Chapter 77 Mycosis Fungoides
Mycosis fungoides (MF) is a low-grade, non-Hodgkin’s lymphoma caused by skin-homing CD4+ T cells that form cutaneous patches, plaques, and tumors.1,2 MF was initially noted in 1806 when Alibert described a patient with cutaneous tumors that he attributed to yaws. Although the disease was initially termed pian fungoides, he later changed the name to mycosis fungoides.3 In 1938, Sézary and Bouvrain described a leukemic variant called Sézary syndrome (SS),4 and Lutzner and Jordan elucidated the ultrastructure of the Sézary cell in 1968.5 The term cutaneous T-cell lymphoma was introduced by Edelson in 1975 and encompasses a variety of cutaneous lymphoproliferative disorders, including MF/SS, adult T-cell leukemia/lymphoma, primary cutaneous CD30+ anaplastic lymphoma, lymphomatoid papulosis, pagetoid reticulosis, and others.6 In clinical practice, the terms MF and cutaneous T-cell lymphoma (CTCL) are often used interchangeably; however, such usage is incorrect.7 MF constitutes only 50% of all CTCLs, and the clinical history and therapy for each subtype of CTCL are different.2
MF is a challenging disorder from all perspectives. Despite improving molecular techniques, diagnosis early in the course of disease is often difficult because of the nonspecific nature of skin lesions and the numerous benign dermatoses that may mimic MF. Once a diagnosis of MF has been correctly established, the optimal initial treatment strategy often remains unclear, given the heterogeneity of clinical presentations and limited data from controlled studies. Although radiotherapy is the most effective single agent in the treatment of MF,8 total skin electron beam therapy (TSEBT) is not readily available at many centers. This chapter provides a summary of the clinically relevant aspects of MF and describes the role and techniques of radiotherapy in patient management.
Etiology and Epidemiology
MF primarily affects adults over the age of 40 years, with incidence rates peaking in the seventh decade. The incidence of CTCL has consistently increased from 1974 through 2002, reaching a current yearly incidence rate of 9.6 cases per million.9 Risk factors for the development of CTCL include black race and male gender. Markers of high socioeconomic status, such as residence in areas with high home values, high level of educational attainment, and high physician density, are associated with an increased incidence CTCL; it is unclear whether these factors are causative or simply increase the likelihood of diagnosis.9
Etiologic agents for the development of MF remain highly speculative.10 Although numerous exposures to toxins, including pesticides, radiation, industrial solvents, tobacco, and alcohol, have been investigated, no consistent causative factors have been identified.11 High rates of seropositivity for both cytomegalovirus12 and human T-cell lymphotrophic virus type I13,14 have been reported, but these studies await further corroboration before a causal relationship can be inferred.15,16,17
The observation that MF is more common in blacks and tends to present in sun-shielded areas (i.e., bathing suit distribution) suggests that sun exposure may protect against the development of MF. Sun exposure may mediate its protective effect by exerting a cytotoxic effect on either the malignant CD4+ T cell of MF or the epidermal antigen-presenting dendritic cell, also known as the Langerhans cell. This cell presents antigens to the malignant CD4+ T cells of MF and may stimulate their growth. The histologic evidence for this interaction is Pautrier’s microabscess, an intraepidermal collection of malignant CD4+ T cells clustered around an antigen-presenting dendritic cell. This finding is considered pathognomonic for MF and suggests that MF may be an antigen-driven malignant disease, though a specific antigen has yet to be identified.18
Prevention and Early Detection
No agents have been identified that will prevent the development of MF. Early diagnosis is critical, however, because local therapy directed against unilesional or oligolesional MF is highly curative.19–21 The most typical presentation of early disease—an erythematous patch with scale arising in a sun-shielded area—may be confused with a number of benign dermatoses, including atopic dermatitis, psoriasis, and tinea corporis.22 At this early stage, most of the lymphocytes noted on histopathologic studies represent reactive inflammatory cells rather than the malignant clone.23 As a result, the histopathologic findings of early MF mimic numerous benign inflammatory conditions,23,24 and a rapid and correct histologic diagnosis is not always possible. In this setting, molecular studies such as the polymerase chain reaction (PCR) for the T-cell receptor will identify a clonal T-cell population in 50% to 80% of patients who ultimately develop overt histologic evidence of MF.25,26 To improve the diagnostic accuracy of early MF, the International Society for Cutaneous Lymphoma proposed a points-based algorithm for early diagnosis27 (Table 77-1).
TABLE 77-1 Proposed Algorithm for Diagnosis of Early Mycosis Fungoides
| Criteria | Score |
|---|---|
| Clinical | 2 points for basic + two additional criteria 1 point for basic + one additional criterion |
| Basic | |
| Persistent and/or progressive patches/thin plaques | |
| Additional | |
| 1. Non–sun-exposed location | |
| 2. Size/shape variation | |
| 3. Poikiloderma* | |
| Histopathologic | 2 points for basic + two additional criteria 1 point for basic + one additional criterion |
| Basic | |
| Superficial lymphoid infiltrate | |
| Additional | |
| 1.Epidermotropism without spongiosis | |
| 2.Lymphoid atypia† | |
| Molecular Biologic | 1 point for clonality |
| Basic | |
| Clonal T-cell receptor gene rearrangement | |
| Immunopathologic | 1 point for one or more criteria |
| 1.<50% CD2+, CD3+, and/or CD5+ T cells | |
| 2.<10% CD7+ T cells | |
| 3.Epidermal/dermal discordance of CD2, CD3, CD5, or CD7‡ | |
| Four or more points satisfy criteria for a diagnosis of early MF. | |
* Poikiloderma is defined as the combination of skin atrophy, telangiectasia, and mottled pigmentation.
† Lymphoid atypia is defined as cells with enlarged, hyperchromatic nuclei and irregular or cerebriform nuclear contours.
‡ T-cell antigen deficiency confined to the epidermis.
Adapted from Pimpinelli N, Olsen EA, Santucci M, et al: Defining early mycosis fungoides. J Am Acad Dermatol 53:1053-1063, 2005.
Biologic Characteristics and Molecular Biology
A malignant clone in patch-plaque MF bears the immunophenotype of activated, skin-homing CD4+ helper T cells.18 When a naïve T cell identifies its cognate antigen in a skin-draining lymph node, activation occurs, and the T cell begins to express cutaneous lymphocyte antigen (CLA) and CC chemokine receptor 4 (CCR4). As these activated T cells pass through the capillaries of inflamed skin, CLA and CCR4 bind to their respective ligands on the dermal capillaries, resulting in extravasation of the activated T cells into the dermal connective tissue. Once outside the circulation, activated T cells migrate to the epidermis and interact with antigen-presenting dendritic (Langerhans) cells (Fig. 77-1).
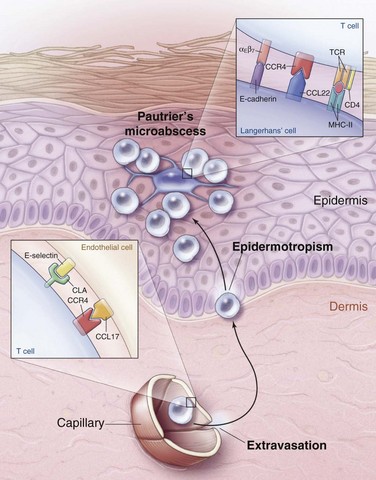
Clinically, progression of MF is associated with loss of epidermotropism and increasing tumor burden. Molecular studies have shown that progression of MF is associated with TP53 mutation,28 numerous chromosomal rearrangements,29 and microsatellite instability.30 In addition, the malignant cells in MF develop mechanisms to escape destruction by the host immune system. For example, although benign activated T cells are eliminated by fas/fas-ligand-mediated apoptosis, the malignant T cells of MF evade fas-mediated apoptosis via fas downregulation, mutation, or alternative splicing.31
As a malignant disease of the immune system, MF results in substantial alteration of host immunity with consequent increased risk of infection32 and possibly secondary malignant disease.33 For example, in patients with Sézary syndrome, the absolute number of normal circulating T cells often drops dramatically, reaching levels typically seen only in acquired immunodeficiency syndrome.34 In addition, malignant CD4+ T cells produce large amounts of interleukin-10 and transforming growth factor-β, resulting in further suppression of cell-mediated immunity.35,36 The malignant cells of MF also produce large amounts of soluble interleukin-2 receptor, which can inactivate interleukin-2,37 a cytokine needed to promote normal T-cell activation. Finally, the malignant cells of Sézary syndrome can elaborate large amounts of interleukin-4 and interleukin-5, producing a syndrome characterized by atopy and eosinophilia.36
Pathology and Pathways of Spread
The diagnosis of MF remains challenging, even for the experienced clinician and dermatopathologist, due to both the absence of a diagnostic gold standard and the number of benign inflammatory dermatoses that may mimic MF, particularly in its early stages. Currently, the diagnosis relies upon integrating the clinical presentation with the histopathologic, immunophenotypic, and genotypic data. The current World Health Organization–European Organization for the Research and Treatment of Cancer (WHO-EORTC)38 pathologic classification scheme for CTCL is presented in Table 77-2.
Histopathology
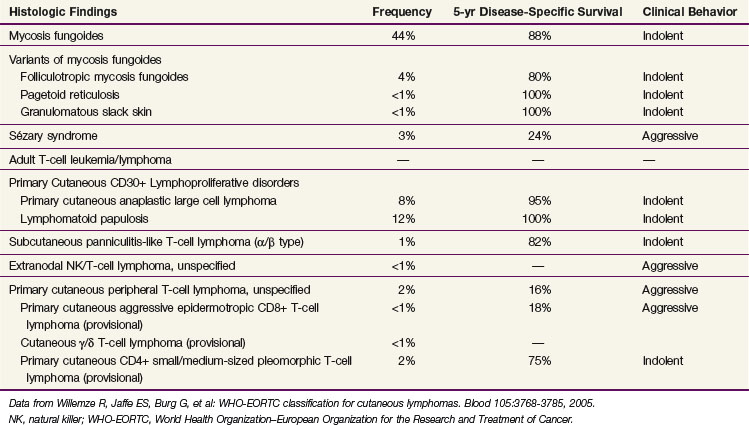
The most striking finding of early MF is profound epidermotropism, characterized by lymphocytes clustered along the basement membrane of the epidermis39 (Fig. 77-2A and B). Microdissection studies have shown that virtually all of the lymphocytes in the epidermis belong to the malignant clone, whereas most dermal lymphocytes are reactive.40,41
Several microscopic findings help to discriminate early MF from benign inflammatory mimics. For example, an EORTC study reported that identification of epidermal lymphocytes with extremely convoluted, medium to large (7 to 9 µm) nuclei enabled a correct diagnosis of MF with 100% sensitivity and 92% specificity24 (see Fig. 77-2B). In contrast, a study from Stanford University found that intraepidermal atypical lymphocytes surrounded by a clear halo (an artifact of fixation) were the most robust indicator of MF in a multivariate model.42 Another finding, Pautrier’s microabscess, is considered pathognomonic but is seen in less than 20% of early lesions42 (see Fig. 77-2D). Efforts are currently under way to establish a grading system to standardize pathology reporting and improve diagnostic accuracy.23,43
As MF progresses from the patch to plaque stage, the lymphoid infiltrate increases in density and begins to invade the deeper reticular dermis (see Fig. 77-2C). Furthermore, due to the increased burden of neoplastic cells, findings such as Pautrier’s microabscesses (see Fig. 77-2D), haloed lymphocytes, and convoluted nuclei are more readily identified, resulting in improved diagnostic accuracy. Tumor formation results from vertical growth of the lymphoid infiltrate and may be associated with complete loss of epidermotropism and sparing of the upper papillary dermis (see Fig. 77-2E). Erythrodermic MF often resembles patch-stage MF, although epidermotropism may be more subtle and neoplastic cells may be quite sparse.44
Immunophenotyping of Skin Lesions
Assessment of T-cell marker expression within the lymphoid infiltrate provides additional information that helps to establish a diagnosis of MF. The hallmark of MF is expression of CD4, the marker of mature helper T cells. A typical immunophenotype for MF is CD2+ (pan T cell), CD3+ (pan T cell), CD4+ (helper T cell), CD5+ (pan T cell), CD45RO+ (memory T cell), CLA+ (cutaneous lymphoid antigen), CD8− (cytotoxic T cell), CD30− (activated T cell).45 Although many benign dermatoses express a similar immunophenotype, two markers, CD7 and Leu-8, are often underexpressed in MF and may be helpful in distinguishing between MF and benign mimics. Finally, rare cases of apparently classic MF that are CD4− but CD3+ and CD8+ have been reported.46
T-Cell Receptor Gene Rearrangement
Clonal T-cell receptor gene rearrangements are frequently identified in MF skin lesions and may help to differentiate between early MF patches and benign mimics. Clinical data indicate that PCR will identify a dominant clonal T-cell receptor-γ rearrangement in 63% to 90% of skin biopsies that show definite histologic evidence of MF.47,48 Furthermore, PCR identifies a clonal T-cell population in 50% to 80% of histologically borderline biopsies obtained from patients who subsequently develop classic MF.25,26 In contrast, T-cell clonality occurs in only 6% to 24% of benign dermatoses that contain a lymphoid infiltrate.47,48 These observations suggest that identification of a clonal T-cell population should always be considered in light of the clinical and histologic context and may help to confirm a diagnosis of MF when it is already suspected on these grounds.
Transformation to Large Cell Histologic Variant
Transformation to a large cell variant occurs in up to 39% of patients initially diagnosed with MF.49 Histologic diagnosis of transformation can be made when large cells (four times or more the size of a small lymphocyte) make up more than 25% of the lymphoid infiltrate or form microscopic nodules.50 Transformation is associated with expression of CD30 in 30% of cases and expression of CD20 in 45% of cases. Transformed MF is typically aggressive, with clinical behavior similar to that of high-grade lymphoma.
Clinical Manifestations
According to the EORTC classification of cutaneous lymphomas, the term mycosis fungoides should be reserved for those CD4+ cutaneous lymphomas that are “characterized by the subsequent evolution of patches to more infiltrated plaques and eventually tumors.”2 Over time, MF may spread to involve lymph nodes, blood, bone marrow, and visceral organs. Symptoms may vary by degree of involvement, but weight loss, night sweats, and fever are uncommon unless infection is present.
Premycotic Phase
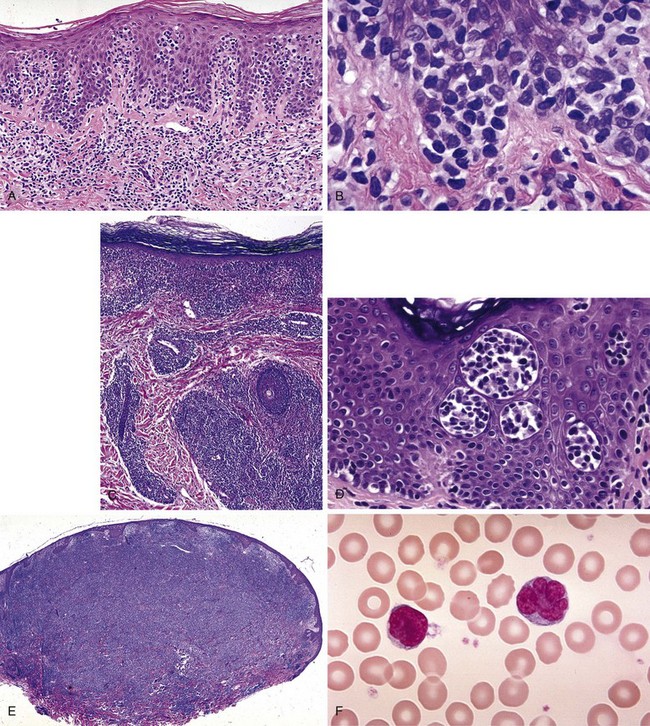
Early MF typically begins with mildly erythematous, slightly scaling, annular or arcuate macules that classically involve sun-shielded areas (Fig. 77-3A). These lesions may wax and wane for years before histologic findings show definitive evidence of MF.
Plaque Phase
If untreated, some patches will progress to form more generalized, deeply infiltrative, scaling plaques that often have well-demarcated, palpable borders and may exhibit central clearance and arcuate morphologic characteristics (see Fig. 77-3B). Associated findings include hyperkeratosis of the palms and soles and fissures.
Tumor Phase
Over 80% of tumors emerge in the setting of established patch-plaque MF (see Fig. 77-3C). The most common sites of tumor involvement include the face, digits, and perineum. Tumors frequently ulcerate and are prone to infection.
Tumeur d’Emblée
The term tumeur d’emblée refers to the rare patient who presents with tumors that arise in the absence of antecedent skin lesions; some cases may be more appropriately classified as CD30− cutaneous large T-cell lymphoma rather than MF.51 The clinical course may be more aggressive than that of classical MF.
Erythroderma
Erythroderma is defined as greater than 80% body surface involvement with confluent patches and/or plaques. It is associated with intense pruritus, hyperkeratosis of the palms and soles, skin atrophy, and lichenification (see Fig. 77-3D). Erythroderma may arise de novo or from progression of patch/plaque MF.
Lymph Nodes
Lymph node involvement is present in 15% of newly diagnosed patients and is associated with advanced cutaneous disease.52 Nodes are typically nontender and mobile and measure from 1 to 4 cm. Bulky adenopathy is uncommon.
Internal Organs
Visceral involvement is typically seen only in patients with advanced cutaneous disease, nodal disease, and blood involvement. The most common sites include the lungs, central nervous system, oral cavity, and oropharynx,52,53 although MF has been observed in other sites such as the breast, thyroid, and pancreas. Visceral involvement is often subclinical and does not routinely precipitate death. Bone marrow involvement at initial staging has been reported in 6% to 28% of patients and is also associated with advanced skin and nodal disease.54,55
Sézary Syndrome
Sézary syndrome (SS) is defined as erythroderma plus evidence of malignant circulating T cells that satisfy any of the five criteria56 listed in Table 77-3. For the purposes of these criteria, the Sézary cell is defined as “any atypical lymphocyte with [a] moderately to highly infolded or grooved nucleus”56 (see Fig. 77-2F). Clinical findings may include edema and tumorous involvement of the face leading to leonine facies, severe fissures of the palms and soles, intense pruritus, and cutaneous pain.
TABLE 77-3 Proposed Hematologic Criteria for Diagnosis of Sézary Syndrome
| Absolute Sézary cell count ≥1000 cells/µL. |
| CD4/CD8 ratio ≥10 due to an increase in CD3+1 or CD4+1 cells by flow cytometry. |
| Aberrant expression of pan-T-cell markers (CD2, CD3, CD4, CD5) by flow cytometry. Deficient CD7 expression on T cells (or expanded CD4+1, CD7− cells ≥40%) is a tentative criterion. |
| Increased lymphocyte count with T-cell clone in blood identified by Southern blot or polymerase chain reaction. |
| A chromosomally abnormal T-cell clone. |
Adapted from Vonderheid EC, Bernengo MG, Burg G, et al: Update on erythrodermic cutaneous T-cell lymphoma. Report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol 46:95-106, 2002.
Variants of Mycosis Fungoides
The progression from subtle patches to indurated plaques, cutaneous tumors, and erythroderma represents classical so-called Alibert-Bazin mycosis fungoides. According to the EORTC classification, variants such as bullous and hyperpigmented or hypopigmented MF manifest similar clinical behavior and should not be considered separately from classical MF.2 Several variants sharing some clinical and pathologic features with MF have been described.
Folliculotropic Mycosis Fungoides
This entity presents with follicular papules, comedo-like lesions, milia-like lesions, patches, and plaques, all of which may produce alopecia.1,38 Although it typically involves the head and neck, any skin site may be affected.2 Pathologically, atypical lymphocytes invade follicles and may deposit acid mucopolysaccharides in the pilosebaceous units. Folliculotropic MF tends to be refractory to topical treatments such as PUVA and nitrogen mustard, and risk of relapse after treatment with TSEBT appears to be higher when compared with that of patients with classical MF.57 The 5-year disease-specific survival (DSS) is approximately 80%.38
Pagetoid Reticulosis or Woringer-Kolopp Disease
Pagetoid reticulosis, also known as Woringer-Kolopp disease, typically presents as a slow-growing, hyperkeratotic or psoriasiform, localized patch or plaque involving a distal extremity. Pathologically, an abundant epidermotropic infiltrate composed of atypical large lymphocytes is noted, along with pagetoid spread of individual lymphocytes interspersed among keratinocytes. Benign-appearing small lymphocytes are found in the upper dermis.1 The prognosis for localized pagetoid reticulosis is excellent with either surgery or radiotherapy, and disease-related deaths have not been reported.2,38 Ketron-Goodman type is a disseminated, more aggressive cutaneous lymphoproliferative disorder that histologically resembles localized pagetoid reticulosis.
Granulomatous Slack Skin
This rare variant presents with lax skin in the axillae, neck, breasts, and inguinal regions. Histologic features include epithelioid or giant cell dermal granulomas and associated destruction of elastin fibers.2 Notably, Hodgkin’s disease has been associated with granulomatous slack skin in roughly one-third of reported cases.58 Because of its rarity, optimal treatment has not been established.
Related Cutaneous T-Cell Lymphoproliferative Disorders
Primary Cutaneous Anaplastic Large Cell Lymphoma or CD30+ Large Cell Cutaneous T-Cell Lymphoma
This entity typically presents as a red or flesh-colored nodule or tumor that frequently ulcerates. Histopathologic testing shows sheets of CD30+ large lymphocytes without epidermotropism. In contrast to systemic CD30+ anaplastic large cell lymphoma, overexpression of anaplastic lymphoma kinase (ALK) is not found in primary cutaneous CD30+ large cell lymphoma. Patients with localized disease are typically treated with radiotherapy alone, and the prognosis is excellent, with a 5-year DSS of approximately 90% to 95%.2,59,60
Lymphomatoid Papulosis
Lymphomatoid papulosis (LyP) presents with grouped erythematous or violaceous papules and/or nodules at different stages of development. Lesions typically resolve spontaneously within 2 to 8 weeks, but scarring is common.61 Three different histologic subtypes have been described, with types A and C consisting of malignant CD30+ T cells, often with an extensive inflammatory infiltrate, and type B simulating classical plaque-stage MF. For type C lesions, discrimination between LyP and CD30+ large cell lymphoma may be difficult on histologic grounds, and assessment of the clinical context may be required to ensure the proper diagnosis.2 Although cytologically malignant, it should be emphasized that LyP is clinically benign, with a 5-year survival rate of 100%.2,38,59 Neither aggressive chemotherapy nor radiotherapy are therefore indicated.62 Treatment options include PUVA or low-dose methotrexate, but neither is considered curative. In the long term, at least 15% to 20% of patients with LyP will develop a secondary malignant tumor, most commonly MF, CD30+ large cell lymphoma, or Hodgkin’s disease.62,63 In patients undergoing TSEBT for MF, a history of LyP is associated with an increased risk of relapse.57
Adult T-Cell Lymphoma or Leukemia
Adult T-cell lymphoma/leukemia (ATLL) develops in 2% to 4% of individuals infected with human T-cell lymphotrophic virus type I (HTLV-I), a retrovirus endemic in southern Japan and the Caribbean.1,61,64,65 Skin findings are present in up to 60% of patients with ATLL and strongly resemble those of MF, including plaques, tumors, and erythroderma. The characteristic immunophenotype is CD2+ CD3+ CD4+ CD5+ CD7− CD8− and the malignant cells strongly express the high-affinity interleukin-2 receptor (CD25, CD122, and CD132). Clinical features vary from an acute form presenting with B symptoms, hypercalcemia, metabolic bone disease, hepatosplenomegaly, generalized adenopathy, and leukemic infiltration to a smoldering or chronic form presenting with skin infiltration and little or no systemic involvement. Although patients often respond to conventional chemotherapy, long-term survival is rare. Newer promising agents include denileukin diftitox66 and interferon-α in combination with zidovudine.67
Subcutaneous Panniculitis-like T-Cell Lymphoma (α/β Type)
In prior years, subcutaneous panniculitis-like T-cell lymphoma was thought to run two markedly different clinical courses—an indolent course and an aggressive course. Recently, molecular studies have indicated that the indolent form of subcutaneous panniculitis-like T-cell lymphoma is caused by CD8+ T cells that express the α/β T-cell receptor. This entity typically presents with subcutaneous nodules or plaques involving the legs or trunk and has a 5-year DSS of approximately 82%.38 In contrast, the more aggressive form of subcutaneous panniculitis-like T-cell lymphoma is caused by CD8− T cells that express the γ/δ T-cell receptor and is now classified as cutaneous γ/δ T-cell lymphoma in the updated WHO-EORTC system.38 This entity is often fatal and complicated by hemophagocytic syndrome. Treatment for the α/β type is often observation, whereas treatment for the γ/δ type includes combination chemotherapy and possibly cyclosporine.1,2
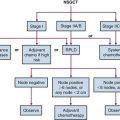
Prognosis and Staging
The seventh edition of the American Joint Committee on Cancer (AJCC) staging system for MF identifies the extent and character of skin lesions, extracutaneous disease, and leukemic transformation as the major predictors of poor prognosis68 (Tables 77-4 and 77-5). Shortcomings of this system include (1) difficulty in assigning T category for patients who fall on the border between T1 and T2; (2) failure to discriminate between the prognosis of patients with extensive patches as compared with extensive plaques; (3) similarity in prognosis between patients with tumors and erythroderma; (4) the questionable prognostic relevance of enlarged, pathologically uninvolved lymph nodes; and (5) the rarity of the N2 descriptor because biopsies of nonpalpable lymph nodes are seldom performed.69,70 To address these shortcomings, the EORTC, in conjunction with the International Society for Cutaneous Lymphoma, recently proposed a new staging system for MF, though it remains to be seen how this proposal will affect subsequent iterations of the AJCC staging system.71
| T1 | Patches and/or plaques involving <10% body surface area |
| T2 | Patches and/or plaques involving ≥10% body surface area |
| T3 | One or more cutaneous tumors |
| T4 | Generalized erythroderma |
| N0 | Lymph nodes clinically uninvolved |
| N1 | Lymph nodes clinically enlarged but histologically uninvolved |
| N2 | Lymph nodes clinically nonpalpable but histologically involved |
| N3 | Lymph nodes clinically enlarged and histologically involved |
| M0 | No visceral disease present |
| M1 | Visceral disease present |
| B0 | No circulating atypical cells (<1000 Sézary cells [CD4+1 CD7–]/µL) |
| B1 | Circulating atypical cells present (≥1000 Sézary cells [CD4+1 CD7–]/µL) |
Adapted from Edge SB, Byrd DR, Compton CC, et al: AJCC Cancer Staging Handbook, 7th ed. New York, Springer, 2010.
TABLE 77-5 Staging Classification for Mycosis Fungoides*
| IA | T1N0M0 |
| IB | T2N0M0 |
| IIA | T1-2N1M0 |
| IIB | T3N0-1M0 |
| IIIA | T4N0M0 |
| IIIB | T4N1M0 |
| IVA | T1-4N2-3M0 |
| IVB | T1-4N0-3M1 |
* The B descriptor is not considered in stage classification.
Adapted from Edge SB, Byrd DR, Compton CC, et al: AJCC Cancer Staging Handbook, 7th ed. New York, Springer, 2010.
Data from a cohort of 468 patients with newly diagnosed MF evaluated at Stanford University52 are presented in Table 77-6. Over 60% of patients presented with patches and/or plaques without nodal or visceral disease. Such patients rarely developed disseminated disease, even at 20 years of follow-up. In contrast, 50% to 60% of patients presenting with tumors or erythroderma will develop extracutaneous disease.
TABLE 77-6 Initial Stage and Natural History of 468 Patients with Mycosis Fungoides Evaluated at Stanford University at the Time of Initial Diagnosis
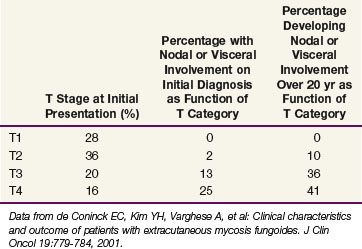
Data from Stanford University showed that patients with limited patches and plaques (IA, T1N0M0) experience 10-year survival rates similar to those of a matched control population. In contrast, the median survival time for patients with extensive patches and plaques (T2), tumors (T3), and erythroderma (T4) was 11, 3.2, and 4.6 years, respectively.72 Patients with either pathologically documented lymph node involvement or visceral involvement experienced a median survival time of roughly 1 year.52
A multi-institutional study showed that survival time is also related to the pattern and extent of lymph node involvement.73 For example, partial or complete effacement of nodal architecture by malignant lymphocytes conferred a median survival time of 2.3 years. In contrast, lymph nodes with aggregates of atypical lymphocytes but preserved nodal architecture resulted in a median survival time of 6 years and lymph nodes with only dermatopathic changes or few atypical lymphocytes resulted in a median survival time of 9 years.
Other factors that may herald a poor prognosis include patient age of 60 years or more,74 elevated LDH levels,74 elevated soluble interleukin-2 receptor levels,37 a low percentage of CD8+ tumor infiltrating lymphocytes,75 extent of skin involvement in those with T3 disease,76 T-cell clonality within the cutaneous infiltrate detected by PCR,77,78 an identical T-cell clone in the skin and peripheral blood,79 and T-cell clonality in dermatopathic lymph nodes.80
The AJCC also included a blood descriptor, B0 versus B1, to document the absence or presence of more than 1000 Sézary cells (CD4+, CD7− per µL). Among patients with erythroderma, the presence of B1 disease is a significant, independent predictor of survival time, doubling the risk of death.81
For those patients with transformation to a large cell variant of MF, the median survival time ranges from 19 months to 36 months.49,50 Factors predictive of poor survival after transformation include a short interval between the diagnosis of MF and transformation (<2 years) and the presence of IIB to IV disease.49
Patient Evaluation
History
All patients should have a thorough history performed by the evaluating dermatologists, radiation oncologists, and any other consultants (Table 77-7). Careful attention should be given to the duration, change in appearance, and distribution of the eruption according to the patient. Inquiry related to a history of pruritus, pain, exfoliation, fissures, bullae, and perineal discomfort should be made. Previous diagnostic considerations, procedures, and therapies should be recorded in detail to establish temporal relationships. The clinical interview, along with the examination, are the two most important aspects of the workup because they can help exclude other diagnoses and establish whether the narrative and findings are consistent with a diagnosis of MF.
TABLE 77-7 Recommended Workup for Patients with Suspected Mycosis Fungoides
CBC, complete blood count; CT, computed tomography; LDH, lactate dehydrogenase; PCR, polymerase chain reaction; PET, positron emission tomography.
Diagnostic Testing
Pathology
Routine bone marrow examination is generally unnecessary but may be considered in patients with blood or other visceral involvement. Bone marrow involvement at initial staging has been reported in 6% to 28% of patients and is associated with advanced skin and nodal disease.54,55 There is no evidence to suggest that marrow involvement is an independent predictor of outcome, however.55,74 Furthermore, although a clonal T-cell population within the marrow is identified in 75% of patients with identical skin and peripheral blood T-cell clones, this finding does not appear to alter the prognosis.54
Hematology and Chemistry
A thorough examination of the peripheral blood for evidence of malignant T cells is indicated for those with IIB to IV MF and may be considered for those with IA to IIA disease. Peripheral blood flow cytometry should be performed to assess expression of CD2, CD3, CD4, CD5, CD7, CD8, CD20, and CD45RO. Findings suggestive of blood involvement include an elevated CD4 : CD8 ratio (normal range, 0.5 to 3.5), or an expanded population of CD4+D7− or CD45RO+ lymphocytes.56 If initially abnormal, findings on peripheral blood flow cytometric testing should be followed to assess the response to therapy. If flow cytometry is unremarkable, peripheral blood PCR for T-cell receptor gene rearrangement should be considered. PCR reveals a clonal T-cell population identical to that found in the cutaneous infiltrate in 40% of patients with erythroderma and 14% of patients with patches, plaques, or tumors.47
Diagnostic Imaging
Chest radiography should be completed for all patients via posteroanterior and lateral views. CT of the chest, abdomen, and pelvis may be obtained for patients with tumors, erythroderma, or nodal involvement but is not necessary for those with IA or IB disease given the low yield.82 FDG-PET (fluorodeoxyglucose–positron emission tomography) may also be beneficial for patients with advanced skin disease; for example, Stanford University reported that CT alone identified pathologic adenopathy by size criteria in only 5 of 13 patients, whereas the addition of PET identified hypermetabolic adenopathy in 13 of 13 patients. The standardized uptake value (SUV) of the FDG correlated with the extent of nodal involvement, with the highest SUVs noted in patients with complete nodal effacement and large cell transformation.83 To confirm nodal involvement and document the presence or absence of transformation, excisional biopsy should be strongly considered for selected abnormal lymph nodes that are radiographically identified.
Primary Therapy
Because MF is a disease of cutaneous (i.e., skin-homing) lymphocytes, therapy is quite distinct from that of nodal lymphomas. For patients with disease limited to the skin, topical therapy alone produces high rates of remission and even cure. Topical therapies include mechlorethamine (nitrogen mustard), carmustine (BCNU), steroids, bexarotene gel, psoralen plus ultraviolet A (PUVA), ultraviolet B (UVB), and either localized or total skin electron radiotherapy. For patients with localized unilesional MF, topical therapies alone produce long-term DFS in excess of 85%.19–21 For those with more than one patch or plaque but with less than 10% body surface area involvement (T1N0 and T1N1), topical therapies alone produce long-term DFS ranging from 30% to 50%. For patients with more extensive patches or plaques, topical therapies may produce remission, but long-term cure is unlikely. Such patients should receive intensive topical therapy to induce a complete remission followed by less intensive adjuvant topical therapy to sustain a remission.84,85
Those with tumors or erythroderma experience severe cutaneous symptoms and are at high risk for extracutaneous dissemination. Of all the topical therapies, total skin electron beam therapy (TSEBT) is associated with the highest rates of complete response for this patient subgroup.85–87 Therefore it is recommended that TSEBT be administered to induce cutaneous remission and that adjuvant systemic and/or topical therapy be administered to sustain remission. Systemic therapies include interferon, retinoids, oral bexarotene, denileukin diftitox, vorinostat, extracorporeal photochemotherapy (photopheresis), and cytotoxic chemotherapy.
Skin-Directed Therapy
Mechlorethamine
Topical mechlorethamine hydrochloride (HN2), also known as nitrogen mustard, is an alkylating agent with proven activity in the treatment of MF patches and plaques. Mechlorethamine is typically applied daily and continued for at least 6 months after a complete response.69 Cutaneous intolerance, manifested by erythema and pruritus, occurs in roughly 50% of patients treated with aqueous HN2,88 but is reduced to less than 10% in patients treated with HN2 dissolved in ointment such as Aquaphor.69 Other cutaneous side effects of HN2 may include xerosis, hyperpigmentation, and, rarely, bullous reactions, urticaria, and Stevens-Johnson syndrome.88 Bone marrow suppression is not observed because of minimal systemic absorption. The carcinogenicity of HN2 remains debated because one series reported no increased risk of secondary skin cancers in patients treated with HN2 monotherapy,69 but another series reported an eightfold increase in the risk of nonmelanoma skin cancers attributable to HN2 monotherapy.89 HN2 may also potentiate the carcinogenicity of other topical therapies such as total skin irradiation or PUVA.90
Carmustine
Topical carmustine (BCNU) is another alkylating agent with activity in MF. Because of systemic absorption that may produce bone marrow suppression, the drug should be applied to no more than 10% of the body surface area, the duration of treatment should be limited to 4 months, and complete blood counts should be monitored. Cutaneous hypersensitivity is uncommon (7% in one series), but chronic skin telangiectasias and hyperpigmentation may occur.91
Topical Steroids
High-potency topical glucocorticoids are an important component in the treatment of MF due to their ability to alleviate cutaneous symptoms and induce lesion regression. Typically, steroids are applied to active lesions only because widespread application can induce reversible depression of serum cortisol in 10% to 15% of patients.92 Persistent application can lead to skin atrophy.
Topical Rexinoids
Bexarotene belongs to a new class of agents called rexinoids that bind to the retinoid X receptor, resulting in transcription of various genes that control cellular differentiation and proliferation.93 Topical bexarotene gel was recently approved after phase I and II trials demonstrated a 44% to 54% response rate in refractory cutaneous MF.94,95 Most patients will develop an irritant dermatitis and thus require close observation and dose titration. Due to its irritant effects, bexarotene gel is not indicated for patients with more than 15% body surface area involvement. As with systemic retinoids, bexarotene in both its topical and systemic forms should be avoided in pregnant women due to possible teratogenicity.
Psoralen Plus Ultraviolet A (PUVA) and Ultraviolet B
Ultraviolet light used in the treatment of MF includes ultraviolet B (wavelength, 320 to 290 nm), narrow-band ultraviolet B (wavelength, 311 nm), or psoralen plus ultraviolet A (PUVA, wavelength, 400 to 320 nm). Because ultraviolet B has limited penetration, its efficacy is limited to thin patches. PUVA penetrates more deeply and will effectively treat some plaque lesions. PUVA requires ingestion of a photochemotherapeutic agent, 8-methoxypsoralen, prior to ultraviolet A exposure. Ultraviolet A activates 8-methoxypsoralen, resulting in DNA cross-linking and apoptotic cell death.96
Acute side effects of PUVA and ultraviolet B include skin erythema that may be painful, hyperpigmentation, xerosis, pruritus, and blistering. Eye goggles are used to decrease the risk of cataract formation. One side effect unique to PUVA is nausea and vomiting after ingestion of 8-methoxypsoralen (8-MOP). This may be avoided by substituting 8-MOP for either a topical “psoralen bath” or 5-methoxypsoralen, a nonemetogenic analog that is currently available in Europe.97 Long-term toxicity includes photoaging and increased risk of melanoma and nonmelanoma skin cancers.98,99
Systemic Therapy
Interferon
Interferon-α-2a (IFN-α) is an effective agent, particularly for patch and plaque disease, likely due to a direct antitumor effect and/or immunomodulation.100 Interferon-α has been used alone100,101 or in combination with retinoids, PUVA,102,103,104 and extracorporeal photopheresis.105 To date, no randomized trials have compared interferon-α plus another therapy with interferon-α alone, although one prospective trial did show a benefit from combining interferon-α with PUVA as compared with interferon-α plus a retinoid.106 Toxicity may include flulike symptoms, psychiatric disturbances including depression and confusion, elevated transaminases, leukopenia, thrombocytopenia, proteinuria, and myelopathy.103,104 Despite these side effects, in a recent phase II trial of interferon-α and PUVA, only 8% of patients withdrew as a result of toxicity.104
Retinoids
Oral retinoids such as isotretinoin and acitretin influence cellular differentiation and may be particularly beneficial in the treatment of folliculotropic MF. Retinoids can be safely combined with other therapies such as PUVA,107 interferon-α, and TSEBT.108 Side effects include photosensitivity, xerosis, myalgias, arthralgias, headaches, impaired night vision, corneal opacities, teratogenicity, elevated transaminases, hyperlipidemia, and pancreatitis.72
Rexinoids
Oral bexarotene has been approved by the U.S. Food and Drug Administration (FDA) for use in all stages of treatment-refractory MF.109,110 Preliminary data suggest that bexarotene can be combined safely with other therapies, including PUVA, extracorporeal photopheresis, interferon-alpha, and HN2.
Hypertriglyceridemia, the most common adverse event, occurs in 80% of treated patients and may result in reversible pancreatitis if triglyceride levels exceed 800 mg/dL.93 Therefore atorvastatin or fenofibrate should be initiated if triglyceride levels exceed 350 mg/dL. Gemfibrozil increases serum bexarotene concentrations, resulting in a paradoxic elevation of triglycerides, and should therefore be avoided.93 Another side effect, central hypothyroidism, affects roughly 75% of patients but responds well to levothyroxine and resolves when treatment is discontinued.93 Patients taking bexarotene therefore require monitoring of estimated free thyroxine because levels of thyroid-stimulating hormone (TSH) will always be low. Other side effects include self-limited headaches, mild neutropenia, mild transaminase elevations, skin peeling, and pruritus. In the phase II to III trial of oral bexarotene for advanced, refractory CTCL, only 10% of patients receiving the optimal dose withdrew as a result of an adverse event.109
Denileukin Diftitox
Denileukin diftitox is a recombinant fusion protein that contains portions of interleukin-2 and diphtheria toxin and has proven activity in refractory MF, stages IB to IVA.111 It selectively targets T cells that express the high-affinity interleukin-2 receptor (a complex of CD25, CD122, and CD132), resulting in endocytosis of diphtheria toxin, inhibition of protein synthesis, and cell death. In the pivotal phase III clinical trial leading to its approval, only patients with neoplastic T cells expressing the high-affinity interleukin-2 receptor received denileukin diftitox, and controversy currently exists as to whether or not patients without significant expression of the high-affinity interleukin-2 receptor benefit from this therapy.112 Denileukin diftitox is typically administered by a 30-minute venous infusion given on five consecutive days and repeated every 3 weeks for up to eight cycles.
Toxicities were commonly encountered in the phase III clinical trial of denileukin diftitox. Acute hypersensitivity reactions, including dyspnea, back pain, hypotension, and chest pain, occurred in 60% of patients. Furthermore, a vascular leak syndrome characterized by hypotension, hypoalbuminemia, and edema was encountered in 25% of patients. Other toxicities may include constitutional symptoms, thrombotic events, infections, transaminase elevations, renal impairment, and lymphopenia. In total, 21% of the patients in this trial withdrew due to adverse events.111 Subsequent investigations have suggested that pretreatment with corticosteroids substantially reduces the risk of acute toxicity.113
Vorinostat
Vorinostat is a histone deacetylase inhibitor (HDAC) approved for use in patients with MF who have progressive, persistent, or recurrent disease following treatment with at least two systemic therapies. HDAC inhibitors increase accumulation of acetylated histones, resulting in decreased availability of nuclear DNA to bind to transcription factors. The decrease in transcription decreases intracellular protein levels, ultimately producing cell cycle arrest and apoptosis. In preclinical studies of MF cell lines, vorinostat has been shown to induce apoptosis and downregulate stat6.114 Phase II clinical studies have reported clinical response rates of 25% to 30% in pretreated patients, with a modest median response duration of 15 to 26 weeks.115,116 Common toxicities include diarrhea, nausea, fatigue, and anorexia.
Extracorporeal Photochemotherapy
Extracorporeal photochemotherapy (EP), also known as photopheresis, is a novel immune therapy that has shown activity in the treatment of erythrodermic MF.96,117 Peripheral blood leukocytes are harvested by leukapheresis, mixed with 8-methoxypsoralen, exposed to 2 J/cm2 of ultraviolet A, and then reinfused into the patient. This results in DNA cross-linking and gradual apoptotic death of the circulating MF cells that were exposed to psoralen plus UVA. For unclear reasons, monocytes are resistant to the apoptotic effects of EP but instead are stimulated to become immature antigen-presenting dendritic cells due to the physical process of leukapheresis. Therefore, once treated leukocytes are reinfused into the bloodstream, activated dendritic cells may phagocytize remnants of the apoptotic MF cells, present their antigens on major histocompatibility class I, and stimulate expansion of antitumor CD8+ T cells.118
EP is typically administered on two consecutive days every 4 weeks, although the frequency may be increased in patients with extensive disease.119 For patients who achieve a complete response, therapy should be maintained for roughly 6 months and then gradually tapered. In general, EP is well tolerated, although hypotension, arrhythmias, and heart failure may occur due to fluid shifts. Patients with a history of cardiac disease therefore require close monitoring.117 EP has been safely combined with interferon-α and TSEBT.105,120,121
Chemotherapy
Systemic chemotherapy is typically reserved for refractory cutaneous disease, visceral disease, or large cell transformation. Chemotherapy is not typically used in the initial management of patients with MF because a randomized phase III trial comparing concurrent TSEBT and systemic chemotherapy to sequential topical therapy failed to show an improvement in DFS or OS.122
For patients who may benefit from chemotherapy, two main strategies exist. The first relies on oral agents such as methotrexate,123 etoposide, or chlorambucil. This strategy avoids the need for central venous lines, which are associated with a high risk of infection due to frequent bacteremia caused by open skin lesions. The second strategy relies on intravenously administered chemotherapy. One exciting new prospect is pegylated liposomal doxorubicin, an agent that tends to remain intravascular but will extravasate into the inflamed lesional skin of MF. A pilot study conducted on 34 patients with CTCL reported a complete response in 15, a partial response in 15, and 6 severe adverse events.124 Another notable agent is the nucleoside analog gemcitabine, which has been shown to induce complete response rates of approximately 10% and overall response rates of 70% without an increased risk of infection.125 Attention has also focused on purine analogs such as fludarabine, 2′-deoxycoformycin, and 2-chlorodeoxyadenosine. Clinical trials of these agents, however, have produced response rates ranging from only 28% to 51% and have documented substantial toxicity, including myelosuppression, infection, and pulmonary dysfunction.126–129 Other agents with activity in MF include the lipophilic antifolate trimetrexate130; 5-fluorouracil; cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP)131; cisplatin; etoposide; bleomycin; and vinblastine.125
High-Dose Chemotherapy with Autologous or Allogeneic Bone Marrow Transplant
Limited experience has been reported using high-dose chemotherapy with either autologous or allogeneic transplant in the treatment of advanced refractory MF. Autologous stem cell rescue has resulted in several complete responses; however, relapses within 1 year are the rule rather than the exception.132–134 In contrast, myeloablative chemotherapy with allogeneic bone marrow transplant may result in prolonged DFS times. One series of three patients treated with cytoreductive chemotherapy and total body irradiation (TBI) found that two of the three patients remained disease free at 4.5 years and 15 months after transplant. The third patient had recurrent disease after allogeneic transplant but developed a second complete response upon withdrawal of prophylactic cyclosporine, suggesting a graft-versus-tumor effect.135,136 In another series, six of eight patients with heavily pretreated, refractory MF who underwent allogeneic transplant with either TBI or non-TBI preparative regimens were alive without evidence of lymphoma at a median of 56 months following transplant, but two deaths secondary to toxicity were reported.137 Nonmyeloablative transplants have also been reported, with one series reporting clearance of clonal T cells and durable complete remissions in three out of three patients, although one died from infectious complications.138 Collectively, these experiences strongly support the use of allogeneic transplant for younger patients with treatment-refractory advanced MF.
Radiotherapy
Radiation therapy is the most effective single modality in the treatment of MF8 and plays an important role in the treatment of localized or disseminated cutaneous disease and in the palliation of nodal and visceral metastases.
Dose
A dose response has been reported both for radiation of single MF lesions and for TSEBT. For example, in a study of 110 lesions from 14 patients with at least 1 year of follow-up, Cotter and colleagues139 reported an infield recurrence rate of 42% for those treated to a total dose of 10 Gy or less, 32% for 10.01 to 20 Gy, 21% for 20.01 to 30 Gy, and 0% for more than 30 Gy. In a study of 30 lesions from patients with stage IA MF treated with localized radiotherapy, Wilson and associates20 reported local failure in 20% (4 of 20 patients) treated with 20 Gy or less, compared with 0% (0 of 10 patients) treated with more than 20 Gy.20 Kim and coauthors140 treated different lesions from the same patient with various total doses and fraction sizes, finding that division of the total dose into two fractions separated by 1 or 7 days did not alter local control rates. This suggests that the malignant cells of MF have minimal ability to execute sublethal damage repair and provides justification for using low daily fraction sizes to minimize normal tissue toxicity without sacrificing local control. Collectively, these experiences demonstrate that curative treatment of localized MF requires total doses of at least 20 Gy and perhaps up to 30 Gy. To spare normal tissue toxicity and to ensure that patients can receive TSEBT in the future, a fraction size of 1.2 to 2 Gy is recommended.
A dose-response relationship has also been demonstrated with TSEBT. A meta-analysis of published results reported higher rates of complete remission when using higher prescribed doses and more penetrating electrons.86 Historically, the Stanford University experience with 176 patients undergoing TSEBT from 1958 to 1975 revealed that complete response rates increased with total dose: 18% for 8 to 9.9 Gy, 55% for 10 to 19.9 Gy, 66% for 20 to 24.9 Gy, 75% for 25 to 29.9 Gy, and 94% for 30 to 36 Gy.141 Similarly, patients treated with higher doses experienced improved OS, regardless of T category. The experience at Hamilton Regional Cancer Center between 1977 and 1992 further supported the importance of high-dose TSEBT. From 1977 to 1980, 25 consecutive patients received 30 Gy of TSEBT. From 1980 to 1992, 121 consecutive patients received 35 Gy of TSEBT.142 Treatment with high-dose TSEBT was an independent predictor of response, with a complete response rate of 64% for the 30-Gy group and 85% for the 35-Gy group. Given these data, the European Organization for the Research and Treatment of Cancer (EORTC) consensus statement recommended that modern TSEBT deliver a total dose of 31 to 36 Gy to the skin surface in order to produce a dose of 26 Gy at a depth of 4 mm in truncal skin along the central axis.143
Clinical Results of Limited Superficial Radiotherapy
Approximately 5% of patients with stage IA disease present with a single skin lesion or with two or three lesions in close proximity, such that all clinically apparent disease can be encompassed by either one field or several abutting fields.20 Radiotherapy is the treatment of choice in this situation, based on results from three institutions that have reported long-term DFS in excess of 85%.20,21,144 Wilson and colleagues20 published a series of 21 patients with minimal IA disease managed with local, superficial radiotherapy. Ten were treated with 100 to 280 kV and 11 with 4- to 12-MeV electrons with appropriate bolus. The median dose was 20 Gy, and 17 of 21 patients received 20 Gy or more. With a median follow-up of 36 months, the rate of complete clinical remission was 97% and the long-term DFS was 91% among patients receiving 20 Gy or more. Similarly, Micaily and associates21 reported an 86% 10-year DFS of in 18 patients with unilesional MF treated with localized radiotherapy to a median dose of 30.6 Gy. In summary, radiotherapy for unilesional IA disease is an excellent first-line therapy given minor acute and chronic toxicities and excellent long-term results. Because very few patients with two to four lesions have been reported in the literature, optimal treatment for this subgroup remains unclear. Although some advocate limited superficial radiotherapy, others advocate a strategy such as TSEBT or PUVA in order to treat all skin surfaces.
Clinical Results of Total Skin Electron Beam Therapy
Limited Patches and/or Plaques: T1N0M0 (IA) and T1N1M0 (IIA) Disease
For example, patients with T1 disease treated with modern TSEBT experience a complete response rate of at least 90%86,145 (Table 77-8), compared with a complete response rate of 65% to 70% for topical mechlorethamine.69,89 Furthermore, the 10-year relapse-free survival rate is roughly 50% for TSEBT146 compared with 34% for mechlorethamine.69 In this group of patients, however, whose long-term survival rate is similar to that of healthy controls, initial treatment with TSEBT has not been associated with an OS advantage.145 The administration of TSEBT for patients with IA MF remains controversial, and although some reserve TSEBT for those refractory to standard therapies, others recommend TSEBT as first-line therapy for even paucilesional IA disease.
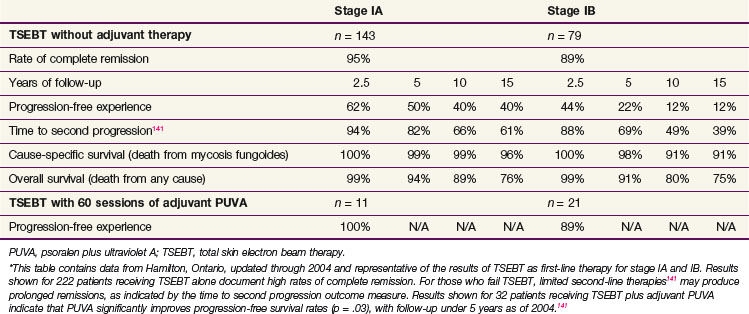
Other first-line therapies for T1 MF include phototherapy and carmustine. For patients with limited patches, ultraviolet B therapy results in a complete response rate of roughly 80%, with a median response duration of 2 years.147,148 PUVA is appropriate for both patches and plaques, producing complete response rates of at least 80%. Although relapse is common, most patients will respond to additional PUVA.149,150 Similar to mechlorethamine, topical BCNU produced a complete response rate of 86%, partial response rate of 12%, and 5-year relapse-free survival rate of 35%.91
For T1 disease that recurs after mechlorethamine, BCNU, ultraviolet B, or PUVA, retreatment with these agents may result in another response. Other options for recurrent or refractory T1 disease include TSEBT,143 class I steroids (complete response, 63%; partial response, 31%),92 interferon-α monotherapy (complete response, 50%; partial response, 35%),100 topical bexarotene (complete response, 21%; partial response, 42%),94 and oral bexarotene (complete response, 7%; partial response, 47%).110
For patients whose disease recurs after initial treatment with TSEBT, relapse is typically limited to less than 5% of the skin surface. Salvage treatments including spot radiation, topical steroids, mechlorethamine, or PUVA have all resulted in prolonged second remissions. For example, data from Hamilton showed that 70% of patients were free of disease 15 years after first-line TSEBT and second-line salvage treatments as needed.146
Extensive Patches and/or Plaques: T2N0M0 (IB) and T2N1M0 (IIA) Disease
For patients in this group with either severe cutaneous symptoms or deeply infiltrative plaques, we favor TSEBT as initial treatment due to superior response rates that produce rapid palliation. For example, complete response rates for T2 disease range from 76% to 90% (see Table 77-8), compared with a complete response rate of 34% for topical mechlorethamine.69,85,143 Adjuvant therapy following TSEBT is crucial for patients with T2 disease because the 10-year relapse-free survival rate is only 10% in those treated with TSEBT alone.146 Retrospective data from Yale University found that patients receiving adjuvant PUVA experienced a 5-year DFS of 85% versus 50% for those not receiving adjuvant PUVA.84 A prospective pilot study at Hamilton documented a similar benefit (see Table 77-8). Retrospective data from Stanford University showed that patients treated with TSEBT and adjuvant topical mechlorethamine experienced a 10-year relapse-free survival rate of roughly 40% compared with 10% for TSEBT alone.85
Patients with T2 disease who are relatively asymptomatic can receive treatment similar to that recommended for T1 disease. Again, topical mechlorethamine is a reasonable choice for these patients, resulting in a complete response rate of 34%, partial response rate of 38%, and 10-year relapse-free survival rate of 20%.69 Similarly, BCNU produced a complete response rate of 47%, partial response rate of 37%, and 5-year relapse-free survival rate of 10%.91 Topical corticosteroids may also be helpful for patients with extensive patches, with data suggesting a complete response rate of 25% and a partial response rate of 57%.92
As an alternative to topical chemotherapy, topical phototherapy may be considered for extensive patch/plaque disease. Ultraviolet B should be reserved for extensive patch disease only, due to its superficial penetration. In contrast, PUVA may be considered for patients with patches or thin plaques and has resulted in complete response rates of 60% to 100%.92,149,150 Phase II trials suggest promising improvements in both the complete response rate and the duration of remission when adding interferon-α to PUVA for the treatment of T2 disease.103,104,106
Cutaneous Tumors: T3N0-1M0 (IIB) Disease
Radiotherapy is an important treatment modality for cutaneous tumors due to its ability to treat the full thickness of deeply infiltrative lesions. For the rare patient with asymptomatic tumors involving less than 10% of the skin, either topical HN2 with local radiotherapy or TSEBT are reasonable first-line treatment options, with both producing a similar 5-year OS of roughly 50%.85 Most patients with T3 disease present with extensive, symptomatic tumors, however. Such patients often benefit from TSEBT as a first-line, palliative treatment, due to a superior complete response rate as compared with topical HN2 plus localized radiotherapy (44% to 54% vs. 8%).85,86
Adjuvant therapy should be strongly considered for patients who achieve a complete response to TSEBT. Retrospective data suggest that adjuvant topical mechlorethamine may improve the duration of response, resulting in a 5-year relapse-free survival rate of 55% compared with 30% with TSEBT alone.85 Adjuvant photopheresis is another reasonable option, with retrospective data reporting a 5-year OS of 100% for patients receiving this modality compared with 50% in patients who did not receive adjuvant therapy.120 Other adjuvant therapies worthy of consideration include interferon-α, bexarotene, and denileukin diftitox.151
Erythroderma: T4N0-1M0 (III) Disease
TSEBT is an appropriate initial therapy for erythrodermic MF due to its ability to produce a rapid and sustained response, thereby ameliorating the severe cutaneous symptoms experienced by such patients. Further, limited early data suggest that TSEBT can result in a substantial reduction in the number of malignant cells circulating in the peripheral blood, potentially altering the natural history of the disease.152 Retrospective data indicate that TSEBT monotherapy produced a 100% complete response rate and 5-year PFS of 69% for patients with T4N0M0B0 MF disease.87 When including those with blood or visceral involvement, however, the complete response rate dropped to 74% and only 36% remained progression free at 5 years. Patients with erythroderma and blood or visceral involvement may benefit from adjuvant photopheresis, as retrospective data suggest that such treatment improves the 2-year cause-specific survival rate from 69% without EP to 100% with EP.121
For patients without access to TSEBT, several other topical treatment options exist. For example, a series of 10 patients treated with PUVA reported a complete response rate of 70% and a median PFS of 5 months.153 Although not studied in a randomized setting, prospective phase II experiences suggested that addition of interferon-α to PUVA may improve the duration of response.102,103,104 Another option for T4 disease is photopheresis monotherapy, following which 80% of patients will experience at least some cutaneous improvement.96 Patients with a normal peripheral blood CD4/CD8 ratio appear more likely to respond.119
Toxicity of Total Skin Electron Beam Therapy
TSEBT is generally well tolerated, and toxicity is minimized by using low daily fraction sizes154 and a shielding regimen that reduces the dose to eyes, ears, lips, hands, and feet. Common acute toxicities from TSEBT include pruritus, dry desquamation, erythema, alopecia, xerosis, bullae of the feet, edema of the hands and feet, hypohidrosis (diminished perspiration),155 and loss of fingernails and toenails.146,156 Rare acute side effects include gynecomastia in men, mild epistaxis, and mild parotiditis.146 Because of the superficial penetration of electrons, patients do not experience gastrointestinal or hematologic toxicities. In general, TSEBT does not cause serious long-term complications,157 although permanent nail dystrophy, xerosis, telangiectasias, partial scalp alopecia, and fingertip dysesthesias have been described.143 Secondary cutaneous malignant diseases, including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma, have been observed in patients treated with TSEBT, particularly in those exposed to multiple therapies that are themselves known to be mutagenic, such as PUVA and mechlorethamine.90,158
Locally Advanced Disease and Palliation
Young patients with locally advanced disease and a good performance status should be treated with curative intent with an allogeneic transplant when possible. However, for the majority of patients whose age or performance status precludes allogeneic transplant, treatment options include novel biologic agents, cytotoxic chemotherapy, and radiation therapy. For example, oral bexarotene has shown efficacy in patients with relapsed or refractory MF, producing an overall response rate of 57% for IIB, 32% for III, 44% for IVA, and 40% for IVB.109 Another option, denileukin diftitox, resulted in an overall response rate of 30% for patients with treatment-refractory IB to IVA disease.111 Systemic chemotherapy should also be considered and may result in complete response rates of 20% to 60%.131
Radiotherapy plays an important role in the palliation of both cutaneous and extracutaneous disease. For those patients with extensive skin disease recurrent after TSEBT, a second course of TSEBT may produce substantial palliation with acceptable toxicity. At Yale University, 14 patients have received two courses of TSEBT and 5 have received three courses of TSEBT.159 The median dose was 36 Gy for the first course, 18 Gy for the second, and 12 Gy for the third. After the second course, 86% achieved a complete response, with a median disease-free interval of 11.5 months. In a similar experience from Stanford University, 15 patients received a second course of TSEBT to a median dose of 23 Gy.160 The complete response rate was 40% and the partial response rate was 60%. Toxicity was limited to xerosis, telangiectasias, pigment changes, and alopecia. Criteria for retreatment include a complete response to the initial course, an extended disease-free interval after the initial course, diffuse cutaneous involvement at relapse, and failure of other modalities.
For patients with localized, symptomatic skin disease, radiotherapy options include higher doses on the order of 20 to 30 Gy, or lower doses on the order of 8 Gy in two fractions. Recent evidence suggests that the lower-dose approach produces complete response rates in excess of 90%, and that retreatment with higher doses of irradiation is safe and effective for those patients whose disease recurs following low-dose irradiation.161 Finally, patients with symptomatic nodal or visceral disease often benefit from a course of palliative megavoltage radiotherapy to a total dose of 20 to 30 Gy delivered in 2- to 3-Gy fractions.
Irradiation Techniques
Historical Development
Localized irradiation was first used to treat MF in 1902, shortly after the discovery of x-rays.162 In 1939, Summerville reported treatment of extensive cutaneous MF with an “x-ray bath” of kilovoltage photons delivered with two large fields.163 A total air exposure of 900 R delivered in daily fractions of 10 R produced a complete response. In 1945, Levin and Behrman164 proposed that the total air exposure at the skin surface should be 600 to 800 R for patches or plaques and 1000 to 1600 R for tumors.164
The development of TSEBT began in the early 1950s. Trump and colleagues165 at the Massachusetts Institute of Technology used a Van de Graaff generator to produce a vertically oriented, stationary beam of 2.5-MeV electrons incident on a motorized couch. By placing the patient in the prone, supine, and lateral decubitus positions and translating the couch through the electron beam, all skin surfaces could receive a meaningful dose. This treatment approach produced complete responses in two patients with extensive MF. In 1960, Stanford University reported the first method for linear accelerator-based TSEBT.166 With a patient standing 10 feet from the end of the accelerator, two fields were treated, one directed above the patient’s head and a second directed below the patient’s feet. This approach is known as the dual-field technique. In addition, patients were treated in two positions with respect to the accelerator: anteroposterior and posteroanterior. Over time, it became clear that increasing the number of treatment positions improved dose homogeneity in the lateral dimension.167,168 Ultimately, the Stanford group adopted a six-treatment position technique in which patients stand in six different orientations with respect to the accelerator: anteroposterior, posteroanterior, right and left anterior oblique, and right and left posterior oblique.169 Although commonly called the six-field technique, it should be remembered that two fields are actually treated for each of the six treatment positions.
The first clinical results of TSEBT were published in 1962 by a group from St. John’s Hospital for Diseases of the Skin in London.170 Using the dual-field, four-treatment position technique developed at Stanford, all five patients treated for MF experienced “very good” responses at total doses ranging from 12 to 18 Gy. Four of five patients relapsed within 8 months, however, and disease was difficult to control in the axillae and perineum due to underdosing. In 1971, Stanford University reported 107 patients with MF who received TSEBT between 1957 and 1968.169 Using a variety of doses, they produced a 52% complete response rate. In addition, among patients with localized patches or plaques, 30% experienced long-term DFS, providing the first evidence that MF could actually be cured.
Target Volumes
The target volume for patients with patch/plaque disease should include the epidermis and dermis.86 The thickness of the epidermis varies from 0.05 to 0.5 mm and is greatest in the distal extremities. The thickness of the dermis varies from 1 to 4 mm and is greatest in the hands and feet. Therefore, the thickness of the skin varies from a minimum of about 2 mm on the trunk to a maximum of about 4.5 mm at the hands and soles of the feet.86 As a result, the EORTC TSEBT consensus statement recommends that the 80% isodose line should be 4 mm deep or more to the skin surface to ensure that the epidermis and dermis fall within the high-dose region. Due to their thickness, the deep margin of cutaneous tumors is often underdosed when treated with TSEBT alone and may require supplemental boosts with appropriately selected electrons or photons to ensure adequate dose delivery at depth.
Limited Superficial Radiotherapy
Minimal IA disease should be treated with a single radiation field where possible, though abutting fields may be required at a convex surface such as the scalp, an axillary fold, breast, hand, or foot. The junction of abutting fields should be shifted during the course of treatment to improve homogeneity. Field margins can be limited by lead cutout to only 1 to 2 cm beyond the visible (or palpable) clinical lesion. Electrons of 6- to 16-MeV energy with appropriate bolus material are sufficient, and the dose should be approximately 30 Gy with the expectation of cure. One or several fields of 6- to 16-MeV electrons can also be used to encompass the limited volumes required to palliate symptomatic skin lesions, including most tumor nodules and skin ulcers. Although a single dose of 4 to 8 Gy may produce a complete response and full symptom relief, a fractionated dose of at least 20 Gy is more likely to achieve a complete and durable clinical remission.20,86,139 Whether irradiating a limited region of the skin for cure or for palliation, dose prescriptions should specify conventional or lower doses per fraction (e.g., 1.2 to 2 Gy) and the total dose should be limited (e.g., 20 to 30 Gy). This preserves as much radiation tolerance of the skin as possible and enables future delivery of local radiotherapy or TSEBT without moderate-to-severe toxicities. Furthermore, in situations of coexisting cancer diagnoses (e.g., Hodgkin’s lymphoma or breast, prostate, or rectal cancers), radiation treatments for those diagnoses must be carefully planned to minimize the superficial skin exposure in order to preserve the option of TSEBT in the future.
Node, Viscera, and Blood Radiotherapy
Several experiences using TSEBT in combination with either low-dose, total body irradiation171 or total nodal irradiation172,173 have been reported. However, such treatment may increase the risk of secondary malignant diseases or myelodysplastic syndrome173 and has not demonstrated superiority over conventional systemic agents. For patients undergoing allogeneic bone marrow transplantation, incorporation of total body irradiation in the preparative regimen may be beneficial.136 A dose of 12 to 15 Gy in 6 to 10 fractions is recommended; single fractions are discouraged because of a lower therapeutic ratio.
Modern TSEBT
Technical advances since the initial Stanford and St. John’s publications have included dose escalation to 35 to 36 Gy, improvements in shielding, and integration of boost treatments to underdosed regions, culminating in the publication of a consensus statement from the EORTC regarding appropriate techniques for TSEBT143 (Table 77-9).
TABLE 77-9 EORTC Guidelines for Total Skin Electron Beam Therapy
Adapted from Jones GW, Kacinski BM, Wilson LD, et al: Total skin electron radiation in the management of mycosis fungoides. Consensus of the European Organization for Research and Treatment of Cancer (EORTC) Cutaneous Lymphoma Project Group. J Am Acad Dermatol 47:364-370, 2002.
The method of TSEBT used clinically at Yale achieves these objectives with a 6-MeV modern linear accelerator using the dual-field, six-treatment position technique. The patient stands 3.8 m from the accelerator head, which rotates 17.5 degrees above and below horizontal to produce the dual fields. A 3.2-mm polycarbonate screen (Lexan) is placed in front of the patient and serves to attenuate and scatter the incident electrons, resulting in an electron energy of 3.9 MeV at the skin surface (Fig. 77-4). A total of six treatment positions are designated: anteroposterior, right and left anterior oblique, posteroanterior, and right and left posterior oblique (Fig. 77-5). These positions maximize skin unfolding, thereby improving dose homogeneity in the lateral dimension. Each position is treated with two fields, an upper field and a lower field, in order to maximize dose homogeneity in the vertical dimension. On treatment day 1, the anteroposterior, right posterior oblique, and left posterior oblique positions are treated. On treatment day 2, the posteroanterior, right anterior oblique, and left anterior oblique positions are treated with the same dose. Over the course of a 2-day treatment cycle, a patient will receive 2 Gy to the entire skin surface. This pattern continues, with patients receiving treatment 4 days a week for a total of 9 weeks, during which time a total dose of 36 Gy is delivered to the skin surface (Table 77-10).
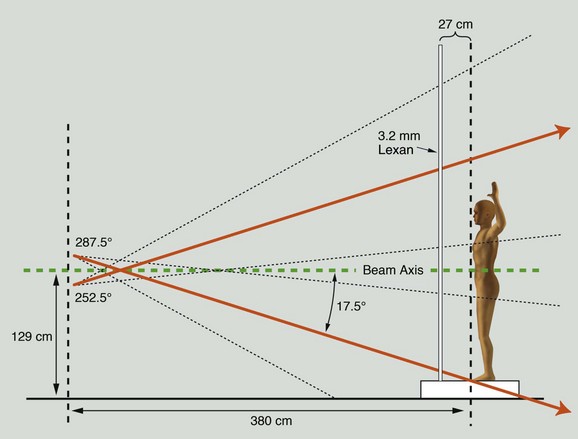

TABLE 77-10 Treatment Protocol at Yale–New Haven Hospital
| Treatment Cycle | |
| Day 1 | AP, RPO, LPO treatment positions |
| Day 2 | PA, RAO, LAO treatment positions |
| Dose | |
| Dose per cycle | 2 Gy |
| Cycles per week | 2 |
| Total cycles | 18 |
| Total dose | 36 Gy |
| Boosts | |
| Perineum | 100 cGy/day, first 9 and last 9 treatment days |
| Soles of feet | 100 cGy/day, first 7 and last 7 treatment days |
| Blocking | |
| External eye shields | First 11 cycles |
| Internal eye shields | Last 7 cycles |
| Lip shield | Cycles 1-4 |
| Lead mitt for hands | Every other cycle |
| Fingernail shield | Every other cycle, alternating with mitts |
| Foot block | Cycles 1-3, 5, 7, 9, 11, 13, 15, 17, 18 |
| Testicular shield | Used with perineal boost only |
AP, anteroposterior; LAO, left anterior oblique; LPO, left posterior oblique; PA, posteroanterior; RAO, right anterior oblique; RPO, right posterior oblique.
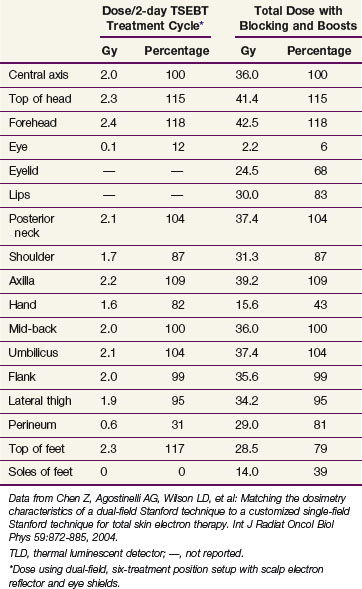
Data from Chen Z, Agostinelli AG, Wilson LD, et al: Matching the dosimetry characteristics of a dual-field Stanford technique to a customized single-field Stanford technique for total skin electron therapy. Int J Radiat Oncol Biol Phys 59:872-885, 2004.
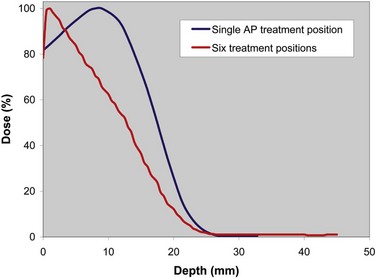
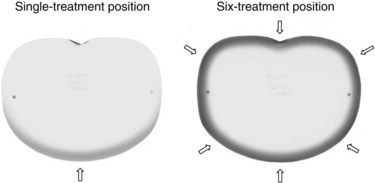
This technique places the dose maximum at 1 mm, the 80% isodose line at 6 mm, and the 20% isodose line at 12 mm,174 thereby satisfying the EORTC criteria for TSEBT.146 Photon contamination due to bremsstrahlung scattering in the machine head, intervening air, scatterers or degraders, and patient, is acceptable at 1.2%. A comparison of the depth-dose curves for a single anteroposterior treatment position and for all six treatment positions is presented graphically in Figure 77-6 and by film dosimetry174 in Figure 77-7. Using six treatment positions shifts the isodose curve toward the skin surface due to the obliquity of the incident electrons.
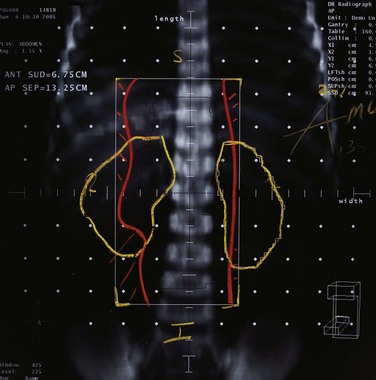
Because of shielding inherent to these treatment positions, the soles of the feet, perineum, and scalp are underdosed and always require supplemental treatment. One can use a dose of 120 kV, HVL 4.2 mm Al, to deliver patch treatments to the soles of the feet (14 Gy in 14 fractions on treatment days 1 to 7 and 30 to 36) and perineum (18 Gy in 18 fractions on treatment days 1 to 9 and 28 to 36). The scalp dose can be supplemented with either orthovoltage patches (6 to 20 Gy over 1 to 3 weeks) or with an electron reflector mounted above the patient’s head175 (see Table 77-10). Other areas of potential underdosing include the ventral penis, upper medial thighs, inframammary folds, folds under any pannus, and lateral and flatter regions of the face and trunk. Supplemental patch fields, as guided by in vivo dosimetry or clinical suspicion, are appropriate for these regions to ensure that the surface dose is at least 50% of the prescribed TSEBT dose.176,177 When determining the total dose given in patch treatments, doses to areas such as the feet and perineum may be reduced as clinically indicated to enhance patient tolerance, provided such areas are uninvolved.
The hands, wrists, ears, ankles, and dorsal penis may be overdosed because of a variety of factors, including tissue heterogeneity (e.g., bone), high convexity, and overlap between more than three of the six primary fields.86 These regions should therefore be shielded for a certain proportion of the treatments to reduce the total dose to 36 Gy or less. Detailed dosimetric measurements are typically required to determine the most appropriate shielding regimen for a particular treatment arrangement and patient geometry (Table 77-11). Lead, bags containing rice, and plywood boards are appropriate methods of shielding. The eyes can be shielded with a combination of external eye shields and lead internal eye shields.178 Although internal eye shields may cause conjunctival irritation and corneal abrasions, the risk is usually less than 1%.146 The shielding regimen used at the Yale–New Haven Hospital and the resulting doses to various anatomic structures are presented in Tables 77-10 and 77-11.
Other Radiation Management Issues
Cutaneous symptoms, including xerosis, pruritus, and pain from fissures or ulceration, are often severe and should be managed aggressively with topical steroids, emollients, oral antihistamines and aggressive wound care with nonocclusive dressings.72 Acute skin toxicity from TSEBT usually responds to the above measures, although treatment breaks on the order of 1 to 2 weeks may occasionally be required.179 Large bullae that develop during TSEBT should be lanced under sterile conditions and require nonocclusive dressings until they reepithelialize.
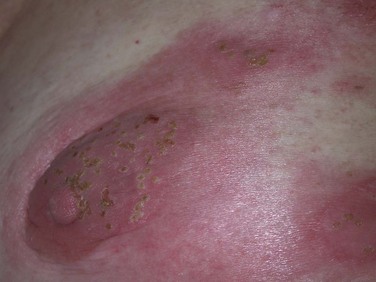
As previously discussed, patients with advanced MF may experience a profound degree of immunosuppression. As a result, cutaneous and systemic infections are an important cause of morbidity and mortality. The most common infections include cellulitis due to Staphylococcus aureus or β-hemolytic streptococci, cutaneous herpes simplex or herpes zoster (both of which may disseminate cutaneously), bacteremia (most commonly with S. aureus), and bacterial pneumonia.32 Signs and symptoms of such infections should be evaluated promptly and treated aggressively. Independent of MF, TSEBT also suppresses cutaneous immunity, likely due to destruction of normal skin-homing lymphocytes and epidermal antigen-presenting dendritic cells. Any new eruption that develops during the course of TSEBT should therefore be evaluated promptly because disseminated cutaneous bacterial, fungal, and viral infections are not uncommon180 (Fig. 77-8).
Treatment Algorithm, Controversies, Problems, Challenges, and Future Possibilities
A treatment algorithm for newly diagnosed and recurrent MF181 is presented in Table 77-12. Multiple treatment options are listed, reflecting different institutional preferences and the paucity of randomized trials comparing different treatment modalities. A major challenge is the need for prospective randomized trials to compare radiation-based and non-radiation-based approaches to initial treatment. Adequately powered trials are problematic given the rarity of MF and the difficulty in accruing patients. In the absence of randomized data, therefore, treatment will continue to be guided by institutional expertise and patient preference.
| Stage | Initial Treatment | Treatment for Relapsed or Refractory Disease |
|---|---|---|
| Unilesional patch T1N0M0 (stage IA) |
Localized, superficial radiotherapy Topical BCNU Topical HN2 Topical bexarotene PUVA UVB |
Same as initial treatment |
| Limited patch-plaque T1NO-1MO (stage LA) |
Topical HN2 Topical BCNU PUVA UVB (if only patches) Topical bexarotene TSEBT ± PUVA or HN2 |
Same as initial treatment TSEBT Topical or oral bexarotene Interferon-α alone PUVA + interferon-α Topical corticosteroids |
| Extensive patch-plaque | Topical HN2 | Same as initial treatment |
| Symptoms controlled | Topical BCNU | TSEBT |
| Minimal plaque thickness T2N0-1M0 (stage IA, IIA) |
PUVA ± interferon-α UVB (if only patches) TSEBT + adjuvant PUVA or HN2 |
Topical or oral bexarotene Interferon-α alone Topical corticosteroids |
| Extensive patch-plaque | TSEBT + adjuvant PUVA or HN2 | Topical HN2 |
| Symptomatic | — | Topical BCNU |
| Indurated plaques T2N0-1M0 (stage IB, IIA) |
— | PUVA + interferon-α Topical or oral bexarotene Topical corticosteroids Repeat TSEBT |
| Cutaneous tumors T3N0-1M0 (stage IIB) |
TSEBT + adjuvant HN2 or photopheresis Bexarotene Denileukin diftitox |
Topical HN2 and local radiotherapy Oral bexarotene Denileukin diftitox Systemic chemotherapy Repeat TSEBT |
| Erythroderma T4N0-1M0 (stage III) |
TSEBT + adjuvant photopheresis PUVA + interferon-α Photopheresis alone Interferon-α |
Methotrexate Oral bexarotene Denileukin diftitox Gemcitabine Fludarabine ± interferon-α Pentostatin ± interferon-α 2-chlorodeoxyadenosine Standard B-cell lymphoma chemotherapy Repeat TSEBT Allogeneic transplant |
| Nodal or visceral disease Stage LV |
Clinical trial Allogeneic transplant Methotrexate TSEBT Topical HN2 Topical BCNU PUVA + interferon-α Topical corticosteroids Localized, superficial radiotherapy |
Same as initial treatment Systemic chemotherapy as listed |
| Transformed to large cell variant | Same as stage IV Consider rituximab if CD20+1 Trimetrexate Allogeneic transplant |
Same as stage IV |
BCNU, bischloroethylnitrosurea; HN2, mechlorethamine; PUVA, psoralen plus ultraviolet A; TSEBT, total skin electron beam therapy; UVB, ultraviolet B.
Adapted with permission from Smith BD, Wilson LD: Management of mycosis fungoides. Part 2. Treatment. Oncology (Huntingt) 17:1419-1433, 2003 (Table 2).
The future will likely bring additional novel systemic therapies for MF. For example, biologic and immunologic agents including monoclonal antibodies directed against CD4 and CD52 (alemtuzumab, Campath-1H), topical tazorotene (a novel retinoid), interleukin-2, interleukin-12, and individualized dendritic cell-based tumor vaccines are under investigation and may continue to improve the treatment of recurrent or refractory disease.182,183 Strategies for integrating novel biologic agents with standard therapies such as TSEBT are needed to maximize the benefits of each. For those with advanced disease, a combination of TSEBT to induce cutaneous remission and biologic therapy to induce systemic remission shows the greatest promise for providing long-term disease control.
15 Wood GS, Salvekar A, Schaffer J, et al. Evidence against a role for human T-cell lymphotrophic virus type I (HTLV-I) in the pathogenesis of American cutaneous T-cell lymphoma. J Invest Dermatol. 1996;107:301-307.
16 Li G, Vowels BR, Benoit BM, et al. Failure to detect human T-lymphotropic virus type-I proviral DNA in cell lines and tissues from patients with cutaneous T-cell lymphoma. J Invest Dermatol. 1996;107:308-313.
31 van Doorn R, Dijkman R, Vermeer MH, et al. A novel splice variant of the Fas gene in patients with cutaneous T-cell lymphoma. Cancer Res. 2002;62:5389-5392.
37 Wasik MA, Vonderheid EC, Bigler RD, et al. Increased serum concentration of the soluble interleukin-2 receptor in cutaneous T-cell lymphoma. Clinical and prognostic implications. Arch Dermatol. 1996;32:42-47.
49 Diamandidou E, Colome-Grimmer M, Fayad L, et al. Transformation of mycosis fungoides/Sezary syndrome. Clinical characteristics and prognosis. Blood. 1998;92:1150-1159.
50 Vergier B, de Muret A, Beylot-Barry M, et al. Transformation of mycosis fungoides. Clinicopathological and prognostic features of 45 cases. French Study Group of Cutaneous Lymphomas. Blood. 2000;95:2212-2218.
53 Stein M, Farrar N, Jones GW, et al. Central neurologic involvement in mycosis fungoides. Ten cases, actuarial risk assessment, and predictive factors. Cancer J. 2006;12:55-62.
58 LeBoit PE. Granulomatous slack skin. Dermatol Clin. 1994;12:375-389.
59 Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders. The Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
60 Yu JB, Blitzblau RC, Decker RH, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
61 Siegel RS, Pandolfino T, Guitart J, et al. Primary cutaneous T-cell lymphoma. Review and current concepts. J Clin Oncol. 2000;18:2908-2925.
63 Beljaards RC, Willemze R. The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas. Br J Dermatol. 1992;126:596-602.
64 Bunn PAJr, Schechter GP, Jaffe E, et al. Clinical course of retrovirus-associated adult T-cell lymphoma in the United States. N Engl J Med. 1983;309:257-264.
71 Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome. A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722.
72 Kim YH, Hoppe RT. Mycosis fungoides and the Sezary syndrome. Semin Oncol. 1999;26:276-289.
73 Vonderheid EC, Diamond LW, van Vloten WA, et al. Lymph node classification systems in cutaneous T-cell lymphoma. Evidence for the utility of the Working Formulation of Non-Hodgkin’s Lymphomas for Clinical Usage. Cancer. 1994;73:207-218.
74 Diamandidou E, Colome M, Fayad L, et al. Prognostic factor analysis in mycosis fungoides/Sezary syndrome. J Am Acad Dermatol. 1999;40:914-924.
75 Hoppe RT, Medeiros LJ, Warnke RA, et al. CD8-positive tumor-infiltrating lymphocytes influence the long-term survival of patients with mycosis fungoides. J Am Acad Dermatol. 1995;32:448-453.
80 Bakels V, Van Oostveen JW, Geerts ML, et al. Diagnostic and prognostic significance of clonal T-cell receptor beta gene rearrangements in lymph nodes of patients with mycosis fungoides. J Pathol. 1993;170:249-255.
95 Heald P, Mehlmauer M, Martin AG, et al. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma. Results of the phase III clinical trial. J Am Acad Dermatol. 2003;49:801-815.
96 Edelson R, Berger C, Gasparro F, et al. Treatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy. Preliminary results. N Engl J Med. 1987;316:297-303.
101 Jumbou O, N’Guyen JM, Tessier MH, et al. Long-term follow-up in 51 patients with mycosis fungoides and Sezary syndrome treated by interferon-alfa. Br J Dermatol. 1999;140:427-431.
102 Roenigk HHJr, Kuzel TM, Skoutelis AP, et al. Photochemotherapy alone or combined with interferon alpha-2a in the treatment of cutaneous T-cell lymphoma. J Invest Dermatol. 1990;95:198S-205S.
103 Kuzel TM, Roenigk HHJr, Samuelson E, et al. Effectiveness of interferon alfa-2a combined with phototherapy for mycosis fungoides and the Sezary syndrome. J Clin Oncol. 1995;13:257-263.
114 Zhang C, Richon V, Ni X, et al. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells. Relevance to mechanism of therapeutic action. J Invest Dermatol. 2005;125:1045-1052.
122 Kaye FJ, Bunn PAJr, Steinberg SM, et al. A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med. 1989;321:1784-1790.
148 Diederen PV, van Weelden H, Sanders CJ, et al. Narrowband UVB and psoralen-UVA in the treatment of early-stage mycosis fungoides. A retrospective study. J Am Acad Dermatol. 2003;48:215-219.
156 Desai KR, Pezner RD, Lipsett JA, et al. Total skin electron irradiation for mycosis fungoides. Relationship between acute toxicities and measured dose at different anatomic sites. Int J Radiat Oncol Biol Phys. 1988;15:641-645.
158 Stein ME, Anacak Y, Zaidan J, et al. Second primary tumors in mycosis fungoides patients. Experience at the Northern Israel Oncology Center (1979-2002). J Buon. 2006;11:175-180.
161 Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy for cutaneous B- and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
165 Trump G, Wright KA, Evans WW, et al. High energy electrons for the treatment of extensive superficial malignant lesions. Am J Roentgenol Rad Ther. 1953;69:623-629.
166 Karzmark CJ, Loevinger R, Steele RE, et al. A technique for large-field, superficial electron therapy. Radiology. 1960;74:633-643.
1 Sander CA, Flaig MJ, Jaffe ES. Cutaneous manifestations of lymphoma. A clinical guide based on the WHO classification. Clin Lymphoma. 2001;2:86-100. discussion Clin Lymphoma 2:101-102, 2001
2 Willemze R, Kerl H, Sterry W, et al. EORTC classification for primary cutaneous lymphomas. A proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood. 1997;90:354-371.
3 Alibert JLM. Tableau du Pian Fungoide Description des Maladies de la Peau, Observees a l’Hopital Saint-Louis et Exposition des meilleurs Methodes Suivies pour leur Traitement. Paris: Barrois L’Aine & Fils; 1806.
4 Sezary A, Bouvrain Y. Erythrodermie avec presence de cellules monstreuses dans derme et sang circulant. Bull Soc Fr Dermatol Syph. 1938;45:254-260.
5 Lutzner MA, Jordan HW. The ultrastructure of an abnormal cell in Sezary’s syndrome. Blood. 1968;31:719-726.
6 Edelson RL, Berger CL, Raafat J, et al. Karyotype studies of cutaneous T cell lymphoma. Evidence for clonal origin. J Invest Dermatol. 1979;73:548-550.
7 Jones GW, Thorson B. Cutaneous T-cell lymphoma (mycosis fungoides). Lancet. 1996;348:130-131.
8 Hoppe RT. Mycosis fungoides. Radiation therapy. Dermatol Ther. 2003;16:347-354.
9 Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-859.
10 Weinstock MA, Horm JW. Mycosis fungoides in the United States. Increasing incidence and descriptive epidemiology. JAMA. 1988;260:42-46.
11 Morales Suarez-Varela MM, Llopis Gonzalez A, Marquina Vila A, et al. Mycosis fungoides. Review of epidemiological observations. Dermatology. 2000;201:21-28.
12 Herne KL, Talpur R, Breuer-McHam J, et al. Cytomegalovirus seropositivity is significantly associated with mycosis fungoides and Sezary syndrome. Blood. 2003;101:2132-2136.
13 Pancake BA, Wassef EH, Zucker-Franklin D. Demonstration of antibodies to human T-cell lymphotropic virus-I tax in patients with the cutaneous T-cell lymphoma, mycosis fungoides, who are seronegative for antibodies to the structural proteins of the virus. Blood. 1996;88:3004-3009.
14 Shohat M, Hodak E, Hannig H, et al. Evidence for the cofactor role of human T-cell lymphotropic virus type 1 in mycosis fungoides and Sezary syndrome. Br J Dermatol. 1999;141:44-49.
15 Wood GS, Salvekar A, Schaffer J, et al. Evidence against a role for human T-cell lymphotrophic virus type I (HTLV-I) in the pathogenesis of American cutaneous T-cell lymphoma. J Invest Dermatol. 1996;107:301-307.
16 Li G, Vowels BR, Benoit BM, et al. Failure to detect human T-lymphotropic virus type-I proviral DNA in cell lines and tissues from patients with cutaneous T-cell lymphoma. J Invest Dermatol. 1996;107:308-313.
17 Boni R, Davis-Daneshfar A, Burg G, et al. No detection of HTLV-I proviral DNA in lesional skin biopsies from Swiss and German patients with cutaneous T-cell lymphoma. Br J Dermatol. 1996;134:282-284.
18 Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides. N Engl J Med. 2004;350:1978-1988.
19 Heald PW, Glusac EJ. Unilesional cutaneous T-cell lymphoma. Clinical features, therapy, and follow-up of 10 patients with a treatment-responsive mycosis fungoides variant. J Am Acad Dermatol. 2000;42:283-285.
20 Wilson LD, Kacinski BM, Jones GW. Local superficial radiotherapy in the management of minimal stage IA cutaneous T-cell lymphoma (Mycosis Fungoides). Int J Radiat Oncol Biol Phys. 1998;40:109-115.
21 Micaily B, Miyamoto C, Kantor G, et al. Radiotherapy for unilesional mycosis fungoides. Int J Radiat Oncol Biol Phys. 1998;42:361-364.
22 Zackheim HS, McCalmont TH. Mycosis fungoides. The great imitator. J Am Acad Dermatol. 2002;47:914-918.
23 Glusac EJ. Of cells and architecture. New approaches to old criteria in mycosis fungoides. J Cutan Pathol. 2001;28:169-173.
24 Santucci M, Biggeri A, Feller AC, et al. Efficacy of histologic criteria for diagnosing early mycosis fungoides: an EORTC cutaneous lymphoma study group investigation. European Organization for Research and Treatment of Cancer. Am J Surg Pathol. 2000;24:40-50.
25 Ashton-Key M, Diss TC, Du MQ, et al. The value of the polymerase chain reaction in the diagnosis of cutaneous T-cell infiltrates. Am J Surg Pathol. 1997;21:743-747.
26 Dadej K, Gaboury L, Lamarre L, et al. The value of clonality in the diagnosis and follow-up of patients with cutaneous T-cell infiltrates. Diagn Mol Pathol. 2001;10:78-88.
27 Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
28 Li G, Chooback L, Wolfe JT, et al. Overexpression of p53 protein in cutaneous T cell lymphoma. Relationship to large cell transformation and disease progression. J Invest Dermatol. 1998;110:767-770.
29 Mao X, Lillington D, Scarisbrick JJ, et al. Molecular cytogenetic analysis of cutaneous T-cell lymphomas. Identification of common genetic alterations in Sezary syndrome and mycosis fungoides. Br J Dermatol. 2002;147:464-475.
30 Scarisbrick JJ, Mitchell TJ, Calonje E, et al. Microsatellite instability is associated with hypermethylation of the hMLH1 gene and reduced gene expression in mycosis fungoides. J Invest Dermatol. 2003;121:894-901.
31 van Doorn R, Dijkman R, Vermeer MH, et al. A novel splice variant of the Fas gene in patients with cutaneous T-cell lymphoma. Cancer Res. 2002;62:5389-5392.
32 Axelrod PI, Lorber B, Vonderheid EC. Infections complicating mycosis fungoides and Sezary syndrome. JAMA. 1992;267:1354-1358.
33 Kantor AF, Curtis RE, Vonderheid EC, et al. Risk of second malignancy after cutaneous T-cell lymphoma. Cancer. 1989;63:1612-1615.
34 Heald P, Yan SL, Edelson R. Profound deficiency in normal circulating T cells in erythrodermic cutaneous T-cell lymphoma. Arch Dermatol. 1994;130:198-203.
35 Bagot M, Nikolova M, Schirm-Chabanette F, et al. Crosstalk between tumor T lymphocytes and reactive T lymphocytes in cutaneous T cell lymphomas. Ann N Y Acad Sci. 2001;941:31-38.
36 Saed G, Fivenson DP, Naidu Y, et al. Mycosis fungoides exhibits a Th1-type cell-mediated cytokine profile whereas Sezary syndrome expresses a Th2-type profile. J Invest Dermatol. 1994;103:29-33.
37 Wasik MA, Vonderheid EC, Bigler RD, et al. Increased serum concentration of the soluble interleukin-2 receptor in cutaneous T-cell lymphoma. Clinical and prognostic implications. Arch Dermatol. 1996;132:42-47.
38 Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
39 Smith BD, Wilson LD. Management of mycosis fungoides. Part 1. Diagnosis, staging, and prognosis. Oncology (Huntingt). 2003;17:1281-1288.
40 Yazdi AS, Medeiros LJ, Puchta U, et al. Improved detection of clonality in cutaneous T-cell lymphomas using laser capture microdissection. J Cutan Pathol. 2003;30:486-491.
41 Gellrich S, Lukowsky A, Schilling T, et al. Microanatomical compartments of clonal and reactive T cells in mycosis fungoides. Molecular demonstration by single cell polymerase chain reaction of T cell receptor gene rearrangements. J Invest Dermatol. 2000;115:620-624.
42 Smoller BR, Bishop K, Glusac E, et al. Reassessment of histologic parameters in the diagnosis of mycosis fungoides. Am J Surg Pathol. 1995;19:1423-1430.
43 Guitart J, Kennedy J, Ronan S, et al. Histologic criteria for the diagnosis of mycosis fungoides. Proposal for a grading system to standardize pathology reporting. J Cutan Pathol. 2001;28:174-183.
44 Kohler S, Kim YH, Smoller BR. Histologic criteria for the diagnosis of erythrodermic mycosis fungoides and Sezary syndrome. A critical reappraisal. J Cutan Pathol. 1997;24:292-297.
45 Fung MA, Murphy MJ, Hoss DM, et al. Practical evaluation and management of cutaneous lymphoma. J Am Acad Dermatol. 2002;46:325-357.
46 Lu D, Patel KA, Duvic M, et al. Clinical and pathological spectrum of CD8-positive cutaneous T-cell lymphomas. J Cutan Pathol. 2002;29:465-472.
47 Delfau-Larue MH, Laroche L, Wechsler J, et al. Diagnostic value of dominant T-cell clones in peripheral blood in 363 patients presenting consecutively with a clinical suspicion of cutaneous lymphoma. Blood. 2000;96:2987-2992.
48 Wood GS, Tung RM, Haeffner AC, et al. Detection of clonal T-cell receptor gamma gene rearrangements in early mycosis fungoides/Sezary syndrome by polymerase chain reaction and denaturing gradient gel electrophoresis (PCR/DGGE). J Invest Dermatol. 1994;103:34-41.
49 Diamandidou E, Colome-Grimmer M, Fayad L, et al. Transformation of mycosis fungoides/Sezary syndrome. Clinical characteristics and prognosis. Blood. 1998;92:1150-1159.
50 Vergier B, de Muret A, Beylot-Barry M, et al. Transformation of mycosis fungoides. Clinicopathological and prognostic features of 45 cases. French Study Group of Cutaneous Lymphomas. Blood. 2000;95:2212-2218.
51 O’Quinn RP, Zic JA, Boyd AS. Mycosis fungoides d’emblee. CD30-negative cutaneous large T-cell lymphoma. J Am Acad Dermatol. 2000;43:861-863.
52 de Coninck EC, Kim YH, Varghese A, et al. Clinical characteristics and outcome of patients with extracutaneous mycosis fungoides. J Clin Oncol. 2001;19:779-784.
53 Stein M, Farrar N, Jones GW, et al. Central neurologic involvement in mycosis fungoides. Ten cases, actuarial risk assessment, and predictive factors. Cancer J. 2006;12:55-62.
54 Sibaud V, Beylot-Barry M, Thiebaut R, et al. Bone marrow histopathologic and molecular staging in epidermotropic T-cell lymphomas. Am J Clin Pathol. 2003;119:414-423.
55 Graham SJ, Sharpe RW, Steinberg SM, et al. Prognostic implications of a bone marrow histopathologic classification system in mycosis fungoides and the Sezary syndrome. Cancer. 1993;72:726-734.
56 Vonderheid EC, Bernengo MG, Burg G, et al. Update on erythrodermic cutaneous T-cell lymphoma. Report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol. 2002;46:95-106.
57 Wilson LD, Cooper DL, Goodrich AL, et al. Impact of non-CTCL dermatologic diagnoses and adjuvant therapies on cutaneous T-cell lymphoma patients treated with total skin electron beam radiation therapy. Int J Radiat Oncol Biol Phys. 1994;28:829-837.
58 LeBoit PE. Granulomatous slack skin. Dermatol Clin. 1994;12:375-389.
59 Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders. The Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
60 Yu JB, Blitzblau RC, Decker RH, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
61 Siegel RS, Pandolfino T, Guitart J, et al. Primary cutaneous T-cell lymphoma: review and current concepts. J Clin Oncol. 2000;18:2908-2925.
62 Cabanillas F, Armitage J, Pugh WC, et al. Lymphomatoid papulosis. A T-cell dyscrasia with a propensity to transform into malignant lymphoma. Ann Intern Med. 1995;122:210-217.
63 Beljaards RC, Willemze R. The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas. Br J Dermatol. 1992;126:596-602.
64 Bunn PAJr, Schechter GP, Jaffe E, et al. Clinical course of retrovirus-associated adult T-cell lymphoma in the United States. N Engl J Med. 1983;309:257-264.
65 Hollsberg P, Hafler DA. Seminars in medicine of the Beth Israel Hospital, Boston. Pathogenesis of diseases induced by human lymphotropic virus type I infection. N Engl J Med. 1993;328:1173-1182.
66 Di Venuti G, Nawgiri R, Foss F. Denileukin diftitox and hyper-CVAD in the treatment of human T-cell lymphotropic virus 1-associated acute T-cell leukemia/lymphoma. Clin Lymphoma. 2003;4:176-178.
67 Hermine O, Bouscary D, Gessain A, et al. Brief report. Treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N Engl J Med. 1995;332:1749-1751.
68 Edge SB, Byrd DR, Compton CC, et al. AJCC Cancer Staging Handbook, 7th Ed. New York: Springer; 2010.
69 Kim YH, Martinez G, Varghese A, et al. Topical nitrogen mustard in the management of mycosis fungoides: update of the Stanford experience. Arch Dermatol. 2003;139:165-173.
70 Kashani-Sabet M, McMillan A, Zackheim HS. A modified staging classification for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2001;45:700-706.
71 Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome. A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722.
72 Kim YH, Hoppe RT. Mycosis fungoides and the Sezary syndrome. Semin Oncol. 1999;26:276-289.
73 Vonderheid EC, Diamond LW, van Vloten WA, et al. Lymph node classification systems in cutaneous T-cell lymphoma. Evidence for the utility of the Working Formulation of Non-Hodgkin’s Lymphomas for Clinical Usage. Cancer. 1994;73:207-218.
74 Diamandidou E, Colome M, Fayad L, et al. Prognostic factor analysis in mycosis fungoides/Sezary syndrome. J Am Acad Dermatol. 1999;40:914-924.
75 Hoppe RT, Medeiros LJ, Warnke RA, et al. CD8-positive tumor-infiltrating lymphocytes influence the long-term survival of patients with mycosis fungoides. J Am Acad Dermatol. 1995;32:448-453.
76 Quiros PA, Kacinski BM, Wilson LD. Extent of skin involvement as a prognostic indicator of disease free and overall survival of patients with T3 cutaneous T-cell lymphoma treated with total skin electron beam radiation therapy. Cancer. 1996;77:1912-1917.
77 Delfau-Larue MH, Dalac S, Lepage E, et al. Prognostic significance of a polymerase chain reaction-detectable dominant T-lymphocyte clone in cutaneous lesions of patients with mycosis fungoides. Blood. 1998;92:3376-3380.
78 Guitart J, Camisa C, Ehrlich M, et al. Long-term implications of T-cell receptor gene rearrangement analysis by Southern blot in patients with cutaneous T-cell lymphoma. J Am Acad Dermatol. 2003;48:775-779.
79 Beylot-Barry M, Sibaud V, Thiebaut R, et al. Evidence that an identical T cell clone in skin and peripheral blood lymphocytes is an independent prognostic factor in primary cutaneous T cell lymphomas. J Invest Dermatol. 2001;117:920-926.
80 Bakels V, Van Oostveen JW, Geerts ML, et al. Diagnostic and prognostic significance of clonal T-cell receptor beta gene rearrangements in lymph nodes of patients with mycosis fungoides. J Pathol. 1993;170:249-255.
81 Kim YH, Bishop K, Varghese A, et al. Prognostic factors in erythrodermic mycosis fungoides and the Sezary syndrome. Arch Dermatol. 1995;131:1003-1008.
82 Bass JC, Korobkin MT, Cooper KD, et al. Cutaneous T-cell lymphoma: CT in evaluation and staging. Radiology. 1993;186:273-278.
83 Tsai EY, Taur A, Espinosa L, et al. Staging accuracy in mycosis fungoides and sezary syndrome using integrated positron emission tomography and computed tomography. Arch Dermatol. 2006;142:577-584.
84 Quiros PA, Jones GW, Kacinski BM, et al. Total skin electron beam therapy followed by adjuvant psoralen/ultraviolet-A light in the management of patients with T1 and T2 cutaneous T-cell lymphoma (mycosis fungoides). Int J Radiat Oncol Biol Phys. 1997;38:1027-1035.
85 Chinn DM, Chow S, Kim YH, et al. Total skin electron beam therapy with or without adjuvant topical nitrogen mustard or nitrogen mustard alone as initial treatment of T2 and T3 mycosis fungoides. Int J Radiat Oncol Biol Phys. 1999;43:951-958.
86 Jones GW, Hoppe RT, Glatstein E. Electron beam treatment for cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9:1057-1076.
87 Jones GW, Rosenthal D, Wilson LD. Total skin electron radiation for patients with erythrodermic cutaneous T-cell lymphoma (mycosis fungoides and the Sezary syndrome). Cancer. 1999;85:1985-1995.
88 Esteve E, Bagot M, Joly P, et al. A prospective study of cutaneous intolerance to topical mechlorethamine therapy in patients with cutaneous T-cell lymphomas. French Study Group of Cutaneous Lymphomas. Arch Dermatol. 1999;135:1349-1353.
89 Vonderheid EC, Tan ET, Kantor AF, et al. Long-term efficacy, curative potential, and carcinogenicity of topical mechlorethamine chemotherapy in cutaneous T cell lymphoma. J Am Acad Dermatol. 1989;20:416-428.
90 Licata AG, Wilson LD, Braverman IM, et al. Malignant melanoma and other second cutaneous malignancies in cutaneous T-cell lymphoma. The influence of additional therapy after total skin electron beam radiation. Arch Dermatol. 1995;131:432-435.
91 Zackheim HS, Epstein EHJr, Crain WR. Topical carmustine (BCNU) for cutaneous T cell lymphoma. A 15-year experience in 143 patients. J Am Acad Dermatol. 1990;22:802-810.
92 Zackheim HS, Kashani-Sabet M, Amin S. Topical corticosteroids for mycosis fungoides. Experience in 79 patients. Arch Dermatol. 1998;134:949-954.
93 Talpur R, Ward S, Apisarnthanarax N, et al. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2002;47:672-684.
94 Breneman D, Duvic M, Kuzel T, et al. Phase 1 and 2 trial of bexarotene gel for skin-directed treatment of patients with cutaneous T-cell lymphoma. Arch Dermatol. 2002;138:325-332.
95 Heald P, Mehlmauer M, Martin AG, et al. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma. Results of the phase III clinical trial. J Am Acad Dermatol. 2003;49:801-815.
96 Edelson R, Berger C, Gasparro F, et al. Treatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy. Preliminary results. N Engl J Med. 1987;316:297-303.
97 British Photodermatology Group guidelines for PUVA. Br J Dermatol. 1994;130:246-255.
98 Young AR. Carcinogenicity of UVB phototherapy assessed. Lancet. 1995;345:1431-1432.
99 Abel EA, Sendagorta E, Hoppe RT. Cutaneous malignancies and metastatic squamous cell carcinoma following topical therapies for mycosis fungoides. J Am Acad Dermatol. 1986;14:1029-1038.
100 Ross C, Tingsgaard P, Jorgensen H, et al. Interferon treatment of cutaneous T-cell lymphoma. Eur J Haematol. 1993;51:63-72.
101 Jumbou O, N’Guyen JM, Tessier MH, et al. Long-term follow-up in 51 patients with mycosis fungoides and Sezary syndrome treated by interferon-alfa. Br J Dermatol. 1999;140:427-431.
102 Roenigk HHJr, Kuzel TM, Skoutelis AP, et al. Photochemotherapy alone or combined with interferon alpha-2a in the treatment of cutaneous T-cell lymphoma. J Invest Dermatol. 1990;95:198S-205S.
103 Kuzel TM, Roenigk HHJr, Samuelson E, et al. Effectiveness of interferon alfa-2a combined with phototherapy for mycosis fungoides and the Sezary syndrome. J Clin Oncol. 1995;13:257-263.
104 Chiarion-Sileni V, Bononi A, Fornasa CV, et al. Phase II trial of interferon-alpha-2a plus psolaren with ultraviolet light A in patients with cutaneous T-cell lymphoma. Cancer. 2002;95:569-575.
105 Wollina U, Looks A, Meyer J, et al. Treatment of stage II cutaneous T-cell lymphoma with interferon alfa-2a and extracorporeal photochemotherapy: a prospective controlled trial. J Am Acad Dermatol. 2001;44:253-260.
106 Stadler R, Otte HG, Luger T, et al. Prospective randomized multicenter clinical trial on the use of interferon -2a plus acitretin versus interferon -2a plus PUVA in patients with cutaneous T-cell lymphoma stages I and II. Blood. 1998;92:3578-3581.
107 Thomsen K, Hammar H, Molin L, et al. Retinoids plus PUVA (RePUVA) and PUVA in mycosis fungoides, plaque stage. A report from the Scandinavian Mycosis Fungoides Group. Acta Derm Venereol. 1989;69:536-538.
108 Jones G, McLean J, Rosenthal D, et al. Combined treatment with oral etretinate and electron beam therapy in patients with cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome). J Am Acad Dermatol. 1992;26:960-967.
109 Duvic M, Hymes K, Heald P, et al. Bexarotene is effective and safe for treatment of refractory advanced- stage cutaneous T-cell lymphoma. Multinational phase II-III trial results. J Clin Oncol. 2001;19:2456-2471.
110 Duvic M, Martin AG, Kim Y, et al. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T- cell lymphoma. Arch Dermatol. 2001;137:581-593.
111 Olsen E, Duvic M, Frankel A, et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J Clin Oncol. 2001;19:376-388.
112 Talpur R, Jones DM, Alencar AJ, et al. CD25 expression is correlated with histological grade and response to denileukin diftitox in cutaneous T-cell lymphoma. J Invest Dermatol. 2006;126:575-583.
113 Foss FM, Bacha P, Osann KE, et al. Biological correlates of acute hypersensitivity events with DAB(389)IL-2 (denileukin diftitox, ONTAK) in cutaneous T-cell lymphoma. Decreased frequency and severity with steroid premedication. Clin Lymphoma. 2001;1:298-302.
114 Zhang C, Richon V, Ni X, et al. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells. Relevance to mechanism of therapeutic action. J Invest Dermatol. 2005;125:1045-1052.
115 Duvic M. Systemic monotherapy vs combination therapy for CTCL. Rationale and future strategies. Oncology (Williston Park). 2007;21:33-40.
116 Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109-3115.
117 Lim HW, Edelson RL. Photopheresis for the treatment of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9:1117-1126.
118 Berger CL, Hanlon D, Kanada D, et al. Transimmunization, a novel approach for tumor immunotherapy. Transfus Apheresis Sci. 2002;26:205-216.
119 Heald P, Rook A, Perez M, et al. Treatment of erythrodermic cutaneous T-cell lymphoma with extracorporeal photochemotherapy. J Am Acad Dermatol. 1992;27:427-433.
120 Wilson LD, Licata AL, Braverman IM, et al. Systemic chemotherapy and extracorporeal photochemotherapy for T3 and T4 cutaneous T-cell lymphoma patients who have achieved a complete response to total skin electron beam therapy. Int J Radiat Oncol Biol Phys. 1995;32:987-995.
121 Wilson LD, Jones GW, Kim D, et al. Experience with total skin electron beam therapy in combination with extracorporeal photopheresis in the management of patients with erythrodermic (T4) mycosis fungoides. J Am Acad Dermatol. 2000;43:54-60.
122 Kaye FJ, Bunn PAJr, Steinberg SM, et al. A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med. 1989;321:1784-1790.
123 Zackheim HS, Kashani-Sabet M, Hwang ST. Low-dose methotrexate to treat erythrodermic cutaneous T-cell lymphoma. Results in twenty-nine patients. J Am Acad Dermatol. 1996;34:626-631.
124 Wollina U, Dummer R, Brockmeyer NH, et al. Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer. 2003;98:993-1001.
125 Zinzani PL, Baliva G, Magagnoli M, et al. Gemcitabine treatment in pretreated cutaneous T-cell lymphoma. Experience in 44 patients. J Clin Oncol. 2000;18:2603-2606.
126 Foss FM, Ihde DC, Linnoila IR, et al. Phase II trial of fludarabine phosphate and interferon alfa-2a in advanced mycosis fungoides/Sezary syndrome. J Clin Oncol. 1994;12:2051-2059.
127 Foss FM, Ihde DC, Breneman DL, et al. Phase II study of pentostatin and intermittent high-dose recombinant interferon alfa-2a in advanced mycosis fungoides/Sezary syndrome. J Clin Oncol. 1992;10:1907-1913.
128 Greiner D, Olsen EA, Petroni G. Pentostatin (2’-deoxycoformycin) in the treatment of cutaneous T-cell lymphoma. J Am Acad Dermatol. 1997;36:950-955.
129 Kuzel TM, Hurria A, Samuelson E, et al. Phase II trial of 2-chlorodeoxyadenosine for the treatment of cutaneous T-cell lymphoma. Blood. 1996;87:906-911.
130 Sarris AH, Phan A, Duvic M, et al. Trimetrexate in relapsed T-cell lymphoma with skin involvement. J Clin Oncol. 2002;20:2876-2880.
131 Rosen ST, Foss FM. Chemotherapy for mycosis fungoides and the Sezary syndrome. Hematol Oncol Clin North Am. 1995;9:1109-1116.
132 Bigler RD, Crilley P, Micaily B, et al. Autologous bone marrow transplantation for advanced stage mycosis fungoides. Bone Marrow Transplant. 1991;7:133-137.
133 Russell-Jones R, Child F, Olavarria E, et al. Autologous peripheral blood stem cell transplantation in tumor-stage mycosis fungoides: predictors of disease-free survival. Ann N Y Acad Sci. 2001;941:147-154.
134 Sterling JC, Marcus R, Burrows NP, et al. Erythrodermic mycosis fungoides treated with total body irradiation and autologous bone marrow transplantation. Clin Exp Dermatol. 1995;20:73-75.
135 Burt RK, Guitart J, Traynor A, et al. Allogeneic hematopoietic stem cell transplantation for advanced mycosis fungoides: evidence of a graft-versus-tumor effect. Bone Marrow Transplant. 2000;25:111-113.
136 Guitart J, Wickless SC, Oyama Y, et al. Long-term remission after allogeneic hematopoietic stem cell transplantation for refractory cutaneous T-cell lymphoma. Arch Dermatol. 2002;138:1359-1365.
137 Molina A, Zain J, Arber DA, et al. Durable clinical, cytogenetic, and molecular remissions after allogeneic hematopoietic cell transplantation for refractory Sezary syndrome and mycosis fungoides. J Clin Oncol. 2005;23:6163-6171.
138 Soligo D, Ibatici A, Berti E, et al. Treatment of advanced mycosis fungoides by allogeneic stem-cell transplantation with a nonmyeloablative regimen. Bone Marrow Transplant. 2003;31:663-666.
139 Cotter GW, Baglan RJ, Wasserman TH, et al. Palliative radiation treatment of cutaneous mycosis fungoides—a dose response. Int J Radiat Oncol Biol Phys. 1983;9:1477-1480.
140 Kim JH, Nisce LZ, D’Anglo GJ. Dose-time fractionation study in patients with mycosis fungoides and lymphoma cutis. Radiology. 1976;119:439-442.
141 Hoppe RT, Fuks Z, Bagshaw MA. The rationale for curative radiotherapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 1977;2:843-851.
142 Jones GW, Tadros A, Hodson DI, et al. Prognosis with newly diagnosed mycosis fungoides after total skin electron radiation of 30 or 35 GY. Int J Radiat Oncol Biol Phys. 1994;28:839-845.
143 Jones GW, Kacinski BM, Wilson LD, et al. Total skin electron radiation in the management of mycosis fungoides. Consensus of the European Organization for Research and Treatment of Cancer (EORTC) Cutaneous Lymphoma Project Group. J Am Acad Dermatol. 2002;47:364-370.
144 Wong R, Jones G, Farrar N, et al. Local superficial radiotherapy (LSRT) for newly diagnosed stage IA patch-plaque mycosis fungoides (MF) with only one, two, or three presenting lesions. Int J Radiat Oncol Biol Phys. 2003;57(Suppl 1):S289-S290.
145 Kim YH, Jensen RA, Watanabe GL, et al. Clinical stage IA (limited patch and plaque) mycosis fungoides. A long- term outcome analysis. Arch Dermatol. 1996;132:1309-1313.
146 Jones G, Wilson LD, Fox-Goguen L. Total skin electron beam radiotherapy for patients who have mycosis fungoides. Hematol Oncol Clin North Am. 2003;17:1421-1434.
147 Ramsay DL, Lish KM, Yalowitz CB, et al. Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol. 1992;128:931-933.
148 Diederen PV, van Weelden H, Sanders CJ, et al. Narrowband UVB and psoralen-UVA in the treatment of early-stage mycosis fungoides. A retrospective study. J Am Acad Dermatol. 2003;48:215-219.
149 Herrmann JJ, Roenigk HHJr, Hurria A, et al. Treatment of mycosis fungoides with photochemotherapy (PUVA). Long-term follow-up. J Am Acad Dermatol. 1995;33:234-242.
150 Honigsmann H, Brenner W, Rauschmeier W, et al. Photochemotherapy for cutaneous T cell lymphoma. A follow-up study. J Am Acad Dermatol. 1984;10:238-245.
151 Rook AH, Kuzel TM, Olsen EA. Cytokine therapy of cutaneous T-cell lymphoma. Interferons, interleukin-12, and interleukin-2. Hematol Oncol Clin North Am. 2003;17:1435-1448. ix
152 Introcaso CE, Micaily B, Richardson SK, et al. Total skin electron beam therapy may be associated with improvement of peripheral blood disease in Sezary syndrome. J Am Acad Dermatol. 2008;58:592-595.
153 Abel EA, Sendagorta E, Hoppe RT, et al. PUVA treatment of erythrodermic and plaque-type mycosis fungoides. Ten-year follow-up study. Arch Dermatol. 1987;123:897-901.
154 Rosenblatt E, Kuten A, Leviov M, et al. Total skin electron irradiation in mycosis fungoides dose and fractionation considerations. Leuk Lymphoma. 1998;30:143-151.
155 Price NM. Electron beam therapy. Its effect on eccrine gland function in mycosis fungoides patients. Arch Dermatol. 1979;115:1068-1070.
156 Desai KR, Pezner RD, Lipsett JA, et al. Total skin electron irradiation for mycosis fungoides. Relationship between acute toxicities and measured dose at different anatomic sites. Int J Radiat Oncol Biol Phys. 1988;15:641-645.
157 Price NM. Radiation dermatitis following electron beam therapy. An evaluation of patients ten years after total skin irradiation for mycosis fungoides. Arch Dermatol. 1978;114:63-66.
158 Stein ME, Anacak Y, Zaidan J, et al. Second primary tumors in mycosis fungoides patients. Experience at the Northern Israel Oncology Center (1979-2002). J Buon. 2006;11:175-180.
159 Wilson LD, Quiros PA, Kolenik SA, et al. Additional courses of total skin electron beam therapy in the treatment of patients with recurrent cutaneous T-cell lymphoma. J Am Acad Dermatol. 1996;35:69-73.
160 Becker M, Hoppe RT, Knox SJ. Multiple courses of high-dose total skin electron beam therapy in the management of mycosis fungoides. Int J Radiat Oncol Biol Phys. 1995;32:1445-1449.
161 Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy for cutaneous B- and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
162 Scholtz W. Ueber den Einfluess def Rontgenstrahlen auf die Haut in gesundem und krankem Zustande. Arch Dermatol Syph. 1902;59:421-449.
163 Sommerville J. Mycosis fungoides treated with general x-ray “bath. Br J Dermatol Syph. 1939;51:323-324.
164 Levin OL, Behrman HT. Roentgen ray therapy of mycosis fungoides. Arch Dermatol Syph. 1945;51:307-308.
165 Trump G, Wright KA, Evans WW, et al. High energy electrons for the treatment of extensive superficial malignant lesions. Am J Roentgenol Rad Ther. 1953;69:623-629.
166 Karzmark CJ, Loevinger R, Steele RE, et al. A technique for large-field, superficial electron therapy. Radiology. 1960;74:633-643.
167 Page V, Gardner A, Karzmark CJ. Patient dosimetry in the electron treatment of large superficial lesions. Radiology. 1970;94:635-641.
168 Bjarngard BE, Chen GT, Piontek RW, et al. Analysis of dose distributions in whole body superficial electron therapy. Int J Radiat Oncol Biol Phys. 1977;2:319-324.
169 Fuks Z, Bagshaw MA. Total-skin electron treatment of mycosis fungoides. Radiology. 1971;100:145-150.
170 Szur L, Silvester JA, Bewley DK. Treatment of the whole body surface with electrons. Lancet. 1962;1:1373-1377.
171 Halberg FE, Fu KK, Weaver KA, et al. Combined total body X-ray irradiation and total skin electron beam radiotherapy with an improved technique for mycosis fungoides. Int J Radiat Oncol Biol Phys. 1989;17:427-432.
172 Micaily B, Vonderheid EC, Brady LW, et al. Total skin electron beam and total nodal irradiation for treatment of patients with cutaneous T-cell lymphoma. Int J Radiat Oncol Biol Phys. 1985;11:1111-1115.
173 Micaily B, Campbell O, Moser C, et al. Total skin electron beam and total nodal irradiation of cutaneous T-cell lymphoma. Int J Radiat Oncol Biol Phys. 1991;20:809-813.
174 Chen Z, Agostinelli AG, Wilson LD, et al. Matching the dosimetry characteristics of a dual-field Stanford technique to a customized single-field Stanford technique for total skin electron therapy. Int J Radiat Oncol Biol Phys. 2004;59:872-885.
175 Peters VG. Use of an electron reflector to improve dose uniformity at the vertex during total skin electron therapy. Int J Radiat Oncol Biol Phys. 2000;46:1065-1069.
176 Gamble LM, Farrell TJ, Jones GW, et al. Composite depth dose measurement for total skin electron (TSE) treatments using radiochromic film. Phys Med Biol. 2003;48:891-898.
177 Anacak Y, Arican Z, Bar-Deroma R, et al. Total skin electron irradiation. Evaluation of dose uniformity throughout the skin surface. Med Dosim. 2003;28:31-34.
178 Asbell SO, Siu J, Lightfoot DA, et al. Individualized eye shields for use in electron beam therapy as well as low-energy photon irradiation. Int J Radiat Oncol Biol Phys. 1980;6:519-521.
179 Reavely MM, Wilson LD. Total skin electron beam therapy and cutaneous T-cell lymphoma. A clinical guide for patients and staff. Dermatol Nurs. 2004;16(36):39-57.
180 Smith BD, Son CB, Wilson LD. Disseminated herpes simplex after total skin electron beam radiotherapy for mycosis fungoides. J R Soc Med. 2003;96:500-501.
181 Smith BD, Wilson LD. Management of mycosis fungoides. Part 2. Treatment. Oncology (Huntingt). 2003;17:1419-1428. discussion Oncology (Huntingt)17:1430, 1433, 2003
182 Duvic M, Edelson R. Cutaneous T-cell lymphoma. J Am Acad Dermatol. 2004;51:S43-S45.
183 Apisarnthanarax N, Talpur R, Duvic M. Treatment of cutaneous T cell lymphoma. Current status and future directions. Am J Clin Dermatol. 2002;3:193-215.