Multiple sclerosis
Demyelination
Axonal myelination is discussed in Chapter 5. The term demyelination refers to the loss of normally formed myelin and can be classified as primary or secondary:


It is important to distinguish demyelination (loss of structurally normal myelin) from dysmyelination in which the myelin sheath is not normally formed in the first place. Conditions characterized by dysmyelination are usually due to a metabolic abnormality or enzyme deficiency and are often inherited (Clinical Box 14.1).
 Clinical Box 14.1:
Clinical Box 14.1:


Clinical features of MS
Common symptoms
Although plaques can occur anywhere in the brain or spinal cord, including the central visual pathways, some sites are more likely to be affected than others. This means that certain symptoms and signs are more common (Fig. 14.1). The most frequently encountered presenting features are weakness in one or more limbs (40% of cases) and optic neuritis (up to 25% of cases; discussed below).
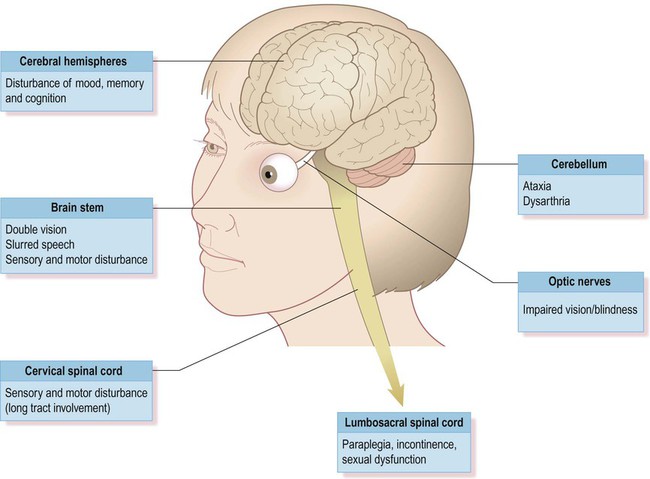
Loss of vision
Inflammatory demyelination of the optic nerve (termed optic neuritis) is common in MS. This causes blurred vision in one eye, with reduced light and colour perception, combined with retrobulbar pain (discomfort behind the affected eye, exacerbated by movement). In some cases there is a blind spot or scotoma (Greek: scotos, darkness). Symptoms usually resolve completely within a few weeks, but there may be a persistent afferent pupillary defect (Clinical Box 14.2). Optic neuritis can occur as an isolated phenomenon, but 75% of affected individuals will eventually develop multiple sclerosis. Another common visual problem in MS is discussed in Clinical Box 14.3.
 Clinical Box 14.2: Relative afferent pupillary defect
Clinical Box 14.2: Relative afferent pupillary defect
The pupillary light reflexes (see Ch. 3, Clinical Box 3.11) are often abnormal in patients with MS. Loss of myelin in the optic nerve may reduce the afferent drive to constrict the pupils when light is shone into the affected eye, causing a relative afferent pupillary defect (RAPD). This is demonstrated using the swinging light test. A pen torch shone into the normal eye causes strong constriction of both pupils. Quickly swinging the light across to the abnormal eye generates a weaker afferent drive to the pupil constrictor muscles (relative to the good eye) and both pupils paradoxically dilate slightly in response to the torch beam.
 Clinical Box 14.3: Internuclear ophthalmoplegia
Clinical Box 14.3: Internuclear ophthalmoplegia
This is an eye movement abnormality that is caused by a small brain stem lesion that interrupts the connection between the lateral gaze centre (close to the abducens nucleus, CN VI, in the pons) and the oculomotor nucleus (CN III, in the midbrain). Interruption of the medial longitudinal fasciculus (MLF) causes failure of adduction in the affected eye on attempted contralateral gaze, illustrated in Fig. 14.2. It is important to note that the figure is a schematic representation and that in reality the two fasciculi lie close to each other on either side of the midline. This means that it is possible for a single lesion to interrupt both sides at once, leading to failure of adduction in both eyes (on attempted left and right lateral gaze).
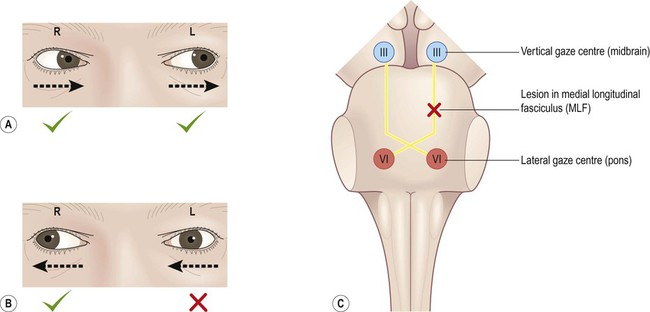
In this person left lateral gaze is normal (A) but on attempted right lateral gaze, the left eye fails to adduct (B). This is due to a lesion in the left MLF (C) which prevents the right lateral gaze centre from communicating with the opposite oculomotor nucleus (which it needs to do to recruit the medial rectus muscle).
Cognitive and emotional changes
Cognitive, emotional and behavioural changes occur in at least 40% of patients with MS. There may be subtle disturbances in frontal executive function (e.g. attention, working memory, decision-making; see Ch. 3) and a small proportion of patients develop more severe cognitive decline or even dementia (Ch. 12). Euphoria is often described, but depression is more common (seen in up to 50% of patients) and the risk of suicide is also increased. Psychotic features (delusions and hallucinations) are rare.
Cerebellar features
Involvement of the cerebellum or its connections with the brain stem may cause dysarthria (slurred speech), ataxia (incoordination) or nystagmus (a rhythmic abnormality of gaze fixation, with a frequency of 1–4 Hz, consisting of a slow drift phase and a brisk corrective ‘snap’). There may also be a cerebellar intention tremor. This is worse towards the end of deliberate or precise movements (in contrast to the ‘rest tremor’ of Parkinson’s disease; see Ch. 13).
Temperature sensitivity
Some MS symptoms are exacerbated (or clinically silent lesions unmasked) by an increase in body temperature. This can occur in a number of situations (e.g. a fever, hot bath or vigorous exercise) and is known as Uhthoff’s phenomenon. It is thought that increased temperature prolongs inactivation of voltage-gated sodium channels (see Ch. 6) and therefore increases the chance of conduction failure in partially myelinated or incompletely remyelinated axons.




Course and progression
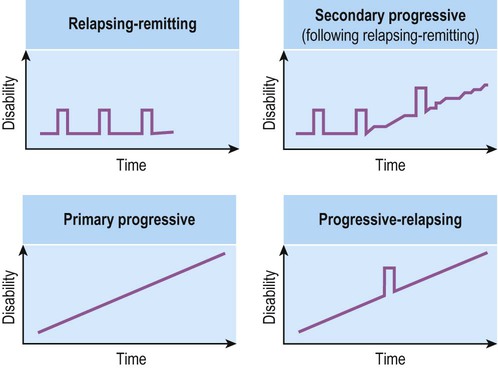
The course of MS is highly variable and difficult to predict in a particular individual. The main clinical patterns are illustrated in Figure 14.3 and discussed further below.



Diagnosis and management
Diagnosis
Neuroimaging
Demyelinating lesions are well-demonstrated on T2-weighted MRI scans, which highlight increased water content or decreased myelin (fat) content. However, since MS plaques tend to be periventricular, the T2 hyperintensity of normal CSF may make them more difficult to see. This is overcome using a fluid attenuation inversion recovery (FLAIR) sequence, which is similar to T2 but with a suppressed CSF signal (Fig. 14.4).
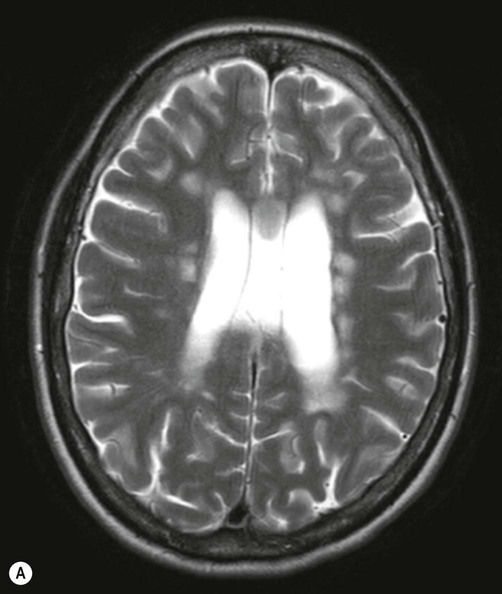
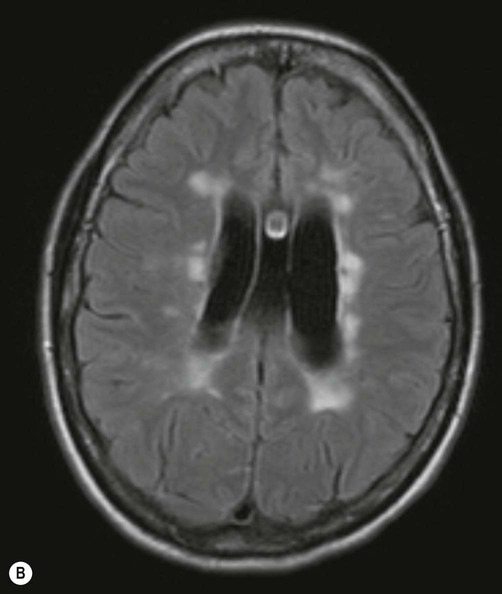
There are numerous plaque-like lesions in the periventricular white matter, best seen on the FLAIR sequence [see text for explanation]. Courtesy of Dr Andrew MacKinnon.
In patients with clinically definite multiple sclerosis, MRI shows multifocal white matter abnormalities in 95% of cases. Administration of the MRI contrast agent gadolinium is useful for demonstrating acute (active) lesions. This correlates with breakdown of the blood–brain barrier (see Ch. 5) in areas of active inflammation and demyelination.
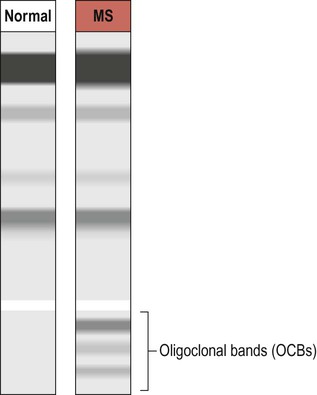
Oligoclonal bands
The CNS inflammatory response in multiple sclerosis is associated with synthesis of antibodies (immunoglobulins) in the brain and spinal cord. It is therefore possible to detect antibodies in the CSF that are not present in peripheral blood. A sample of CSF is obtained by lumbar puncture (see Ch. 1, Clinical Box 1.3) and a specimen of venous blood is taken at the same time, for comparison. The two specimens are run on an electrophoretic gel to look for bands indicating the presence of type G immunoglobulins (IgG) that are only present in the CSF (which is indicative of CNS inflammation). These are known as oligoclonal bands (OCBs) and are found in 90% of people with MS (Fig. 14.5).
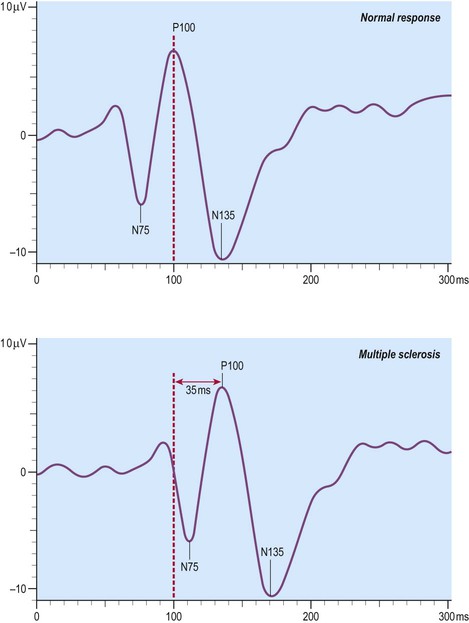
Visual evoked potentials
Decreased conduction speed in the central visual pathways can be demonstrated in the majority of patients with MS by obtaining visual evoked potentials (VEPs). Scalp electrodes record electrical activity in the occipital cortex in response to a changing visual stimulus such as an alternating chequerboard pattern. The stimulus-response sequence is repeated many times and averaged (to increase the signal-to-noise ratio). This reveals a characteristic positive wave in the visual cortex at 100 milliseconds (the P100 wave) which is delayed by 30–40 milliseconds in 95% of people with MS (Fig. 14.6).



Management
Disease-modifying drugs (DMDs)
Natalizumab
This is a monoclonal antibody (immunoglobulin G, IgG) which is given by intravenous injection every 28 days. Clinical trials show that it reduces the number of relapses by about two-thirds. Natalizumab recognizes an adhesion molecule called α4 integrin which binds to a vascular cell adhesion molecule (VCAM-1) on endothelial cells. This is designed to prevent leukocytes from binding to blood vessels, reducing the number of chronic inflammatory cells entering the CNS from the bloodstream. Side effects include headache, nausea, vomiting and skin rash. In rare cases it has been associated with an acute white matter disorder: progressive multifocal leukoencephalopathy (PML) (Clinical Box 14.4).
 Clinical Box 14.4:
Clinical Box 14.4:Long-term supportive care
 Neuropathic pain can be very troublesome and is managed with a number of agents including the anti-epileptic drugs gabapentin and carbamazepine (see Ch. 11).
Neuropathic pain can be very troublesome and is managed with a number of agents including the anti-epileptic drugs gabapentin and carbamazepine (see Ch. 11).
 Extreme tiredness often responds to the Parkinson’s disease drug amantadine (the stimulant effect is mediated by increased dopamine release at central synapses) (see Ch. 13).
Extreme tiredness often responds to the Parkinson’s disease drug amantadine (the stimulant effect is mediated by increased dopamine release at central synapses) (see Ch. 13).

 Muscle spasms, musculoskeletal pain and spasticity may be improved by physiotherapy or a muscle relaxant such as baclofen, gabapentin, dantrolene or a benzodiazepine (see Ch. 7).
Muscle spasms, musculoskeletal pain and spasticity may be improved by physiotherapy or a muscle relaxant such as baclofen, gabapentin, dantrolene or a benzodiazepine (see Ch. 7).


 Epilepsy is also more common in patients with MS and this can be managed with anti-epileptic drugs if necessary (Ch. 11).
Epilepsy is also more common in patients with MS and this can be managed with anti-epileptic drugs if necessary (Ch. 11).
Careful attention to these long-term problems may have a considerable impact on quality of life and co-operation between the neurologist, family doctor and specialist team members (including nurses, physiotherapists and occupational therapists) is highly beneficial.




Pathological features
MS plaques
In fresh post-mortem brain tissue, chronic plaques appear as sharply demarcated areas that lack normal myelin, giving them a salmon-pink appearance (Fig. 14.7). This is in contrast to acute plaques which have a yellowish colour due to the high lipid content. The presence of gliosis (‘glial scarring’; see Ch. 8) gives plaques a firm or sclerotic consistency, from which the name multiple sclerosis is derived (Greek: sklerōs, hard). After the brain has been preserved, plaques have a greyish appearance (Fig. 14.8).
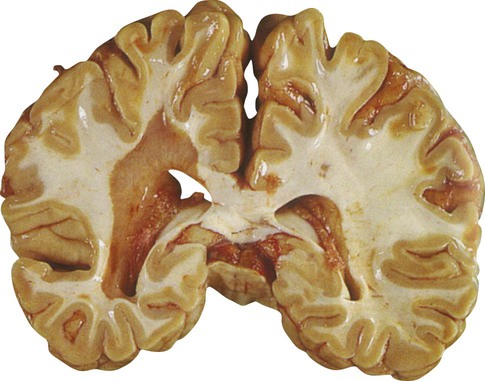
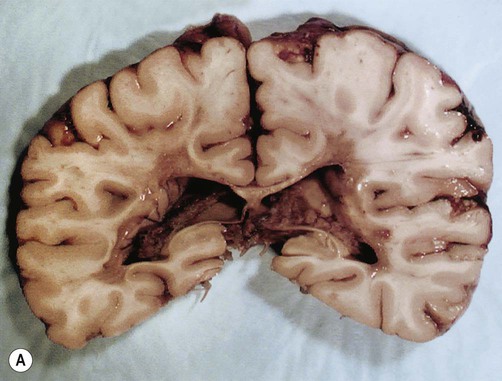
In this case the brain has been preserved in formaldehyde prior to slicing. From Prayson, R: Neuropathology (Chuchill Livingstone 2005) with permission.
Plaque distribution
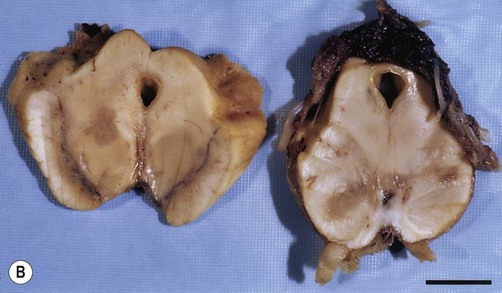
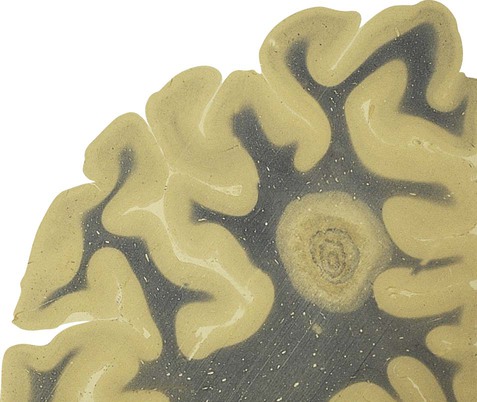
Plaques can occur anywhere in the brain or spinal cord, but they are most often found in the periventricular white matter. They are typically between 2–10 mm in diameter and have a well-defined margin (Fig. 14.9). Plaques also frequently occur at grey–white matter junctions (especially at the border between the cerebral cortex and subcortical white matter) and within the cortex itself, which also contains myelinated axons. In advanced cases extensive white matter loss may be associated with marked compensatory dilation of the ventricles, termed hydrocephalus ex vacuo. In a subset of patients, plaques are found only in the optic pathways and cervical spinal cord (Clinical Box 14.5).
 Clinical Box 14.5:
Clinical Box 14.5: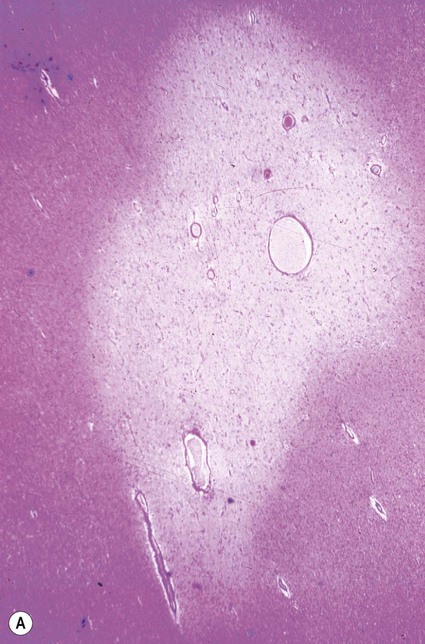
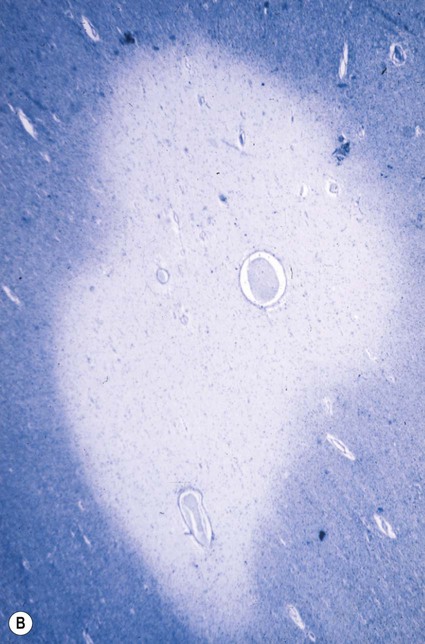
Note the sharp peripheral margin with the surrounding normal white matter. From Stevens, A, Lowe, JS and Young: Wheater’s Basic Histopathology 4e (Churchill Livingstone 2002) with permission.
Acute and chronic plaques
Chronic plaques may be active or inactive. Chronic active plaques have a cell-poor centre which is gliotic (with numerous astrocytic processes) but shows little inflammation. This is surrounded by a peripheral margin of activity at the interface with healthy myelin. The rim of active demyelination contains macrophages and lymphocytes. Chronic inactive plaques are similar, but lack the peripheral rim of demyelination. Some histological patterns are associated with a particular MS variant (Clinical Box 14.6).
 Clinical Box 14.6: Balo’s concentric sclerosis
Clinical Box 14.6: Balo’s concentric sclerosis
Balo’s concentric sclerosis is a histological variant of MS which used to be regarded as a distinct form of the disease. Histological examination of the brain shows sharply defined concentric rings of demyelination alternating with bands of unaffected myelin, giving an ‘onion-skin’ appearance (Fig. 14.10). This was previously only diagnosed at post-mortem examination and was thought to be associated with an invariably aggressive (usually fatal) clinical course. However, it is now possible to identify Balo’s disease in life using modern neuroimaging methods, which suggests that this pattern of demyelination is much more common and not necessarily aggressive.
The presence of multiple concentric rings of demyelinated and myelinated axons creates an ‘onion skin’ appearance [section of cerebral hemisphere; Loyez stain], in which myelin appears black. From Ellison, D and Love, S: Neuropathology 2e (Mosby 2003) with permission.
Remyelination
After the inflammation has subsided, approximately 20% of MS plaques remyelinate to form a ‘shadow plaque’ (Fig. 14.11). However, remyelination is often partial or inadequate, with thinner than normal myelin sheaths and shortened internodal segments.
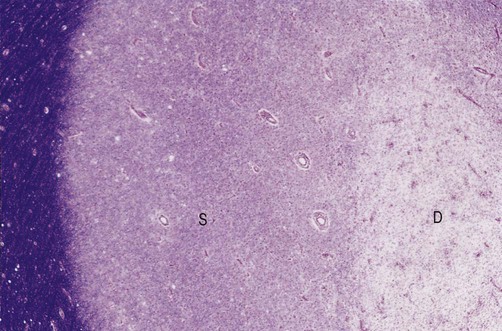
This section shows a shadow plaque (S) next to an area of active demyelination (D). Normal myelin (stained blue) can be seen at the far left-hand side of the image. From Ellison, D and Love, S: Neuropathology 2e (Mosby 2003) with permission.
Failure of remyelination
Remyelination may be prevented by on-going inflammation and continued demyelination. In older plaques, the presence of glial scarring (see Ch. 8) may also be a factor. This is characterized by a dense meshwork of astrocytic processes at the centre of the lesion which may physically prevent inward migration of oligodendrocyte precursor cells.




Aetiology
Environmental factors
The prevalence of multiple sclerosis varies with distance from the equator (Fig. 14.12). Equatorial regions tend to have comparatively low prevalence rates, whereas more temperate areas to the north and south have a progressively greater incidence. Some of the highest recorded rates of MS have been identified in the northern part of Scotland and in North America.
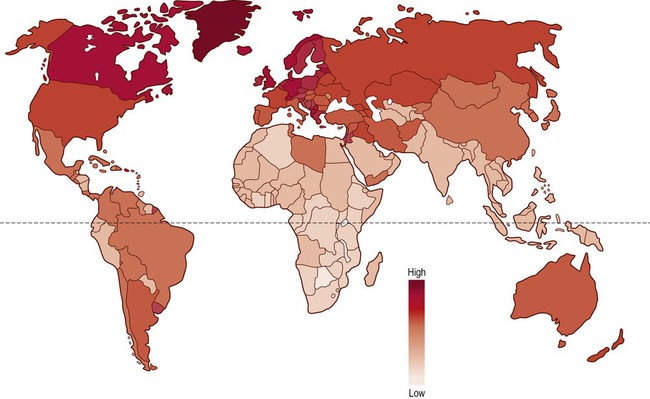
Individuals who are closer to the equator below the age of 15 years are at a lower risk of MS in adulthood, regardless of subsequent migration.
Migration studies
Geographic risk in MS relates to location before puberty (under the age of 15) and individuals who migrate after this age carry the risk of their original location. This may reflect exposure to an environmental agent (such as a virus) during a critical time-window prior to puberty. The time frame coincides with maximal development and involution of the thymus gland. This is the site of T-cell (thymus cell) maturation and the deletion of potentially auto-aggressive cells by apoptosis (see Ch. 8).
Viral and other infections
The bacterial agent Chlamydia pneumoniae has also been implicated – and bacterial infection is known to be associated with some cases of demyelination in the peripheral nervous system (Clinical Box 14.7). Nevertheless, no causative agent has been unequivocally implicated and no microorganisms have been isolated from human tissues.
 Clinical Box 14.7:
Clinical Box 14.7:



Animal models of MS




Autoimmune models
This induces a T-cell-mediated immune response (often in a rat or mouse, but sometimes in other species including non-human primates). This leads to brain and spinal cord inflammation with blood–brain barrier dysfunction, usually accompanied by demyelination. In particular, the immune response is mediated by helper T-cells, which is the same type of immune response seen in MS. This is accompanied by clinical signs and axonal changes, including conduction block. A criticism of this model is that since it produces a monophasic, post-vaccinial demyelinating disease, it has more in common with acute disseminated encephalomyelitis (Clinical Box 14.8).
 Clinical Box 14.8: Acute disseminated encephalomyelitis
Clinical Box 14.8: Acute disseminated encephalomyelitis
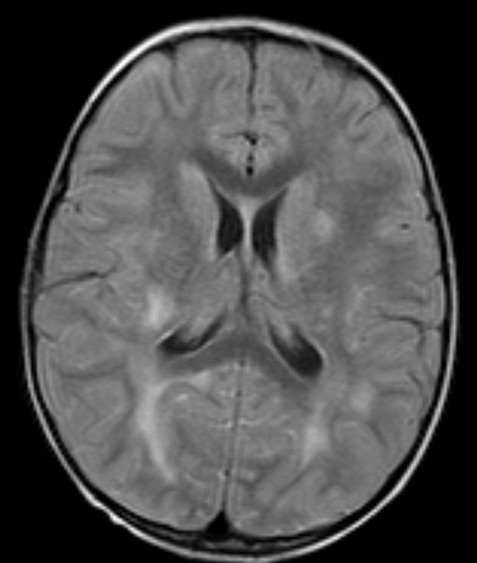
This is a self-limiting CNS disorder of childhood that presents with meningism (headache, fever, neck stiffness) and usually occurs 2–10 days after a virus infection or vaccine. Imaging shows bilateral hemispheric white matter changes corresponding to perivenous demyelination and oedema (Fig. 14.13). The clinical course is usually benign, with complete recovery. It is almost always monophasic. A hyperacute variant termed acute haemorrhagic leukoencephalopathy (AHL) or Hurst disease is characterized by widespread haemorrhagic lesions with brisk inflammatory infiltrates and marked brain oedema. This subtype is much more aggressive and usually fatal.
Viral models
A number of CNS viruses are known to cause demyelination in humans (such as JC virus in PML; see Clinical Box 14.4). Several animal models have been developed that exploit this phenomenon. Many of the viruses used in these models infect oligodendrocytes and are either directly cytotoxic or induce a cell-mediated immune response against oligodendrocytes.
Genetic models
A number of models are available in which the experimental animal (usually a rodent) has a myelin gene mutation. These include animals with mutations in genes for myelin basic protein (e.g. Shiverer) and proteolipid protein (e.g. Rumpshaker and Jimpy). Gene knockout animals for myelin components also exist. However, animals with myelin gene mutations typically exhibit abnormal primary myelination (dysmyelination) and are thus more comparable to human leukodystrophies (see Clinical Box 14.1).


Pathogenesis
Our understanding of disease pathogenesis in MS is derived from a combination of findings in animal models and observations in human disease. Gadolinium-enhanced MRI scans in patients with MS suggest that disruption of the blood–brain barrier (see Ch. 5) may be an early event in acute lesions. Pathological studies confirm increased permeability of cerebral vessels, associated with immune activation of endothelial cells and passage of leukocytes from the bloodstream into the brain tissue.
Inflammatory cells
Macrophages
Macrophages (and activated microglia, which are of similar lineage) are the principal cellular mediators of demyelination. These cells actively strip axons of their myelin sheaths and digest its lipid and protein constituents. Myelin debris accumulates within activated macrophages to give a ‘foamy’ appearance (Fig. 14.14).
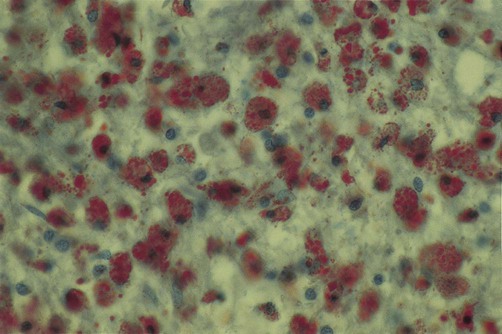
T-lymphocytes
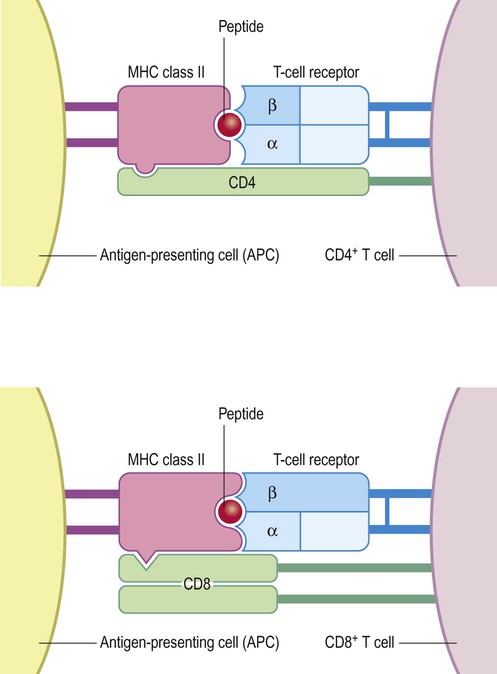
Active multiple sclerosis lesions are rich in T-lymphocytes. These can be divided into: (i) helper T-cells which express the cell-surface molecule CD4; and (ii) cytotoxic T-cells which express CD8. The CD4 and CD8 molecules help T-lymphocytes to recognize myelin constituents that have been processed and ‘presented’ to them by antigen-presenting cells (APCs) including activated microglia and macrophages (Fig. 14.15).




Impact on axonal conduction
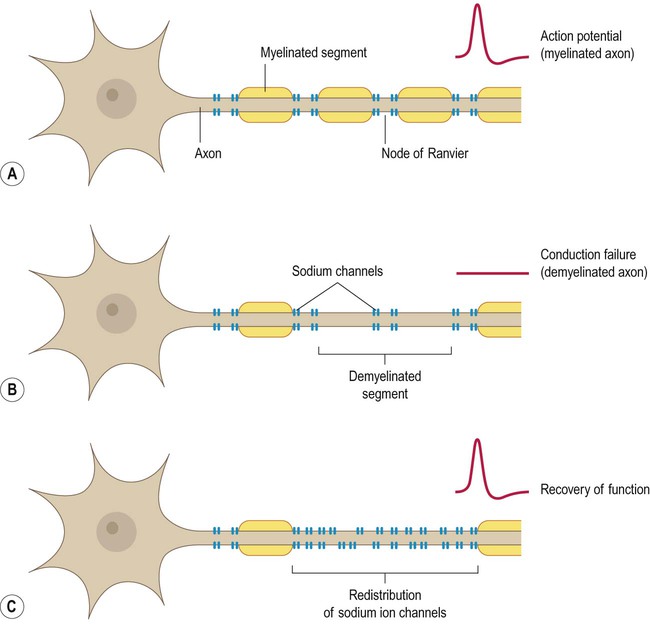
In myelinated fibres, voltage-gated ion channels are concentrated at the nodes of Ranvier (Fig. 14.16A). This means that following demyelination, the denuded portion of the axon is unable to transmit action potentials, leading to conduction block (Fig. 14.16B). Partial recovery of function may be possible due to redistribution or insertion of new voltage-gated sodium channels along the internodal region (Fig. 14.16C). However, this permits only continuous (rather than saltatory) conduction, which is considerably slower and much less energy-efficient (see Ch. 6).
Types of active plaque
Studies of post-mortem brain tissue (and diagnostic brain biopsies in living patients) have provided evidence of four distinct patterns in early (active) MS plaques, each with a different pathophysiological mechanism (Fig. 14.17). In a particular patient, all plaques are of the same type.
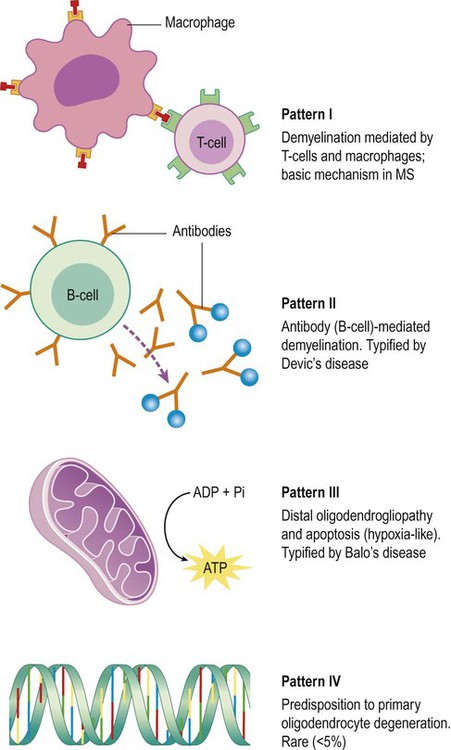
The basic mechanism in multiple sclerosis (pattern I) is characterized by a T-cell-mediated immune response with macrophage-associated demyelination. This accounts for around 20% of MS cases. In many patients, the basic T-cell response is supplemented by a specific antibody and complement-mediated assault on CNS myelin (pattern II). This appears to be the most common type overall, accounting for more than 50% of cases. It is typified by Devic’s disease and in this particular case the antibodies are raised against the water channel aquaporin-4 (discussed above; see Clinical Box 14.5).
Pattern III is referred to as a distal oligodendrogliopathy because the target is the distal (para-axonal) loop of myelin that lies in intimate contact with the axon (see Fig. 14.18); the oligodendrocyte cell body remains intact, but there is a ‘dying back’ of its processes, with consequent demyelination. There is also evidence of mitochondrial dysfunction in this type of plaque, which has features in common with hypoxic or ischaemic cell death (see Ch. 10). Pattern III is typified by Balo’s disease in which multiple waves of hypoxic-type damage create an onion skin appearance (see Clinical Box 14.6) and accounts for approximately 25% of lesions overall.
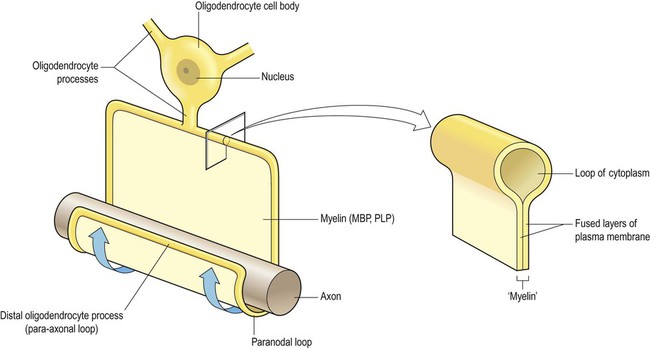
The distal oligodendrocyte process (or para-axonal loop) is indicated. This is the target in ‘dying-back’ oligodendrogliopathy, the characteristic finding in pattern III multiple sclerosis plaques.




Neurodegeneration in MS
In the progressive phase of MS, there is gradual white matter atrophy, with ventricular dilatation and thinning of the cerebral cortex (Fig. 14.19). This is associated with accumulation of permanent neurological disability and is thought to be due to axonal damage and loss of cortical neurons.
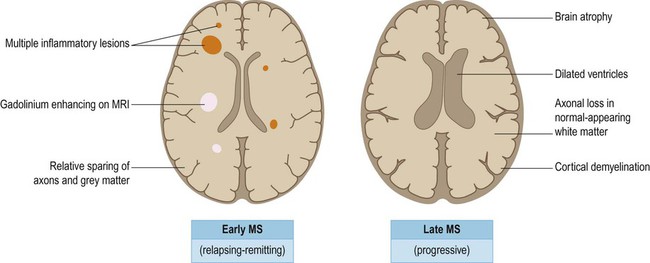
In most patients, the early stages of MS are characterized by a relatively benign relapsing-remitting course with episodes of acute inflammatory demyelination and discrete clinical relapses, often with very good functional recovery. Most patients convert to a progressive phase of neurological decline, characterized by axonal loss (even in the normal-appearing white matter), together with plaques and neuronal loss in the cerebral cortex. The degenerative phase of MS is associated with brain atrophy and dilation of the ventricles.
Axonal damage
Disrupted axons (axon swellings) can be demonstrated using silver stains or by immunohistochemistry for axonal proteins such as neurofilament protein. Immunolabelling for beta amyloid precursor protein (β-APP) is a sensitive method for demonstrating axonal disruption, which is also used in traumatic head injury (see Ch. 9).
Mechanism of axonal injury
Glutamate-mediated excitotoxicity (see Ch. 8) has also been implicated, with axonal calcium overload as a final common pathway. In experimental models of inflammation, axonal degeneration can be triggered by nitric oxide (which is known to be present in MS plaques and is generated in response to excess glutamatergic stimulation). In addition, the presence of antineuronal antibodies in the CSF of patients with MS raises the possibility of more direct, immune-mediated axonal damage.
Loss of demyelinated axons
Metabolically active demyelinated axons appear to be most at risk and energy depletion is considered to be an important factor. This may be due in part to redistribution of sodium channels along the internodal segments as a result of demyelination (see Fig. 14.16). This permits impulse conduction across the denuded axonal segments, but makes axonal conduction less efficient and increases axonal energy demands.
Cortical demyelination
The cerebral cortex also contains myelinated axons and cortical plaques are present in the majority of patients with MS (Fig. 14.20). Pathological studies in longstanding MS show that more than 25% of the cortex may contain plaques, which are of three types:
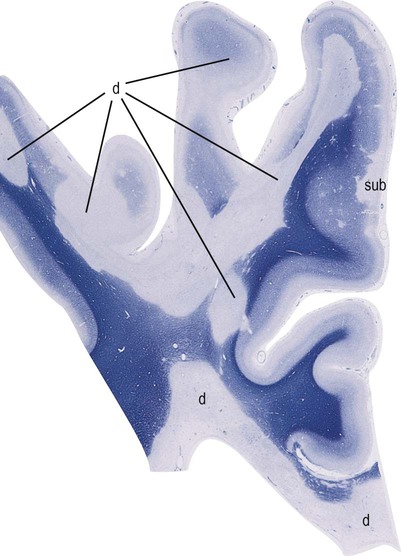
This image shows numerous plaques of demyelination (labelled “d”) at the cortical–subcortical junction, within the cerebral cortex and in the subcortical white matter. A prominent area of subpial demyelination (labelled “sub”) is also indicated. [Micrograph of the cerebral hemisphere; the tissue has been stained with Luxol fast blue, which is used to demonstrate myelin]. From Ellison, D and Love, S: Neuropathology 2e (Mosby 2003) with permission.



Cortical plaques are not associated with chronic inflammation, which makes them less obvious in pathological preparations. They are also difficult to identify using clinical imaging because there is no associated compromise of the blood–brain barrier (so they do not appear on T2-weighted MRI scans or with gadolinium contrast enhancement).



