Chapter 601 Muscular Dystrophies
601.1 Duchenne and Becker Muscular Dystrophies
Clinical Manifestations
Infant boys are only rarely symptomatic at birth or in early infancy, although some are mildly hypotonic. Early gross motor skills, such as rolling over, sitting, and standing, are usually achieved at the appropriate ages or may be mildly delayed. Poor head control in infancy may be the first sign of weakness. Distinctive facies are not an early feature because facial muscle weakness is a late event; in later childhood, a “transverse” or horizontal smile may be seen. Walking is often accomplished at the normal age of about 12 mo, but hip girdle weakness may be seen in subtle form as early as the 2nd year. Toddlers might assume a lordotic posture when standing to compensate for gluteal weakness. An early Gowers sign is often evident by age 3 yr and is fully expressed by age 5 or 6 yr (see Fig. 584-5). A Trendelenburg gait, or hip waddle, appears at this time. Common presentations in toddlers include delayed walking, falling, toe walking and trouble running or walking upstairs, developmental delay, and, less often, malignant hyperthermia after anesthesia.
Diagnosis
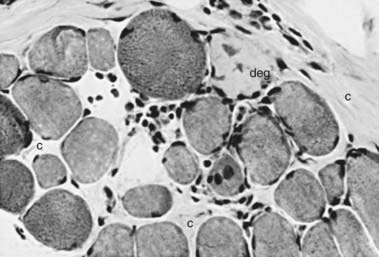
The muscle biopsy is diagnostic and shows characteristic changes (Figs. 601-1 and 601-2). Myopathic changes include endomysial connective tissue proliferation, scattered degenerating and regenerating myofibers, foci of mononuclear inflammatory cell infiltrates as a reaction to muscle fiber necrosis, mild architectural changes in still-functional muscle fibers, and many dense fibers. These hypercontracted fibers probably result from segmental necrosis at another level, allowing calcium to enter the site of breakdown of the sarcolemmal membrane and trigger a contraction of the whole length of the muscle fiber. Calcifications within myofibers are correlated with secondary β-dystroglycan deficiency.
Genetic Etiology and Pathogenesis
Despite the X-linked recessive inheritance in DMD, about 30% of cases are new mutations, and the mother is not a carrier. The female carrier state usually shows no muscle weakness or any clinical expression of the disease, but affected girls are occasionally encountered, usually having much milder weakness than boys. These symptomatic girls are explained by the Lyon hypothesis in which the normal X chromosome becomes inactivated and the one with the gene deletion is active (Chapter 75). The full clinical picture of DMD has occurred in several girls with Turner syndrome in whom the single X chromosome must have had the Xp21 gene deletion.
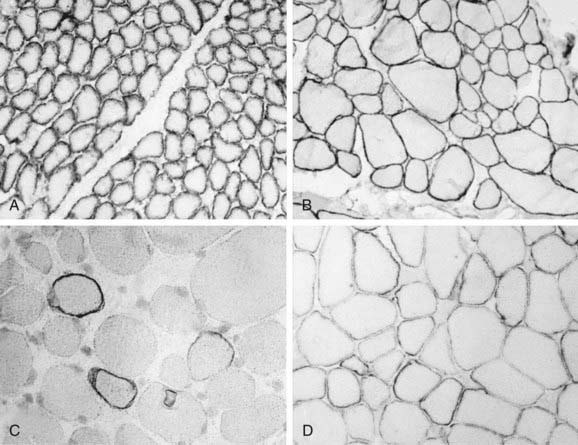
Analysis of the dystrophin protein requires a muscle biopsy and is demonstrated by Western blot analysis or in tissue sections by immunohistochemical methods using either fluorescence or light microscopy of antidystrophin antisera (see Fig. 601-2). In classic DMD, levels of <3% of normal are found; in BMD, the molecular weight of dystrophin is reduced to 20-90% of normal in 80% of patients, but in 15% of patients the dystrophin is of normal size but reduced in quantity, and 5% of patients have an abnormally large protein caused by excessive duplications or repeats of codons. Selective immunoreactivity of different parts of the dystrophin molecule in sections of muscle biopsy material distinguishes the Duchenne and Becker forms (Fig. 601-3). The demonstration of deletions and duplications also can be made from blood samples by the more rapid PCR, which identifies as many as 98% of deletions by amplifying 18 exons but cannot detect duplications. The diagnosis can thus be confirmed at the molecular genetic level from either the muscle biopsy material or from peripheral blood, although as many as 30% of boys with DMD or BMD have a false-normal blood PCR; all cases of dystrophinopathy are detected by muscle biopsy.
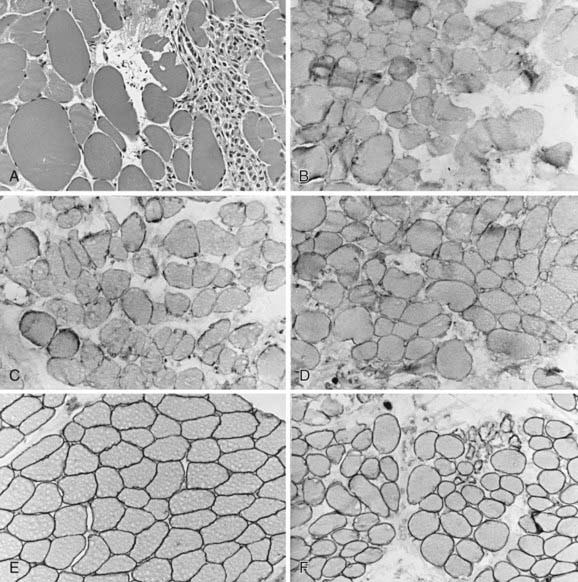
Figure 601-3 Quadriceps femoris muscle biopsy specimens from a 4 yr old boy with Becker muscular dystrophy. A, Myofibers vary greatly in size, with both atrophic and hypertrophic forms; at the right is a zone of degeneration and necrosis infiltrated by macrophages, similar to Duchenne muscular dystrophy (H&E, ×250). Immunoreactivity using antibodies against the dystrophin molecule in the rod domain (B), carboxyl-terminus (C), and amino-terminus (D) all show deficient but not totally absent dystrophin expression; most fibers of all sizes retain some dystrophin in parts of the sarcolemma but not around the entire circumference in cross section. Alternatively, the prominence of dystrophin is less, appearing weak, when compared with the simultaneously incubated normal control from another child of similar age (E). F, Merosin expression is normal in this patient with Becker dystrophy, in both large and small myofibers, and is lacking only in frankly necrotic fibers. Compare with classic Duchenne muscular dystrophy illustrated in Figure 601-2C and with Figure 601-5.
Blau HM. Cell therapies for muscular dystrophy. N Engl J Med. 2008;359:1403-1405.
Beenakker EAC, Fock JM, Van Tol MJ, et al. Intermittent prednisone therapy in Duchenne muscular dystrophy. Arch Neurol. 2005;62:128-132.
Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet. 2010;9:77-93.
Bushby K, Lochmüller H, Lynn S, Straub V. Interventions for muscular dystrophy: molecular medicines entering the clinic. Lancet. 2009;374:1849-1856.
Centers for Disease Control and Prevention (CDC). Prevalence of Duchenne/Becker muscular dystrophy among males aged 5–24 years—four states, 2007. MMWR Morb Mortal Wkly Rep. 2009;58:1119-1122.
Chamberlain JR, Chamberlain JS. Muscling in. Nat Med. 2010;16(2):170-171.
Ciafaloni E, Fox DJ, Pandya S, et al. Delayed diagnosis in Duchenne muscular dystrophy: data from the muscular dystrophy surveillance, tracking, and research network (MD STARnet). J Pediatr. 2009;155:380-385.
Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Engl J Med. 2011;364(10):939-946.
Kley RA, Tarnopolsky MA, Vorgerd M: Creatine for treating muscle disorders, Cochrane Database Syst Rev (2):CD004760, 2011.
Manzur AY, Kinali M, Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child. 2008;93:986-990.
Manzur AY, Kuntzer T, Pike M, Swan A: Glucocorticoid corticosteroids for Duchenne muscular dynstrophy, Cochrane Database Syst Rev (1):CD003725, 2008.
Mendell JR, Campbell K, Rodino-Klapac L, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363(15):1429-1436.
Moxley RT, AShwal S, Pandya S, et al. Practice parameter: corticosteroid treatment of Duchenne muscular dystrophy (report on the Quality Standards subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society). Neurology. 2005;64:13-20.
van Deutekom JC, Janson AA, Giniaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677-2686.
601.2 Emery-Dreifuss Muscular Dystrophy
Colomer J, Iturriaga C, Bonne G, et al. Autosomal dominant Emery-Dreifuss muscular dystrophy; a new family with late diagnosis. Neuromuscul Disord. 2002;12:19-25.
Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mjutations. N Engl J Med. 2006;12:209-210.
Mutoni F, Bonne G, Goldfarb LG, et al. Disease severity in dominant Emery Dreifuss is increased by mutations in both emery and desmin proteins. Brain. 2006;129:1260-1268.
601.3 Myotonic Muscular Dystrophy
Clinical Manifestations
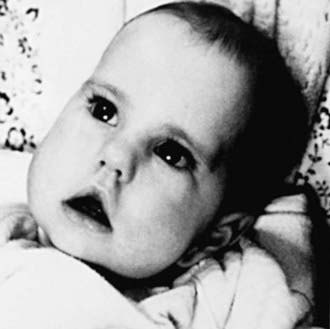
In the usual clinical course, excluding the severe neonatal form, infants can appear almost normal at birth, or facial wasting and hypotonia can already be early expressions of the disease. The facial appearance is characteristic, consisting of an inverted V-shaped upper lip, thin cheeks, and scalloped, concave temporalis muscles (Fig. 601-4). The head may be narrow, and the palate is high and arched because the weak temporal and pterygoid muscles in late fetal life do not exert sufficient lateral forces on the developing head and face.
Treatment
Myotonia may be diminished, and function may be restored by drugs that raise the depolarization threshold of muscle membranes, such as mexiletine, phenytoin, carbamazepine, procainamide, and quinidine sulfate. These drugs also have cardiotropic effects; thus, cardiac evaluation is important before prescribing them. Phenytoin and carbamazepine are used in doses similar to their use as anticonvulsants (Chapter 586.6); serum concentrations of 10-20 µg/mL for phenytoin and 5-12 µg/mL for carbamazepine should be maintained. If a patient’s disability is caused mainly by weakness rather than by myotonia, these drugs will be of no value.
Other Myotonic Syndromes
Myotonic chondrodystrophy (Schwartz-Jampel disease) is a rare congenital disease characterized by generalized muscle hypertrophy and weakness. Dysmorphic phenotypical features and the radiographic appearance of long bones are reminiscent of Morquio disease (Chapter 82), but abnormal mucopolysaccharides are not found. Dwarfism, joint abnormalities, and blepharophimosis are present. Several patients have been the products of consanguinity, suggesting autosomal recessive inheritance. The muscle protein perlecan, encoded by the SJS1 gene, a large heparan sulfate proteoglycan of basement membranes and cartilage, is defective in some cases of Schwartz-Jampel disease and explains both the muscular hyperexcitability and the chondrodysplasia.
Myotonia congenita (Thomsen disease) is a channelopathy (Table 601-1) and is characterized by weakness and generalized muscular hypertrophy so that affected children resemble bodybuilders. Myotonia is prominent and can develop at age 2-3 yr, earlier than in myotonic dystrophy. The disease is clinically stable and is apparently not progressive for many years. Muscle biopsy specimens show minimal pathologic changes, and the EMG demonstrates myotonia. Various families are described as showing either autosomal dominant (Thomsen disease) or recessive (Becker disease, not to be confused with BMD or DMD) inheritance. Rarely, myotonic dystrophy and myotonia congenita coexist in the same family. The autosomal dominant and autosomal recessive forms of myotonia congenita have been mapped to the same 7q35 locus. This gene is important for the integrity of chloride channels of the sarcolemmal and T-tubular membranes.
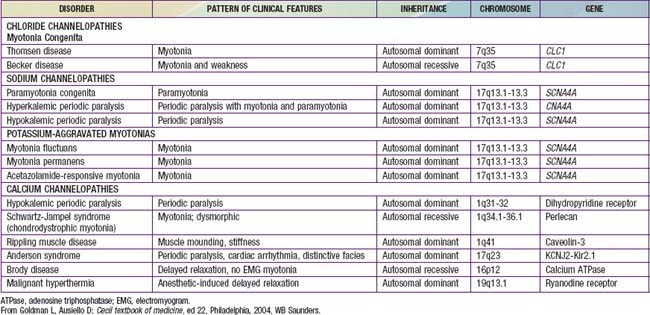
Paramyotonia is a temperature-related myotonia that is aggravated by cold and alleviated by warm external temperatures. Patients have difficulty when swimming in cold water or if they are dressed inadequately in cold weather. Paramyotonia congenita (Eulenburg disease) is a defect in a gene at the 17q13.1-13.3 locus, the identical locus identified in hyperkalemic periodic paralysis. By contrast with myotonia congenita, paramyotonia is a disorder of the voltage-gated sodium channel caused by a mutation in the α subunit. Myotonic dystrophy also is a sodium channelopathy (see Table 601-1).
Campbell C, Sherlock R, Jacob P, Blayney M. Congenital myotonic dystrophy: assisted ventilation duration and outcome. Pediatrics. 2004;113:811-816.
Day JW, Richater K, Jacobsen JP, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657-664.
Modoni A, Silvestri G, Pomponi MG, et al. Characterization of the pattern of cognitive impairment in myotonic dystrophy type 1. Arch Neurol. 2004;61:1943-1947.
Rose MR. Neurological channelopathies. BMJ. 1998;316:1104-1105.
601.4 Limb-Girdle Muscular Dystrophies
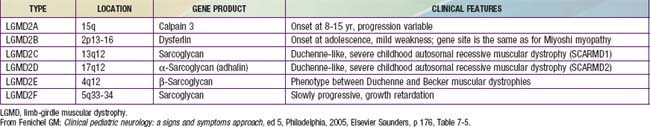
Limb-girdle muscular dystrophies encompass a group of progressive hereditary myopathies that mainly affect muscles of the hip and shoulder girdles (Table 601-2). Distal muscles also eventually become atrophic and weak. Hypertrophy of the calves and ankle contractures develop in some forms, causing potential confusion with BMD. Sixteen genetic forms of LGMD are now described, each at a different chromosomal locus and expressing different protein defects. Some include diseases classified with other traditional groups, such as the lamin-A/C defects of the nuclear membrane (see Emery-Dreifuss muscular dystrophy, earlier), and some forms of congenital muscular dystrophy.
601.5 Facioscapulohumeral Muscular Dystrophy
601.6 Congenital Muscular Dystrophy
Clinical Manifestations
Infants often have contractures or arthrogryposis at birth and are diffusely hypotonic. The muscle mass is thin in the trunk and extremities. Head control is poor. Facial muscles may be mildly involved, but ophthalmoplegia, pharyngeal weakness, and weak sucking are not common. A minority have severe dysphagia and require gavage or gastrostomy. Tendon stretch reflexes may be hypoactive or absent. Arthrogryposis is common in all forms of congenital muscular dystrophy (Chapter 600.10). Congenital contractures of the elbows have a high association with the Ullrich type of congenital muscular dystrophy owing to a defect in 1 or more of the 3 collagen VI genes, each at a different locus.
Diagnosis
Immunocytochemical reactivity for merosin (α2 chain of laminin) at the sarcolemmal region is absent in about 40% of cases and normally expressed in the others (Figs. 601-5 and 601-6). Merosin is a protein that binds the sarcolemmal membrane of the myofiber to the basal lamina or basement membrane. Its defect is a mutation of the LAMA2 gene at the 6q22-q23 locus. Merosin also is expressed in brain and in Schwann cells. The presence or absence of merosin does not always correlate with the severity of the myopathy or predict its course, but cases with merosin deficiency tend to have more severe cerebral involvement and myopathy. Adhalen (α-dystroglycan) may be secondarily reduced in some cases. Collagen VI is selectively reduced or absent in Ullrich disease because of a mutation in the COL6A gene. Mitochondrial dysfunction may be a secondary defect.
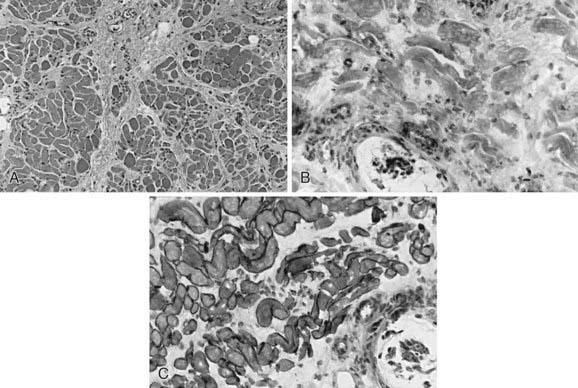
Figure 601-5 Quadriceps femoris muscle biopsy of a 6 mo old girl with congenital muscular dystrophy associated with merosin (α2-laminin) deficiency. A, Histologically, the muscle is infiltrated by a great proliferation of collagenous connective tissue; myofibers vary in diameter, but necrotic fibers are rare. B, Immunocytochemical reactivity for merosin (α2-laminin) is absent in all fibers, including the intrafusal myofibers of a muscle spindle seen at bottom. C, Dystrophin expression (rod domain) is normal. Compare with Figures 601-2, 601-3, and 601-6.
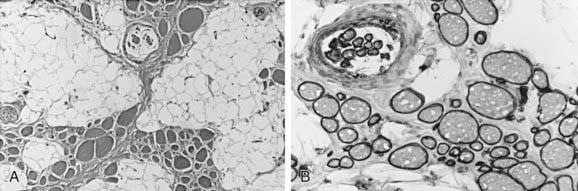
Figure 601-6 Quadriceps femoris muscle biopsy specimen of a 2 yr old girl with congenital muscular dystrophy. A, The fascicular architecture of the muscle is severely disrupted, and muscle is replaced by fat and connective tissue; the remaining small groups of myofibers of variable size are seen, including a muscle spindle at top. B, Merosin expression is normal in both extrafusal fibers of all sizes and in intrafusal spindle fibers. The severity of the myopathy does not relate to the presence or absence of merosin in congenital muscular dystrophy. Compare with Figure 601-5.
Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033-1043.
de Bernabe DB, van Bokhoven H, van Beusekom E, et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome pheonotype. J Med Genet. 2003;40:845-848.
Jiménez-Mallebrera C, Maioli MA, Kim J, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromusc Disord. 2006;16:571-582.
Le Ber I, Martínez M, Campion D, et al. A non-DM1, non-DM2 multisystem myotonic disorder with frontotemporal dementia: Phenotype and suggestive mapping of the DM3 locus to chromosome 15q21–24. Brain. 2004;127:1979-1992.
Ruggieri V, Lubieniecki F, Meli F, et al. Merosin-positive congenital muscular dystrophy with mental retardation, microcephaly and central nervous system abnormalities unlinked to the Fukuyama muscular dystrophy and muscular-eye-brain loci: report of three siblings. Neuromuscul Disord. 2001;11:570-578.





