr Canc Netw. 2008;6(4):436-442.
57 Bunn PAJr, Hoffman SJ, Norris D, et al. Systemic therapy of cutaneous T-cell lymphomas (mycosis fungoides and the Sézary syndrome). Ann Intern Med. 1994;121(8):592-602.
58 Rosen ST, Foss FM. Chemotherapy for mycosis fungoides and the Sézary syndrome. Hematol Oncol Clin North Am. 1995;9(5):1109-1116.
59 Grozea PN, Jones SE, McKelvey EM, et al. Combination chemotherapy for mycosis fungoides: a Southwest Oncology Group study. Cancer Treat Rep. 1979;63(4):647-653.
60 Braverman IM, Yager NB, Chen M, et al. Combined total body electron beam irradiation and chemotherapy for mycosis fungoides. J Am Acad Dermatol. 1987;16(1 Pt 1):45-60.
61 Case DCJr. Combination chemotherapy for mycosis fungoides with cyclophosphamide, vincristine, methotrexate, and prednisone. Am J Clin Oncol. 1984;7(5):453-455.
62 Tsimberidou AM, Giles F, Duvic M, et al. Phase II study of pentostatin in advanced T-cell lymphoid malignancies: update of an M.D. Anderson Cancer Center series. Cancer. 2004;100(2):342-349.
63 Kuzel TM, Hurria A, Samuelson E, et al. Phase II trial of 2-chlorodeoxyadenosine for the treatment of cutaneous T-cell lymphoma. Blood. 1996;87(3):906-911.
64 Quaglino P, Fierro MT, Rossotto GL, et al. Treatment of advanced mycosis fungoides/Sézary syndrome with fludarabine and potential adjunctive benefit to subsequent extracorporeal photochemotherapy. Br J Dermatol. 2004;150(2):327-336.
65 Von Hoff DD, Dahlberg S, Hartstock RJ, et al. Activity of fludarabine monophosphate in patients with advanced mycosis fungoides: a Southwest Oncology Group study. J Natl Cancer Inst. 1990;82(16):1353-1355.
66 Mercieca J, Matutes E, Dearden C, et al. The role of pentostatin in the treatment of T-cell malignancies: analysis of response rate in 145 patients according to disease subtype. J Clin Oncol. 1994;12(12):2588-2593.
67 Greiner D, Olsen EA, Petroni G. Pentostatin (2′-deoxycoformycin) in the treatment of cutaneous T-cell lymphoma. J Am Acad Dermatol. 1997;36(6 Pt 1):950-955.
68 Duvic M, Talpur R, Wen S, et al. Phase II evaluation of gemcitabine monotherapy for cutaneous T-cell lymphoma. Clin Lymphoma Myeloma. 2006;7(1):51-58.
69 Zinzani PL, Baliva G, Magagnoli M, et al. Gemcitabine treatment in pretreated cutaneous T-cell lymphoma: experience in 44 patients. J Clin Oncol. 2000;18(13):2603-2606.
70 Di Lorenzo G, Di Trolio R, Delfino M, et al. Pegylated liposomal doxorubicin in stage IVB mycosis fungoides. Br J Dermatol. 2005;153(1):183-185.
71 Duarte RF, Schmitz N, Servitje O, et al. Haematopoietic stem cell transplantation for patients with primary cutaneous T-cell lymphoma. Bone Marrow Transplant. 2008;41(7):597-604.
72 Olavarria E, Child F, Woolford A, et al. T-cell depletion and autologous stem cell transplantation in the management of tumour stage mycosis fungoides with peripheral blood involvement. Br J Haematol. 2001;114(3):624-631.
73 Guitart J, Wickless SC, Oyama Y, et al. Long-term remission after allogeneic hematopoietic stem cell transplantation for refractory cutaneous T-cell lymphoma. Arch Dermatol. 2002;138(10):1359-1365.
74 Molina A, AD, Collins JL, Nademanee A, et al. Clinical, cytogenetic and molecular remission after allogenic hematopoietic stem cell translantation for refractory Sézary syndrome and tumor stage mycoses fungoides. Blood. 2001;98:409a.
75 Soligo D, Ibatici A, Berti E, et al. Treatment of advanced mycosis fungoides by allogeneic stem-cell transplantation with a nonmyeloablative regimen. Bone Marrow Transplant. 2003;31(8):663-666.
76 Herbert KE, Spencer A, Grigg A, et al. Graft-versus-lymphoma effect in refractory cutaneous T-cell lymphoma after reduced-intensity HLA-matched sibling allogeneic stem cell transplantation. Bone Marrow Transplant. 2004;34(6):521-525.
77 Onida F, Saporiti G, Berti E, et al. Reduced-intensity conditioning allogeneic haematopoietic stem cell transplantation in advanced mycosis fungoides and Sézary syndrome. Bone Marrow Transplant. 2007;39:S40.
78 Bigler RD, Crilley P, Micaily B, et al. Autologous bone marrow transplantation for advanced stage mycosis fungoides. Bone Marrow Transplant. 1991;7(2):133-137.
79 Edelson R, Heald P, Perez M. Photopheresis update. Prog Dermatol. 1991;25:1.
80 Holloway KB, Flowers FP, Ramos-Caro FA. Therapeutic alternatives in cutaneous T-cell lymphoma. J Am Acad Dermatol. 1992;27(3):367-378.
81 Lim HW, Edelson RL. Photopheresis for the treatment of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9(5):1117-1126.
82 Heald P, Rook A, Perez M, et al. Treatment of erythrodermic cutaneous T-cell lymphoma with extracorporeal photochemotherapy. J Am Acad Dermatol. 1992;27(3):427-433.
83 Gottlieb SL, Wolfe JT, Fox FE, et al. Treatment of cutaneous T-cell lymphoma with extracorporeal photopheresis monotherapy and in combination with recombinant interferon alfa: a 10-year experience at a single institution. J Am Acad Dermatol. 1996;35(6):946-957.
84 Richardson SK, Lin JH, Vittorio CC, et al. High clinical response rate with multimodality immunomodulatory therapy for Sézary syndrome. Clin Lymphoma Myeloma. 2006;7(3):226-232.
85 Wilson LD, Jones GW, Kim D, et al. Experience with total skin electron beam therapy in combination with extracorporeal photopheresis in the management of patients with erythrodermic (T4) mycosis fungoides. J Am Acad Dermatol. 2000;43(1 Pt 1):54-60.
86 Olsen EA, Rosen ST, Vollmer RT, et al. Interferon alfa-2a in the treatment of cutaneous T cell lymphoma. J Am Acad Dermatol. 1989;20(3):395-407.
87 Vegna ML, Papa G, Defazio D, et al. Interferon alpha-2a in cutaneous T-cell lymphoma. Eur J Haematol Suppl. 1990;52:32-35.
88 Duvic M, Martin AG, Kim Y, et al. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch Dermatol. 2001;137(5):581-593.
89 Duvic M, Lemak NA, Redman JR, et al. Combined modality therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 1996;34(6):1022-1029.
90 Knobler RM, Trautinger F, Radaszkiewicz T, et al. Treatment of cutaneous T cell lymphoma with a combination of low-dose interferon alfa-2b and retinoids. J Am Acad Dermatol. 1991;24(2 Pt 1):247-252.
91 Jones G, McLean J, Rosenthal D, et al. Combined treatment with oral etretinate and electron beam therapy in patients with cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome). J Am Acad Dermatol. 1992;26(6):960-967.
92 Miller VA, Benedetti FM, Rigas JR, et al. Initial clinical trial of a selective retinoid X receptor ligand, LGD1069. J Clin Oncol. 1997;15(2):790-795.
93 Olsen E, Duvic M, Frankel A, et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J Clin Oncol. 2001;19(2):376-388.
94 Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109-3115.
95 Quiros PA, Jones GW, Kacinski BM, et al. Total skin electron beam therapy followed by adjuvant psoralen/ultraviolet-A light in the management of patients with T1 and T2 cutaneous T-cell lymphoma (mycosis fungoides). Int J Radiat Oncol Biol Phys. 1997;38(5):1027-1035.
96 Wilson LD, Licata AL, Braverman IM, et al. Systemic chemotherapy and extracorporeal photochemotherapy for T3 and T4 cutaneous T-cell lymphoma patients who have achieved a complete response to total skin electron beam therapy. Int J Radiat Oncol Biol Phys. 1995;32(4):987-995.
97 Chinn DM, Chow S, Kim YH, et al. Total skin electron beam therapy with or without adjuvant topical nitrogen mustard or nitrogen mustard alone as initial treatment of T2 and T3 mycosis fungoides. Int J Radiat Oncol Biol Phys. 1999;43(5):951-958.
98 Dreno B. Roferon-A (interferon alpha 2a) combined with Tigason (etretinate) for treatment of cutaneous T cell lymphomas. Stem Cells. 1993;11(4):269-275.
99 McGinnis KS, Junkins-Hopkins JM, Crawford G, et al. Low-dose oral bexarotene in combination with low-dose interferon alfa in the treatment of cutaneous T-cell lymphoma: clinical synergism and possible immunologic mechanisms. J Am Acad Dermatol. 2004;50(3):375-379.
100 Winkler CF, Sausville EA, Ihde DC, et al. Combined modality treatment of cutaneous T cell lymphoma: results of a 6-year follow-up. J Clin Oncol. 1986;4(7):1094-1100.
101 Herrmann JJ, Roenigk HHJr, Honigsmann H. Ultraviolet radiation for treatment of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9(5):1077-1088.
102 Scholtz W. Ueber den Einfluss der Rontgenstrahlen auf die Haut in gesunden und krankem Zustande. Arch Dermat u Syph. 1902;59:421.
103 Kim JH, Nisce LZ, D’Anglo GJ. Dose-time fractionation study in patients with mycosis fungoides and lymphoma cutis. Radiology. 1976;119(2):439-442.
104 Cotter GW, Baglan RJ, Wasserman TH, et al. Palliative radiation treatment of cutaneous mycosis fungoides—a dose response. Int J Radiat Oncol Biol Phys. 1983;9(10):1477-1480.
105 Trump JG, Wright KA, Evans WW, et al. High energy electrons for the treatment of extensive superficial malignant lesions. Am J Roentgenol Radium Ther Nucl Med. 1953;69(4):623-629.
106 Karzmark CJ, Loevinger R, Steele RE, et al. A technique for large-field, superficial electron therapy. Radiology. 1960;74:633-644.
107 Page V, Gardner A, Karzmark CJ. Patient dosimetry in the electron treatment of large superficial lesions. Radiology. 1970;94(3):635-641.
108 Spittle MF. Mycosis fungoides: electron beam therapy in England. Cancer Treat Rep. 1979;63(4):639-641.
109 Nisce LZ, Safai B, Kim JH. Effectiveness of once weekly total skin electron beam therapy in mycosis fungoides and Sézary syndrome. Cancer. 1981;47(5):870-876.
110 Hamminga B, Noordijk EM, van Vloten WA. Treatment of mycosis fungoides: total-skin electron-beam irradiation vs topical mechlorethamine therapy. Arch Dermatol. 1982;118(3):150-153.
111 Tadros AA, Tepperman BS, Hryniuk WM, et al. Total skin electron irradiation for mycosis fungoides: failure analysis and prognostic factors. Int J Radiat Oncol Biol Phys. 1983;9(9):1279-1287.
112 Van Vloten WA, de Vroome H, Noordijk EM. Total skin electron beam irradiation for cutaneous T-cell lymphoma (mycosis fungoides). Br J Dermatol. 1985;112(6):697-702.
113 Desai KR, Pezner RD, Lipsett JA, et al. Total skin electron irradiation for mycosis fungoides: relationship between acute toxicities and measured dose at different anatomic sites. Int J Radiat Oncol Biol Phys. 1988;15(3):641-645.
114 Bjarngard BE, Chen GT, Piontek RW, et al. Analysis of dose distributions in whole body superficial electron therapy. Int J Radiat Oncol Biol Phys. 1977;2(3–4):319-324.
115 Hoppe RT, Fuks Z, Bagshaw MA. Radiation therapy in the management of cutaneous T-cell lymphomas. Cancer Treat Rep. 1979;63(4):625-632.
116 Price NM. Electron beam therapy. Its effect on eccrine gland function in mycosis fungoides patients. Arch Dermatol. 1979;115(9):1068-1070.
117 Price NM. Radiation dermatitis following electron beam therapy. An evaluation of patients ten years after total skin irradiation for mycosis fungoides. Arch Dermatol. 1978;114(1):63-66.
118 Sausville EA, Eddy JL, Makuch RW, et al. Histopathologic staging at initial diagnosis of mycosis fungoides and the Sézary syndrome. Definition of three distinctive prognostic groups. Ann Intern Med. 1988;109(5):372-382.
119 Kim YH, Martinez G, Varghese A, et al. Topical nitrogen mustard in the management of mycosis fungoides: update of the Stanford experience. Arch Dermatol. Feb 2003;139(2):165-173.
120 Jones GW, Hoppe RT, Glatstein E. Electron beam treatment for cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9(5):1057-1076.
121 Ramsay DL, Meller JA, Zackheim HS. Topical treatment of early cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9(5):1031-1056.
122 Abel EA, Deneau DG, Farber EM, et al. PUVA treatment of erythrodermic and plaque type mycosis fungoides. J Am Acad Dermatol. 1981;4(4):423-429.
123 Kim YH, Bishop K, Varghese A, et al. Prognostic factors in erythrodermic mycosis fungoides and the Sézary syndrome. Arch Dermatol. 1995;131(9):1003-1008.