CHAPTER 39 Muscles
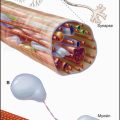
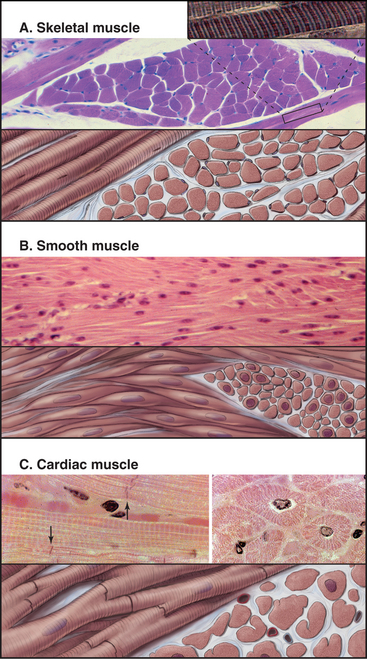
Vertebrates have three types of specialized contractile cells—smooth muscle, skeletal muscle, and cardiac muscle—that use actin and myosin to generate powerful, unidirectional movements (Fig. 39-1). These muscles have much in common but differ in their activation mechanisms, arrangement of contractile filaments, and energy supplies. This provides three options for physiological responses. The nervous system controls the timing, force, and speed of skeletal muscle contraction over a wide range. Cardiac muscle generates its own rhythmic, fatigue-free contractions that spread through the heart in a highly reproducible fashion. Neurotransmitters, acting like hormones, regulate the force and frequency of heartbeats over a narrow range. Nerves, hormones, and intrinsic signals control the activity of smooth muscles, which contract slowly but maintain tension very efficiently.
Skeletal Muscle
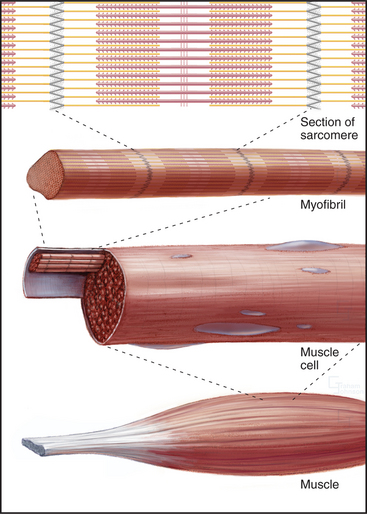
Skeletal muscle cells are optimized for rapid, forceful contractions. Accordingly, they have a massive concentration of highly ordered contractile units composed of actin, myosin, and associated proteins (Fig. 39-2). Actin and myosin filaments are organized into sarcomeres, aligned contractile units that give the cells a striped appearance in the microscope. For this reason, they are called striated muscles. Myosin uses ATP hydrolysis to power contraction, which results from myosin-powered sliding of actin-based thin filaments past myosin-containing thick filaments. Speed is achieved by linking many sarcomeres in series. Force is determined by the number of sarcomeres contracting in parallel.
Although skeletal muscle cells have only two states—inactive (relaxed) or active (contracting)—skeletal muscles produce a wide range of contractions, varying from slow and delicate to rapid and forceful. These graded contractions are achieved by varying the number of muscle cells activated by voluntary or reflex signals from the nervous system (Fig. 39-14). Nerve impulses stimulate a transient rise in cytoplasmic calcium that activates the contractile proteins.
Organization of the Skeletal Muscle Contractile Apparatus
Skeletal muscle cells (also called muscle fibers in the physiological literature) are among the largest cells of vertebrates. During development, mesenchymal stem cells give rise to progenitor cells with single nuclei called myoblasts. A family of master transcription factors, including MyoD and myogenin, coordinates the expression of specialized muscle proteins. As they differentiate, numbers of myoblasts fuse and elongate to form muscle cells with multiple nuclei and lengths of millimeters to tens of centimeters. The number of muscle cells is determined genetically and is relatively stable throughout life even as the size of the cells varies with the level of exercise and nutrition. Mature muscles harbor small numbers of stem cells (called satellite cells [see Fig. 41-15]) with a limited capacity to differentiate and repair damage.
A basal lamina (see Fig. 29-18C) surrounds and supports each muscle cell. At the ends of each cell, actin thin filaments are anchored to the plasma membrane at myotendinous junctions, which are similar to focal contacts (see Fig. 30-11). Integrins spanning the membrane link actin filaments to the basal lamina and to collagen fibrils of tendons. These physical connections transmit contractile force to the skeleton.
Organization of the Actomyosin Apparatus
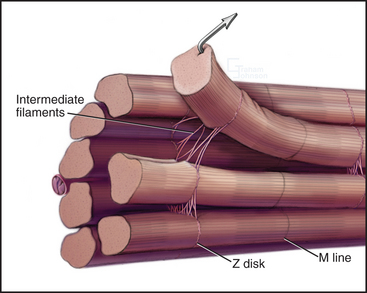
Interdigitation of thick, bipolar, myosin filaments and thin actin filaments in the sarcomeres of living muscle cells is so precise (Fig. 39-3) that it yields an X-ray diffraction pattern (Fig. 39-13) revealing the spacing of the filaments and the helical repeats of their subunits to a resolution of about 3 nm. Z disks at both ends of the sarcomere anchor the barbed ends of the actin filaments, so their pointed ends are near the center of the sarcomere. Myosin heads project from the surface of thick filaments, whereas their tails are anchored in the filament backbone. Thick and thin filaments overlap, with the myosin heads only a few nanometers away from adjacent actin filaments. The alignment and interdigitation of the filaments facilitate the sliding interactions required to produce contraction.
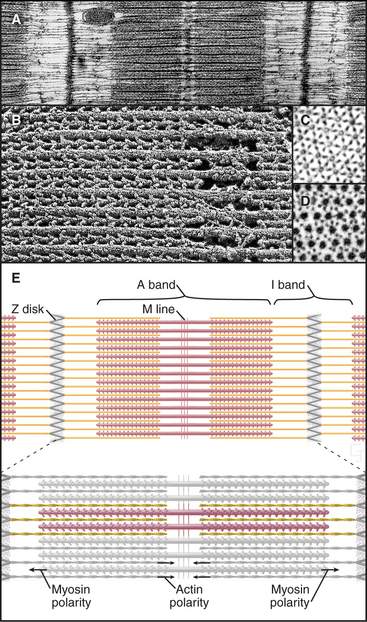
An important, simplifying architectural feature is that sarcomeres are symmetrical about their middles (Fig. 39-3). Consequently, the polarity of myosin relative to the actin filaments is the same in both halves of the sarcomere, allowing the same force-generating mechanism to work at both ends of the bipolar myosin filaments. Sarcomeres are organized end to end into long, cylindrical assemblies called myofibrils (Fig. 39-2) that retain their contractility even after isolation from muscle.
Thin Filaments
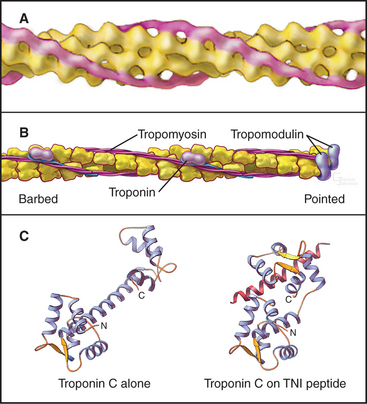
Thin filaments consist of actin and the tightly bound regulatory proteins troponin and tropomyosin (Fig. 39-4). When the concentration of Ca2+ is low in cytoplasm, troponin and tropomyosin inhibit the actin-activated ATPase of myosin. Tropomyosin, a 40-nm long coiled-coil of two α-helical polypeptides (see Fig. 3-10), binds laterally to seven contiguous actin subunits as well as head to tail to neighboring tropomyosins, forming a continuous strand along the whole thin filament. Troponin (TN) consists of three different subunits called TNC, TNI, and TNT (Table 39-1). TNT anchors troponin to tropomyosin. Like calmodulin (see Fig. 3-12 and Chapter 26), TNC is a dumbbell-shaped protein with four EF-hand motifs to bind divalent cations. In resting muscle the C-terminal globular domain of TNC binds two Mg2+ ions and an N-terminal α-helix of TNI, while the two low-affinity sites in the N-terminal globular domain of TNC are empty. Ca2+ binding to the low-affinity sites during muscle activation exposes a new binding site for TNI. The resulting conformational change in TNI allows tropomyosin to expose myosin-binding sites on the actin filament.
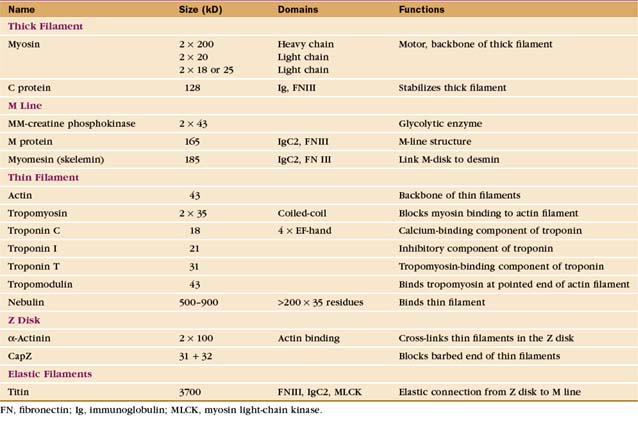
A protein meshwork in the Z disk anchors the barbed end of each thin filament (Fig. 39-5). Some cross-links between actin filaments consist of α-actinin, a short rod with actin-binding sites on each end (see Fig. 33-16). At least a half dozen structural proteins stabilize the Z disk through interactions with α-actinin, actin, and titin in the Z disk.
Proteins cap both ends of thin filaments. Cap-Z, the muscle isoform of capping protein (see Fig. 33-14), binds the barbed ends of thin filaments with high affinity, limiting actin subunit addition or loss. Tropomodulin associates with both tropomyosin and actin to cap and stabilize the pointed end of thin filaments (Fig. 39-4B).
Tropomyosin and a gigantic filamentous protein, nebulin, stabilize thin filaments laterally. Nebulin consists of about 185 imperfect repeats of 35 amino acids that interact with each actin subunit, tropomyosin, and troponin along the length of thin filaments. Interactions with tropomodulin and Z disk proteins anchor nebulin at the two ends of the thin filament. Acting as a ruler and a cap, nebulin and tropomodulin help to set the length of thin filaments.
Thick Filaments
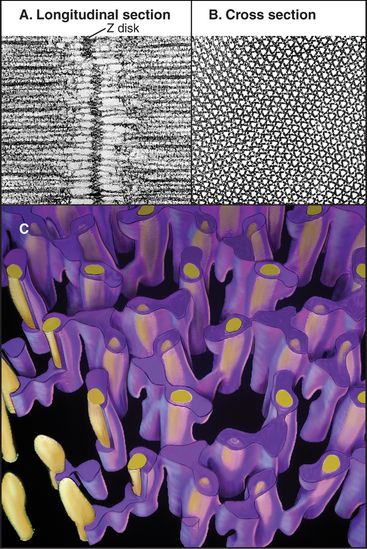
The self-assembly of myosin II (see Fig. 5-7) establishes the bipolar architecture of striated muscle thick filaments (Fig. 39-6). Some features of thick filaments are invariant, such as a backbone consisting of myosin tails, a surface array of myosin heads, the 14.3-nm stagger between rows of heads, and a central bare zone formed by antiparallel packing of tails. Filaments may vary in length, diameter, and organization of the helical array of heads in various species. Invertebrate thick filaments have a core of paramyosin, a second coiled-coil protein, which is not found in vertebrates.
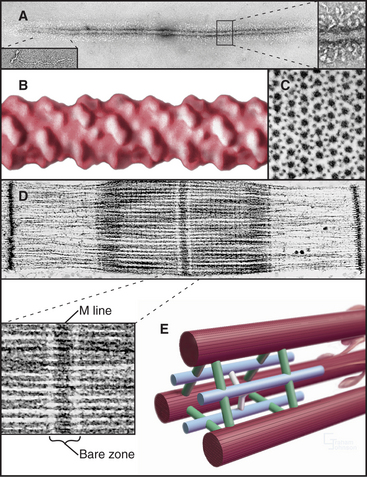
Several accessory proteins stabilize thick filaments (Table 39-1). Thick filaments in most striated muscles are girdled at intervals by semicircular bands of a protein that is, coincidentally, called C-protein. C-protein consists of fibronectin III and immunoglobulin domains. The “M line” in the center of the sarcomere is a three-dimensional array of protein cross-links that maintains the precise registration of thick filaments. At least three structural proteins and the enzyme MM-creatine phosphokinase (which transfers phosphate from creatine-phosphate to ADP) are located in the M line.
Titin Filaments
A third array of protein filaments lies parallel to the thin and thick filaments, connecting the Z disk to the thick filaments and the M line (Fig. 39-7). Hard to preserve for electron microscopy, these diaphanous filaments were neglected for years. Each filament is a single polypeptide named titin (after mythological giants), so named because of its remarkable size: more than 30,000 amino acids folded into a linear array of 300 immunoglobulin and fibronectin II domains measuring more than 1.2 μm long. Titin is thought to be the largest protein encoded by the human genome.
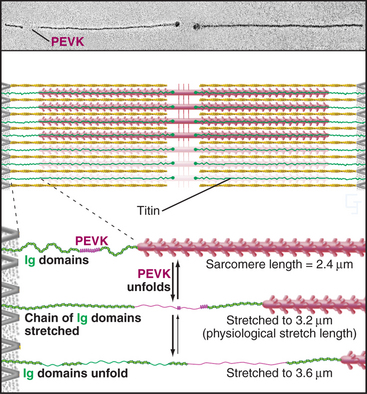
Titin molecules are elastic, and this accounts for the passive resistance to stretching of relaxed muscle. Their connections to the Z disk and thick filaments provide elastic continuity from one sarcomere to the next and keep the thick filaments centered in the sarcomere during contraction. If titin molecules are broken experimentally, thick filaments slide out of register toward one Z disk during contraction. Two features provide the elasticity during short (∼0.3 μm per titin), physiological stretches: The irregular chain of immunoglobulin domains in the I band straightens out, and a segment of the polypeptide rich in proline, glutamic acid, valine, and lysine (the PEVK domain) stretches. Stretching decreases entropy and provides the energy for elastic recoil. (See Fig. 29-12 for another example of an entropic spring in biology.) Extreme stretching unfolds Ig domains one by one.
Intermediate Filaments
Desmin intermediate filaments (see Chapter 35) help to align the sarcomeres laterally (Fig. 39-8) by linking each Z disk to its neighbors and to specialized attachment sites on the plasma membrane. Myofibrils near the cell surface are attached to the plasma membrane at specializations called costameres. In addition to desmin, costameres contain several cytoskeletal proteins (vinculin, talin, spectrin, and ankyrin) found in focal contacts and adherens junctions of nonmuscle cells (see Figs. 30-11 and 31-7). Desmin mutations in humans cause disorganization of myofibrils, resulting in generalized muscle failure.
Organization of the Muscle Membrane System
Structural Proteins of the Plasma Membrane: Defects in Muscular Dystrophies
In addition to providing a permeability barrier, the plasma membrane of the muscle cell must maintain its integrity while being subjected to years of forceful contractions. Accordingly, the membrane is stabilized by a transmembrane complex of proteins, the dystroglycan-sarcoglycan complex, that links the internal membrane skeleton to the basal lamina outside (Fig. 39-9 and Table 39-2). Occasional breaches of the membrane are inevitable, so muscle cells also depend on a repair process that reseals holes. If membrane damage exceeds the repair capacity, muscle cells degenerate locally (segmental necrosis) or globally. Cell death beyond the ability of muscle stem cells to repair the tissue results in muscular dystrophy. The age of onset and clinical features of inherited muscular dystrophies depend on the molecular defect. Patients with severe defects develop progressive muscle weakness as children. Ultimately, failure of respiratory muscles is fatal.
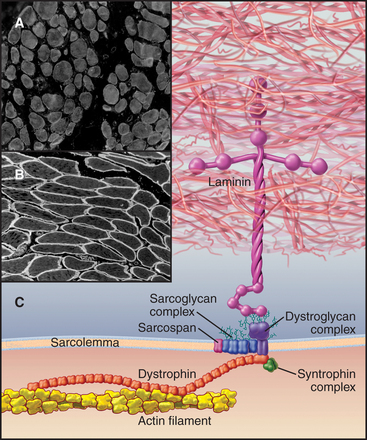
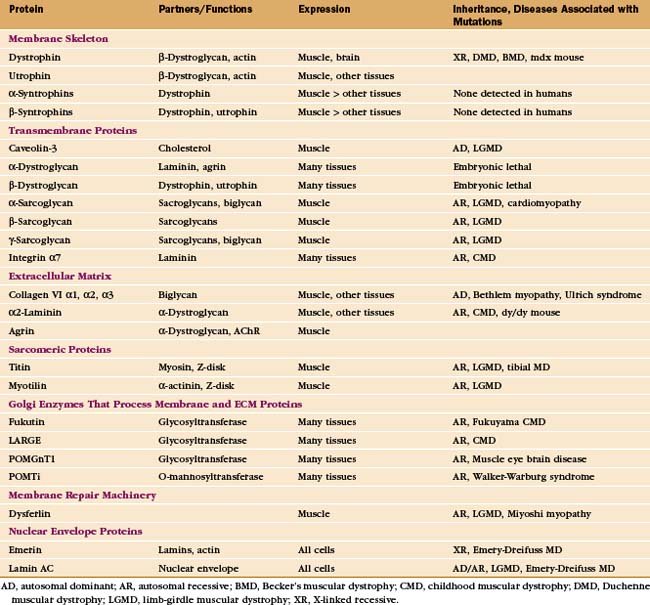
Mutations in more than 40 human genes have been linked to muscular dystrophies (Table 39-2), so it is possible that the malfunction or lack of any molecule in the system that maintains the integrity of the plasma membrane will cause disease. Disease-causing mutations occur in genes for the proteins of the dystroglycan-sarcoglycan complex, extracellular matrix proteins, Golgi apparatus enzymes that process these structural proteins, and the membrane repair machinery. Some of these mutations also affect the nervous system. Mutations are usually autosomal-recessive and are not uncommon. About one in several thousand humans develops some form of muscular dystrophy, because they inherit mutations in both copies of one of the sensitive genes. The mechanism of disease in muscular dystrophies is similar to that in hereditary spherocytosis, in which deficiencies of the membrane skeleton make red blood cells susceptible to mechanical damage (see Fig. 6-10).
The proteins that stabilize muscle membranes escaped detection until the late 1980s, when x-linked mutations in the dystrophin gene were discovered to cause Duchenne’s muscular dystrophy, the most common human form of the disease. Dystrophin is an enormous member of the α-actinin superfamily of actin-binding proteins (see Fig. 33-16). The dystroglycan-sarcoglycan complex was found when it copurified with dystrophin after solubilizing the membrane with detergents. Loss of any protein of the dystrophin-dystroglycan-sarcoglycan complex typically leads to secondary loss of the other proteins from muscles.
Dystroglycans and a dystrophin homolog, utrophin, participate in clustering acetylcholine receptors at the neuromuscular junction, the chemical synapse between motor neurons and skeletal muscle (see Fig. 11-8). When, during development, a motor neuron arrives at the surface of its target muscle cell, the neuron secretes an adhesive protein called agrin, which is incorporated into the basal lamina, immediately adjacent to the nerve terminal. Dystroglycan binds agrin and positions associated acetylcholine receptors at the site where they receive acetylcholine secreted by the nerve in response to an action potential.
Interaction of Plasma Membrane Invaginations with the Smooth Endoplasmic Reticulum
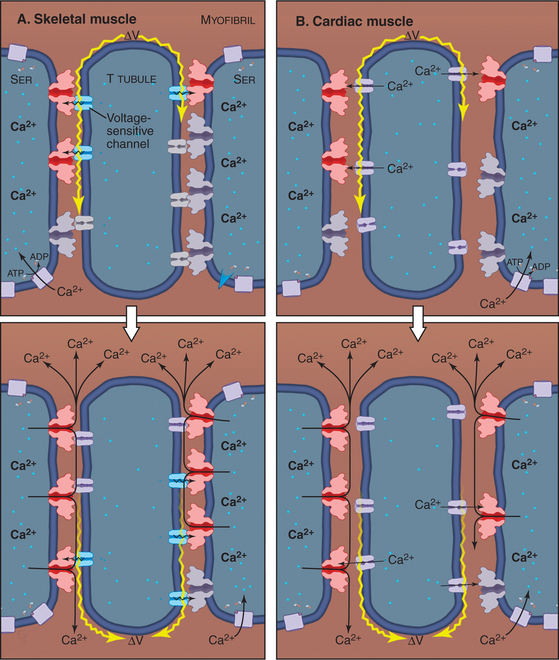
The plasma membrane of skeletal muscle cells, like the plasma membrane of nerve cells, is excitable (see Fig. 11-6) and invaginates deeply to form so-called T tubules that run across the entire cell (Fig. 39-10). Depending on the species and type of striated muscle (skeletal versus cardiac), T tubules may be located either at the level of the Z disks or at the thick fila-ment ends. Inside the muscle cell, T tubules interact extensively with the smooth endoplasmic reticulum (SER) that surrounds each myofibril. Historically, this SER has been called sarcoplasmic reticulum. Terminal cisternae of SER are closely associated with the passing T tubules by foot processes that can be visualized by electron microscopy. Together, T tubules and SER constitute a signal-transducing apparatus that converts depolarizations of the plasma membrane into a spike of cytoplasmic Ca2+ that triggers contraction (Fig. 39-15).
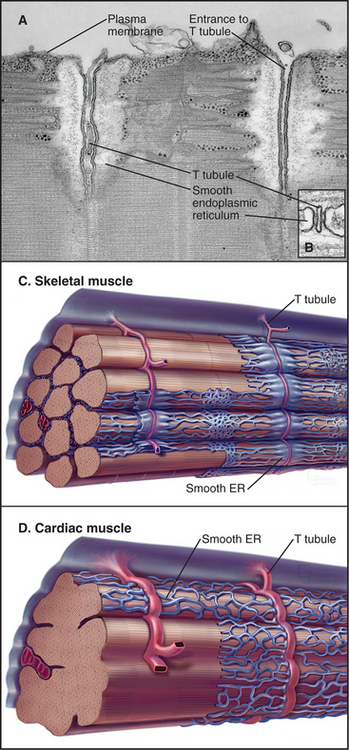
Figure 39-10 plasma membrane specializations of striated muscles. A–B, Electron micrographs of thin sections of fish skeletal muscle showing invaginations called T tubules, which cross the whole muscle cell and associate closely with smooth endoplasmic reticulum (ER). The complex of a T tubule with smooth ER on both sides is called a triad. Foot processes, consisting of voltage-sensitive calcium channels in a T tubule paired with calcium release channels in the ER, connect the T tubule to the smooth ER. (Fig. 39-15 provides molecular details.) C–D, Drawings of the three-dimensional arrangement of T tubules and smooth ER relative to the sarcomeres in skeletal and cardiac muscle.
(A–B, Courtesy of C. Franzini-Armstrong and K. Porter, University of Pennsylvania, Philadelphia.)
Molecular Basis of Skeletal Muscle Contraction
The Sliding Filament Mechanism
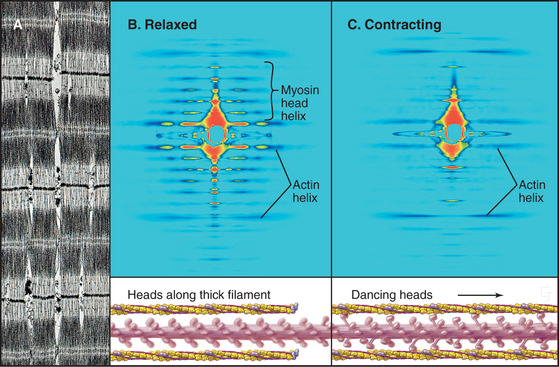
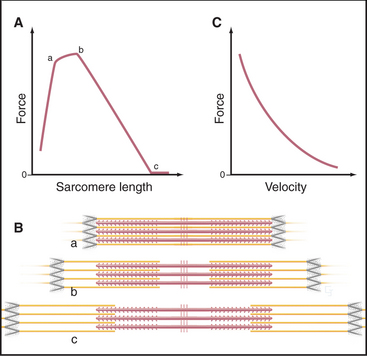
The key to understanding muscle contraction was the discovery that thick and thin filaments maintain constant lengths and slide past each other as sarcomeres (and the muscle) shorten (Fig. 39-11). About the same time, it was appreciated that cross-bridges (now recognized to be myosin heads) can connect actin and myosin filaments and that tension produced during contrac-tion is proportional to the overlap of actin and myosin filaments (Fig. 39-12). Supported by biochemical and ultrastructural evidence for actin-myosin interaction, these pioneering observations led to the theory that cross-bridges between the thick and thin filaments produce force for contraction. Fifty years of research on cross-bridges have yielded a detailed picture of the chemistry and molecular mechanics underlying the force-producing reactions. A review of the steps of the actomyosin-ATPase cycle (see Fig. 36-5) is helpful in understanding the contraction mechanism. Three different physiological states reveal information about cross-bridge mechanisms.
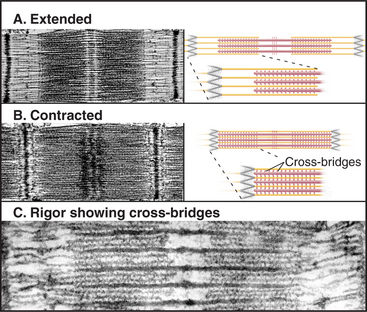
Most myosin heads in contracting muscle have bound ATP or ADP-Pi and oscillate rapidly among the four “weakly bound” states illustrated in Figure 36-5. During some of the transient interactions of myosin-ADP-Pi with actin, phosphate dissociates from myosin, and the light-chain domain rapidly reorients (see Figs. 36-4 and 36-5). This stretches elastic elements in the myosin heads and both thick and thin filaments. Energy in these elastic elements can be used over a period of milliseconds to displace the actin filament relative to the cross-bridge and contract the muscle. When ADP dissociates from the actin-myosin-ADP intermediate, ATP rapidly binds to the actin-myosin complex, dissociating the cross-bridge and starting a new ATPase cycle.
Relationship of Cross-Bridge Behavior to the Mechanical Properties of Muscle
The behavior of cross-bridges explains why the velocity of muscle contractions of an active muscle depends on the external load (Fig. 39-12). Contraction velocity is maximal when opposed by no load. Without a load, the molecular motion stored in elastic elements of each cross-bridge is largely converted into movement of actin filaments relative to myosin filaments. Under these conditions, the filaments in muscle slide past each other at a rate of about 5 μm s−1, the same speed that is observed for free actin filaments moving over myosin heads in vitro (see Fig. 36-6). For this rapid sliding to occur, myosin heads that do not produce force must not impede movement. If bound tightly to actin, they would interfere mechanically with rapid sliding. This is avoided by the rapid equilibrium of the myosin intermediates between being bound to actin and being free. Myosin heads with bound ATP or ADP-Pi do not produce force when they bind transiently to a given actin subunit. Nor do these brief encounters retard sliding driven by force-producing cross-bridges.
Muscle produces maximum force when the contraction rate is zero (Fig. 39-12). The conformational change in the myosin head stretches elastic elements in the cross-bridge, but the force cannot overcome the resistance from the load on the muscle. Consequently, the filaments do not slide, and energy stored in each stretched elastic element is lost as heat when the cross-bridge dissociates at the end of the ATPase cycle. The maximum force depends on the numbers of sarcomeres in parallel, that is, the cross-sectional area of the muscle. Thus, muscles respond to strengthening exercises by growing in diameter.
Regulation of Skeletal Muscle Contraction
Control of Skeletal Muscle by Motor Neurons
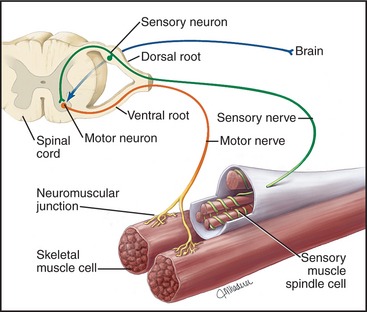
Neural stimuli that activate skeletal muscles arise in two ways (Fig. 39-14). In organisms with well-developed central nervous systems, most neural signals that activate skeletal muscles result from conscious decisions, providing voluntary control over skeletal muscles. Other signals result from reflex responses to stimula-tion of sensory nerves. Specialized muscle cells innervated with both motor and sensory nerves function as stretch receptors, relaying information about length and tension back to the spinal cord, where reflexes coordinate the motor neuron output. Neural inputs from both sources converge on motor neurons located in the brain stem and spinal cord of vertebrates. Axons of these motor neurons branch in the muscle to contact one or more muscle cells. A motor neuron together with its target muscle cells forms a motor unit. In the most precisely controlled muscles, such as the extraocular muscles, some motor neurons innervate single muscle cells.
The contractile activity of a muscle is graded in terms of the speed and force of the contraction, so individual muscles can produce both delicate and powerful movements. Nerve stimulation determines the contractile force in two ways: (1) The number of active motor units determines how many muscle cells produce force, and (2) the rate of stimulation adjusts the force produced by active cells. Every time a muscle cell is stimulated, all of the sarcomeres are activated, but the force that they produce increases as the rate of stimulation increases, up to a maximum of about 200 stimuli per second. The shortening velocity of an active muscle depends simply on force produced and the resistance (Fig. 39-12C). If a large force or high velocity of contraction is required, many motor units are called into action and stimulated repetitively. To sustain contraction, motor nerves fire repeatedly. By varying the number of active cells in a muscle and the rate of stimulation, the nervous system sets the force required for a particular movement.
Synaptic Transmission at Neuromuscular Junctions
The terminal branch of each motor neuron axon forms a synapse called the motor end plate or neuromuscular junction on the muscle surface (see Fig. 11-8). These nerve endings are filled with synaptic vesicles containing the neurotransmitter acetylcholine. Arrival of an action potential at the nerve terminal stimulates fusion of synaptic vesicles with the nerve plasma membrane, releasing acetylcholine into the cleft between nerve and muscle. In less than a millisecond, acetylcholine diffuses across the extracellular space and binds to acetylcholine receptors concentrated in the adjacent muscle plasma membrane. Acetylcholine binding opens the receptor cation channel, initiating a new action potential that spreads over the muscle cell plasma membrane and down into the T tubules.
Coupling Action Potentials to Contraction
An action potential in a T tubule triggers the release of Ca2+ from SER into the cytoplasm (Fig. 39-15). Ca2+ binding to troponin allows myosin to interact with the thin filament, initiating contraction. This signal transduction process is called excitation-contraction coupling. Ca2+ release in skeletal muscle is the best-characterized example of a general regulatory mechanism used by many cells (see Chapter 26).
Three transmembrane proteins located in the T tubule and the terminal cisternae of the SER cooperate to generate the transient Ca2+ signal (Fig. 39-16). The operation of this system is described after its components are introduced:
An action potential in a T tubule results in a transient rise in cytoplasmic Ca2+, from 0.1 μM to about 2 μM (Fig. 39-16), in the following way. The action potential causes a short-lived conformational change in the DHP receptors that is transmitted directly to associated ryanodine receptor Ca2+ release channels. Many Ca2+ channels open transiently, allowing Ca2+ to diffuse down the steep concentration gradient from the SER lumen to cytoplasm. Physical connections between ryanodine receptors may spread their activation laterally, ensuring synchronous activation of a patch of channels. The structural changes in these channels that release Ca2+ are not yet understood.
After a single action potential, the rise in the cytoplasmic Ca2+ level lasts but a few milliseconds for three reasons. First, Ca2+ release channels close quickly. Second, cytoplasmic Ca2+ binds to troponin C and other proteins. Third, Ca2+ pumps efficiently transport cytoplasmic Ca2+ back into the lumen of the smooth endoplasmic reticulum, even before the muscle develops maximum force. Ca2+ pumps are continuously active, keeping the cytoplasmic Ca2+ concentration low. Re-peated action potentials are required to prolong the rise in cytoplasmic Ca2+ (Fig. 39-16B).
Transduction of the Calcium Spike into Contraction
Troponin-tropomyosin on thin filaments cooperates with myosin to turn on contraction in response to a Ca2+ spike. At rest, two Ca2+-binding sites of troponin C are largely unoccupied (owing to their low affinity for Ca2+ and the low Ca2+ concentration), and the troponin-tropomyosin complex partially blocks the binding site for myosin heads on actin (Fig. 39-17). This prevents most of the weak-binding myosin intermediates with ATP or ADP-Pi in the active site from binding the thin filament. When released into cytoplasm, Ca2+ binds troponin C, causing a conformational change that creates a binding site for a helical region of TNI. This interaction attracts the C-terminus of TNI away from actin and tropomyosin, allowing a small shift in the position of tropomyosin on the thin filament. This shift increases the probability that myosin-ADP-Pi heads will bind to the thin filament, dissociating their bound Pi and producing force. The initial binding of the first force-producing heads shifts the long tropomyosin molecule a bit farther away from the myosin-binding sites, allowing adjacent actins to interact freely with other myosin heads. The end-to-end association of tropomyosins facilitates this cooperative switch by exposing the myosin-binding sites on more distant actin subunits in the thin filament.
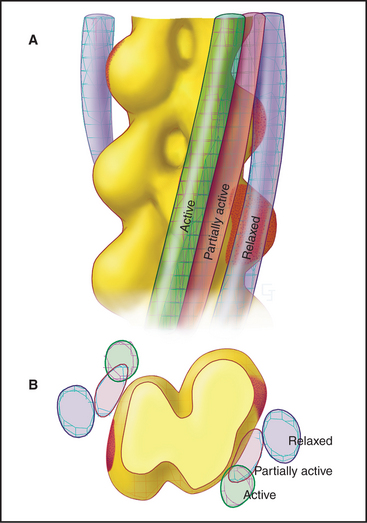
Thus, the combined influence of Ca2+ and myosin makes the thin filament receptive to myosin binding. Activation is cooperative because both Ca2+-binding sites on troponin C must be occupied, because the effects of Ca2+ binding and myosin binding are transmitted to neighboring tropomyosins through their end-to-end attachments, and because every myosin that binds accentuates the response. This cooperativity makes the on-off switch respond very sharply to a relatively small, 10- to 20-fold change in the cytoplasmic Ca2+ concentration. The efficiency of this switch is underscored by the fact that the energy consumption of a muscle cell increases more than 1000-fold when it is activated. Activation of slow skeletal muscle (Table 39-3) and cardiac muscle is less cooperative, as their troponin C has only one Ca2+-binding site.
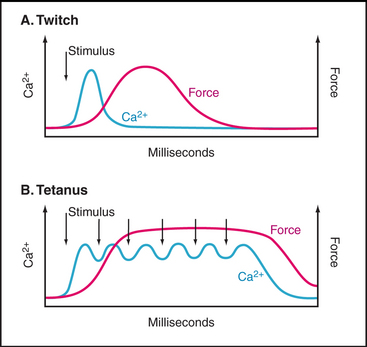
Note the delay between the Ca2+ spike and the onset of tension (Fig. 39-16). The Ca2+-sensitive switch is sharp but slow owing to the slow response of thin filaments to Ca2+ binding. Note also that muscle continues to produce force well after the cytoplasmic Ca2+ concentration returns to resting levels. Ca2+ binds troponin C rapidly (milliseconds) but dissociates slowly (tens of milliseconds). Thus, the Ca2+ spike saturates troponin C, and the muscle remains active even after free Ca2+ has returned to the endoplasmic reticulum lumen. Force declines slowly as Ca2+ dissociates from troponin C and returns to the smooth endoplasmic reticulum without raising the cytoplasmic Ca2+ concentration.
A single action potential produces a short contractile “twitch” (Fig. 39-16). Maximum contractile force is produced by a series of closely spaced action potentials, leading to a sustained rise in cytoplasmic Ca2+ and prolonged activation of actomyosin. The extended contraction is called tetanus.
Specialized Skeletal Muscle Cells
All skeletal muscle cells are built on the same principles, but vertebrates actually have several different types of skeletal muscle cells, each with distinct contractile protein isoforms and metabolic enzymes. The myosin and actin isoforms are coded by different genes, whereas alternative splicing of one primary transcript (see Fig. 16-6) creates more than 50 isoforms of troponin T. Mutations in the genes for myosin, actin, tropomyosin, troponin-T, and nebulin can each cause defects in human skeletal muscles.
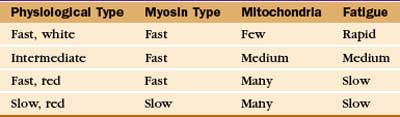
Physiological properties, such as the speed of contraction and the rate of fatigue, provide criteria for classifying muscle cells (Table 39-3). The isoforms of myosin (and probably the other contractile proteins) determine the speed of contraction, whereas the content of mitochondria and myoglobin determines the endurance and overall color of the muscle. White muscle cells depend largely on glycolysis to supply ATP, accounting for their rapid fatigue compared with red muscle cells, which are specialized for oxidative metabolism with abundant mitochondria and myoglobin.
Remarkably, the pattern of nerve stimulation determines the muscle cell type by controlling which genes are expressed (and presumably, how the troponin T messenger RNA is processed). This was demonstrated by transplanting motor nerves between fast and slow muscles. Over a period of weeks, slow isoforms replace fast isoforms and vice versa. Even more surprising, the same result is achieved by stimulating muscles electrically with fast or slow patterns of impulses. Chronic low-level stimulation biases gene expression toward the proteins that are found in slow muscle cells. Calcium and calmodulin provide one prominent link between activity and gene expression. The concentration of active calmodulin tracks with the pattern of stimula-tion, because Ca2+ is released in the cytoplasm each time a muscle contracts. Among other things, calcium-calmodulin activates protein phosphatase PP2b (calcineurin; see Fig. 25-6), which dephosphorylates tran-scription factors (see Fig. 15-21). These activated tran-scription factors move into the nucleus and cooperate with other transcription factors including a nuclear re-ceptor to turn on expression of proteins found in slow muscles, including contractile proteins and enzymes for oxidative metabolism.
Cardiac Muscle
To maintain the circulation of blood, heart muscle is specialized for repetitive (˜100,000 times per day), fatigue-free contractions driven at regular intervals by action potentials from specialized pace-making cells. Gap junctions allow these action potentials to spread from one muscle cell to the next. The arrangement of the contractile apparatus into sarcomeres is similar to that in skeletal muscle.
The Contractile Apparatus of Cardiac Muscle
Cardiac muscle cells have sarcomeres with thick and thin filaments like skeletal muscle (Fig. 39-18), but they have more mitochondria, larger T tubules, less smooth endoplasmic reticulum, and a smaller version of nebulin called nebulette. The atrium has one major myosin isoform. Two ventricular myosin isoforms (one shared with slow skeletal muscle) differ in ATPase activity and speed of contraction. Humans almost exclusively express only one of these isoforms. In rats, thyroid hormone regulates expression of these isoforms. Both are expressed normally, but one predominates in hypothyroidism, and the other predominates in hyperthyroidism. Heart expresses different isoforms of TNI and TNT than does skeletal muscle. If damaged in a heart attack, cardiac cells release TNI and TNT into the blood. Measurement of cardiac TNI and TNT in blood is now the most sensitive chemical test for heart attacks.

Short, modestly branched, cardiac muscle cells have centrally located nuclei and squared-off ends (Fig. 39-1) where neighboring cells attach to each other at specialized adhesive junctions called intercalated disks (Fig. 39-18). These junctions have properties of both adherens junctions (links to actin filaments) and desmosomes (links to intermediate filaments). Ca2+-dependent cadherins form the physical connections between adjacent cells (see Fig. 30-5).
Pacemaker Cells
Modified cardiac muscle cells in the right atrium (sinoatrial node) spontaneously depolarize their plasma membrane at regular intervals to initiate each heartbeat (Fig 39-19; see also Fig. 11-11). Special nonselective cation channels allow Na+ to leak into these cells and K+ to leak out, depolarizing the plasma membrane and triggering an action potential. The action potential spreads from cell to cell through gap junctions (see Fig. 31-6), activating all cells in the atrium within a few hundred milliseconds. After a brief delay in the atrioventricular node, the action potential and contraction spread through the ventricle.
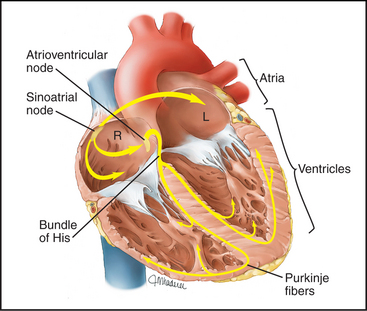
Motor nerves do not stimulate cardiac muscle directly, but the heart is rich in autonomic nerves from the sympathetic and parasympathetic nervous systems. These nerves secrete acetylcholine and norepinephrine, which act as hormones to modulate the rate and force of contraction (see Fig. 11-12). Norepinephrine acting through the β-adrenergic receptor and cAMP, activates PKA (see Fig. 27-3), which phosphorylates TNI. This increases the rate of cross-bridge cycling and the strength of contraction.
Molecular Basis of Inherited Heart Diseases
Because the heart is so vital to survival, relatively minor molecular defects command attention in humans. About 1 of 500 individuals carries a mutation in a gene that compromises cardiac function (Table 39-4). For example, many different point mutations in the myosin heavy chain can compromise its function. Affected individuals are typically heterozygous for these mutations, and the mutant myosin interferes with the function of the normal myosin (i.e., they are dominant negative mutations). Over a period of years, the heart attempts to compensate for the contractility defect through hypertrophy, but the thickened heart wall compromises cardiac relaxation and filling of the chambers with blood. More serious, heart hypertrophy eventually causes defects in activation and abnormal rhythms that can be fatal. The rate of progress of these so-called hypertrophic cardiomyopathies depends on not only the particular mutation but also other factors that vary from person to person. Individuals with defects in C protein develop hypertrophy in their fifties and can live normal life spans. By contrast, those with defects in troponin T can be affected as teenagers and die of arrhythmias in their twenties. These severe mutations of cardiac contractile proteins account for about half of the deaths of apparently healthy young athletes. Myosin mutations are intermediate in severity. Mutations in actin and dystrophin cause a disease of an opposite sort. Individual cells hypertrophy, but many die and are replaced by connective tissue, leading to thinning of the wall of the heart and defective contractility.
Table 39-4 GENETIC DEFECTS IN CARDIAC AND SKELETAL MUSCLE DISEASE
| Gene/Protein | Normal Function | Disease Manifestations |
|---|---|---|
| Contractile Proteins | ||
| Actin | Thin filament | Dilated cardiomyopathy, heart failure, myopathy |
| Titin | Passive elasticity | Dilated cardiomyopathy, heart failure, muscular dystrophy |
| Nebulin | Thin filament | Myopathy |
| Dystrophin | Membrane stabilization | Dilated cardiomyopathy, heart failure, Duchenne muscular dystrophy |
| β-Myosin heavy chain | Thick-filament motor | Hypertrophic cardiomyopathy, arrhythmias, myopathy |
| Myosin essential light chain | Myosin motor | Hypertrophic cardiomyopathy, arrhythmias |
| Myosin regulatory light chain | Myosin motor | Hypertrophic cardiomyopathy, arrhythmias |
| Myosin C protein | Thick-filament structure | Hypertrophic cardiomyopathy, arrhythmias |
| Troponin T | Calcium regulation | Hypertrophic cardiomyopathy, arrhythmias, myopathy |
| Troponin I | Calcium regulation | Hypertrophic cardiomyopathy, arrhythmias, myopathy |
| Tropomyosin | Calcium regulation | Hypertrophic cardiomyopathy, arrhythmias, myopathy |
| Electrophysiology | ||
| HERG | Potassium channel | Long QT syndrome, arrhythmias |
| KVLQT1 | Potassium channel | Long QT syndrome, arrhythmias |
| minK | Potassium channel | Long QT syndrome, arrhythmias |
| SCN5A | Sodium channel | Long QT syndrome, arrhythmias |
| Ankyrin | Membrane scaffold | Long-QT syndrome, arrhythmias |
| Other Functions | ||
| NKX2–5 | Transcription factor | Congenital atrioseptal defects |
| TBX5 | Transcription factor | Multiple congenital defects between heart chambers |
Smooth Muscle
The Contractile Apparatus
Smooth muscle cells are specialized for slow, powerful, efficient contractions under the control of a variety of involuntary mechanisms. Smooth muscle cells are generally confined to internal organs, such as blood vessels (where they regulate blood pressure), the gastrointestinal tract (where they move food through the intestines), and the respiratory system (where their excessive contraction contributes to asthma and other allergic reactions). The cytoplasm of spindle-shaped smooth muscle cells (Fig. 39-1) appears homogeneous by light microscopy because the contractile proteins are not organized in regular arrays like sarcomeres of skeletal and cardiac muscle. A basal lamina and variable amounts of collagen and elastic fibers surround each cell.
In terms of organization and biochemistry, smooth muscle cells (Fig. 39-20) resemble nonmuscle cells more than they do skeletal or cardiac muscle. For example, the gene for smooth muscle myosin arose relatively recently from a cytoplasmic myosin II gene (see Fig. 36-7). These myosins also share the same regulatory light chain. Long myosin thick filaments are interspersed among the thin filaments but not in a regular way like striated muscles. Thin filaments are composed of actin and tropomyosin, along with two regulatory proteins, caldesmon and calponin, rather than troponin. Thin filaments are arranged obliquely in the cell, some with their barbed ends attached to dense plaques on the plasma membrane, others to dense bodies in the cytoplasm. Like Z disks in striated muscles, dense bodies anchor desmin intermediate filaments, forming a continuous, inextensible, internal “tendon” running from end to end into the cell, preventing excess stretching (see Fig. 35-8).
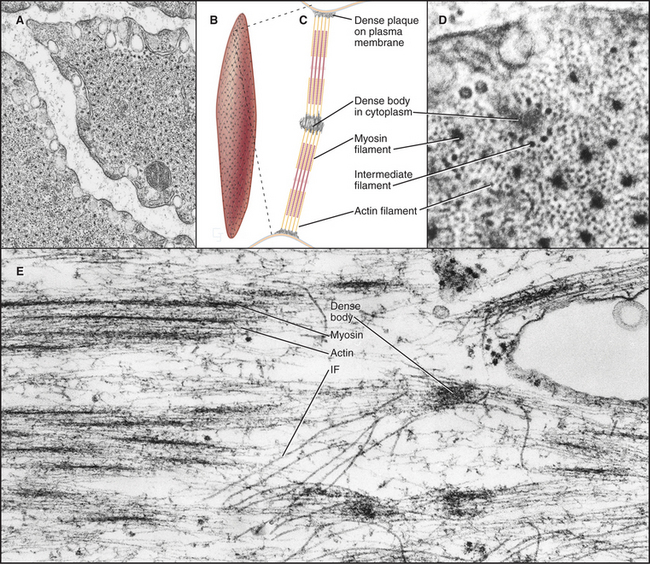
Smooth muscle cells contract like a concertina (Fig. 39-21) because tension generated by myosin and actin is applied to discrete spots on the plasma membrane. This compression can be seen in light micrographs as irregular cells with “corkscrew” nuclei. Given that smooth muscle cells have less myosin than striated muscle cells do, it is remarkable that they develop the same force. This is explained by two factors. First, the force-generating unit, the myosin filament, is larger in smooth muscle than in skeletal muscle. Deploying a given amount of myosin in large, thick filaments in a long sarcomere produces more force than does the same myosin in smaller filaments arranged in a series of short sarcomeres. Second, individual smooth muscle myosin molecules produce a larger force than skeletal muscle myosin, at least in vitro assays.
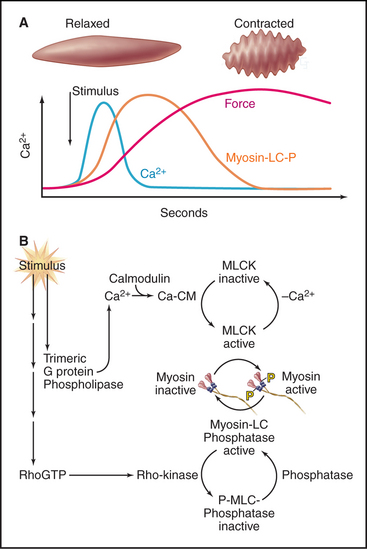
Regulation of Smooth Muscle Contraction
Depending on the muscle, Ca2+ for contraction enters the cytoplasm through either voltage-dependent calcium channels in the plasma membrane or IP3 (inositol, 1,4,5-triphosphate)–induced Ca2+ release from smooth endoplasmic reticulum (Fig. 39-21). Drugs that block plasma membrane calcium channels can distinguish these two pathways experimentally. In intestines, parasympathetic nerves release acetylcholine to stimulate seven-helix muscarinic receptors (see Figs. 11-7 and 11-12). Associated trimeric G-proteins activate cation channels that depolarize the plasma membrane and allow Ca2+ to enter through voltage-sensitive calcium channels. Gap junctions couple gut smooth muscle cells, allowing excitation to spread from cell to cell. Calcium channel blockers strongly inhibit activation of gut smooth muscle. At the other end of the spec-trum, vascular smooth muscle depends on IP3 to release Ca2+ from intracellular stores rather than from outside the cell.
Following stimulation, intracellular Ca2+ increases rapidly but transiently, declining to a value above resting level as the receptors desensitize (see Fig. 24-3). Ca2+ pumps in both smooth endoplasmic reticulum and plasma membrane clear the cytoplasm of Ca2+ so that Ca2+ levels decrease to resting levels and the muscle relaxes when the activating stimulus is removed. Relaxing agents, acting through cyclic guanosine monophosphate (cGMP) or cAMP (see Fig. 26-1), promote clearance of cytoplasmic Ca2+. Epinephrine relaxes smooth muscles of the respiratory system by another method. Stimulation of β-adrenergic receptors activates potassium channels that hyperpolarize the plasma membrane and reduce Ca2+ entry. This approach is widely used to treat asthma.
After a considerable delay (>200 ms) following the Ca2+ spike, contractile force develops slowly. The delay is attributable to the time required for a se-quence of three biochemical reactions: Ca2+ binding to calmodulin, calcium-calmodulin activation of myosin light-chain kinase (see Fig. 25-4), and phosphorylation of myosin regulatory light chains, turning on the myosin-actin ATPase cycle (Fig. 39-21). Unphosphorylated myosin-II from smooth muscle and vertebrate nonmuscle cells is inactive.
Phosphorylation of myosin light chains is required to initiate but not maintain contraction, so slowly cycling, unphosphorylated myosins maintain peak force with little expenditure of energy. Regulation of unphosphorylated cross-bridges is not well understood, but they appear to be activated cooperatively by a small population of phosphorylated myosin heads. Caldesmon, a calcium-calmodulin-binding protein associated with tropomyosin on actin filaments, may contribute to activation and/or allow myosin heads to cycle very slowly even in the presence of ATP.
The sensitivity of light-chain phosphorylation to Ca2+ depends on a parallel signaling pathway that partially inhibits myosin phosphatase, thus increasing the number of phosphorylated myosin cross-bridges and force at any given Ca2+ concentration (Fig. 39-21). Receptors coupled to trimeric G-proteins activate the small GTPase RhoA, which stimulates a protein kinase that inhibits myosin light-chain phosphatase. Malfunction of this Ca2+-sensitizing mechanism may contribute to some forms of high blood pressure, since in hypertensive animals, drugs that inhibit Rho-activated kinase relax smooth muscle and lower blood pressure.
Agarkova I, Perriard J-C. The M-band: An elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol. 2005;15:477-485.
Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206-213.
Bassel-Duby R, Olson EN. Role of calcineurin in striated muscle: Development, adaptation and disease. Biochem Biophys Res Comm. 2003;311:1133-1141.
Bolton TB, Prestwich SA, Zholos AV, Gordienko DV. Excitation-contraction coupling in gastrointestinal and other smooth muscles. Annu Rev Physiol. 1999;61:85-115.
Clark KA, McElhinny AS, Beckerle MC, Gregorio CC. Striated muscle cytoarchitecture: An intricate web of form and function. Annu Rev Cell Dev Biol. 2002;18:637-706.
Dalkilic I, Kunkel LM. Muscular dystrophies, genes to pathogenesis. Curr Opin Gen Dev. 2003;13:231-238.
Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: Old and new players. Nature Rev Mol Cell Biol. 2006;7:762-773.
Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82:945-980.
Fischer RS, Fowler VM. Tropomodulin: Life at the slow end. Trends Cell Biol. 2003;13:593-601.
Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann N Y Acad Sci. 1998;853:20-30.
Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu Rev Biochem. 1999;68:687-728.
Goody RS. The missing link in the muscle cross-bridge cycle. Nat Struct Biol. 2003;10:773-775.
Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853-924.
Horowits R. Nebulin regulation of actin filament lengths: New angles. Trends Cell Biol. 2005;15:121-124.
Kiriazis H, Kranias EG. Genetically engineered models with alterations in cardiac membrane calcium-handling proteins. Annu Rev Physiol. 2000;62:321-351.
McElhinny AS, Kazmierski ST, Labeit S, Gregorio CC. Nebulin, the nebulous, multifunctional giant of striated muscle. Trends Cardiovasc Med. 2003;13:195-201.
Olson EN, Williams RS. Remodeling muscles with calcineurin. Bioessays. 2000;22:510-519.
Pownall ME, Gustafsson MK, Emerson CPJr. Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol. 2002;18:747-783.
Severs NJ. The cardiac muscle cell. Bioessays. 2000;22:188-199.
Somlyo AP, Somlyo AV. Signal transduction by G-proteins, Rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin-II. J Physiol (Lond). 2000;522:177-185.
Squire JM, Morris EP. A new look at thin filament regulation in vertebrate striated muscle. FASEB J. 1998;12:761-771.
Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35-41.
Tskhovrebova L, Trinick J. Titin: Properties and family relationships. Nat Rev Mol Cell Biol. 2003;4:679-689.
Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69-98.