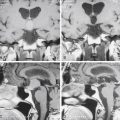
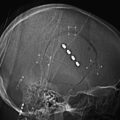
Chapter 16 Multimodal Assessment of Pituitary and Parasellar Lesions
Background and Epidemiology
The most common lesion encountered in the sella will be a pituitary adenoma. Estimates of the prevalence of pituitary adenomas vary widely between studies, which are performed using either MRI or autopsy findings. A recent meta-analysis found a prevalence of 22.5% in imaging studies (range 1%–40%) and 14.4% in postmortem studies (range 1%–35%), for an overall prevalence of 16.7%.1 The vast majority of pituitary adenomas are microadenomas (diameter <1 cm). Macroadenomas are far less common, and their prevalence in the population is estimated to be approximately 0.2%.1 Macroadenomas make up a much larger proportion of adenomas that come to clinical attention, however. A cross-sectional study of 81,149 patients in Banbury, UK found the overall prevalence of known pituitary tumors to be 77.6 cases per 100,000 inhabitants (or 63 total cases). Of these, only 7 were found incidentally. The others presented with some sort of clinical symptomatology, and of those, 41% were macroadenomas.2
The prevalence of “functional tumors”—adenomas that hypersecrete hormones resulting in a clinical syndrome—is in the range of 50% to 60% of all clinically apparent pituitary tumors. The most common functional or secretory tumors are prolactinomas (approximately 40%), followed by ACTH-secreting tumors (14%), growth hormone (GH)–secreting tumors (5%), and TSH-secreting tumors and mixed tumors (both under 1%).3 Many “nonfunctional” tumors are clinically silent gonadotroph adenomas, while a small proportion of these produce but do not secrete ACTH, GH, or TSH.
The majority of pituitary tumors remain stable after their initial detection. In one meta-analysis of eight studies including 144 patients who were followed from 2 to 8 years, 84% of microadenomas of the pituitary gland remained stable in size. A few (6%) regressed and approximately 10% increased in size. In contrast, 20% of macroadenomas grew (11% of those were due to apoplexy), 11% diminished in size, and 69% remained stable.4
Pathogenesis
A great deal has been learned about the pathogenesis of some pituitary lesions but the pathogenesis of the majority of pituitary and parasellar lesions is still unknown. It is clear there are a variety of molecular mechanisms responsible for neoplastic transformation. Pituitary adenomas arise from adenohypophysial cells of the anterior pituitary. The vast majority of pituitary tumors are thought to be benign. Adenomas can, however invade local structures including the dura of the sella, local bony structures, and the sphenoid sinus cavity. Pituitary carcinoma, defined by the presence of local discontinuous or systemic spread, is extremely rare and probably represents approximately 0.2% to 0.5% of all pituitary lesions.5 Pituitary neoplasms are monoclonal, and outside of defined genetic syndromes, their cause is not well understood.
At least five genes have been identified as causes of pituitary adenomas with four of them comprising familial pituitary tumor syndromes: MEN1, PRKAR1A, CDKN1B, and AIP. Genetic disorders such as MEN1 (a mutation in the MENIN protein that reverses function of tumor suppressor gene), and Carney’s complex (PRKAR1A, insufficient activity of protein kinase-A regulatory subunit 1) are examples of models for pituitary tumor development. The CDKN1B gene causes a MEN-like syndrome (MEN4) associated with hyperparathyroidism and other rare tumors.6 Another recently discovered mutation is located in the AIP (aryl hydrocarbon receptor–interacting protein) gene (11q13).7,8 This appears to act as a tumor suppressor gene and mutations predispose to GH-secreting pituitary tumors.7,9,10 Approximately 40% of sporadic GH-secreting adenomas are associated with a somatic mutation in the Gs alpha gene.11,12 The mutation results in constitutive activation of the cAMP/PKA signal transduction pathway and leads to neoplastic transformation of somatotroph cells of the pituitary.13,14
Laboratory Investigations
The most appropriate screening tests for an incidentally discovered pituitary lesion are the topic of much debate.15,16 A survey of endocrinologists in the United States and United Kingdom suggested that, when confronted with a microadenoma, a range of 0 to 16 tests would be ordered (median 7).16,17 Most experts would advocate screening for both pituitary insufficiency and hormone excess as both states are relevant in the initial decision-making and also the long term follow-up of patients. This information is particularly important in the context of a preoperative evaluation. Clinical findings should guide medical decision-making. For example, patients who are obviously Cushingoid will require specific testing of adrenal function that may not be conducted in those who appear to have acromegaly.
Hypopituitarism
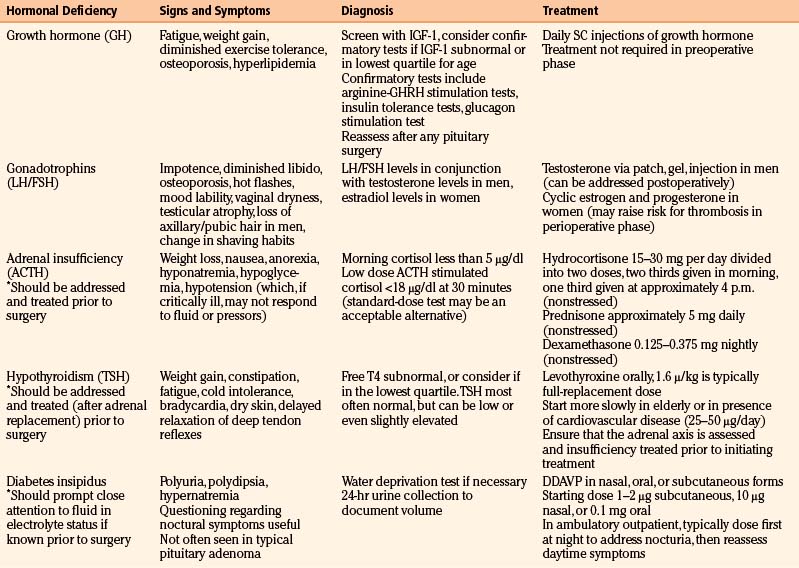
One or more pituitary deficiencies will be present in 70% to 90% of patients with macroadenomas.18 Microadenomas rarely cause pituitary insufficiency. Other tumors in the area of the sella, including tuberculum sellae and cavernous sinus meningiomas, craniopharyngiomas, and Rathke’s cleft cysts may also cause pituitary insufficiency. A uniform approach to screening for pituitary dysfunction is useful since symptoms and signs can be vague as many patients have adapted over time to a state of chronic pituitary dysfunction and may not perceive their symptoms as abnormal. Pituitary deficiency tends to manifest in the following order of frequency: loss of GH, followed by gonadotrophins, followed by loss of adrenal and then thyroid function.4 Diabetes insipidus is the least common deficiency in a typical pituitary adenoma. A typical screening panel therefore will include the following, with interpretation discussed below: serum IGF-1, estradiol, or testosterone levels (depending on sex) in conjunction with an FSH and LH level, TSH and free T4, and a cortisol stimulation test. A prolactin level should always be drawn as well and is discussed in the section on pituitary hypersecretion. Table 16-1 summarizes typical signs and symptoms of pituitary insufficiency.
Central Hypothyroidism
It is critical to ensure adequate adrenal function (see below) prior to the initiation of thyroid hormone replacement. Thyroid hormone administration will increase metabolic demand for cortisol while at the same time increasing its clearance, and might precipitate overt adrenal insufficiency in a patient deficient in cortisol. Chronic replacement can be accomplished in most adults with approximately 1.6 μg of levothyroxine per kilogram lean body weight per day. In the elderly, or those with significant cardiac morbidities, most experts recommend initiating levothyroxine replacement with 25 to 50 μg daily. To address severe hypothyroidism in the preoperative setting, in the absence of contraindications, 200 to 400 μg of levothyroxine may be administered IV, followed by oral administration of a usual replacement dose. When patients are unable to take their medication orally the intravenous administration of 85% of a typical oral dose daily until the patient can take oral medications will meet their needs. There is no clear evidence that T3 replacement is beneficial.
Central Adrenal Insufficiency
Undiagnosed central adrenal insufficiency represents the most significant danger to the patient with hypopituitarism. The best test to assess the integrity of pituitary-adrenal function has been the subject of significant controversy. While insulin-induced hypoglycemia is the gold standard, the procedure is time consuming and sometimes risky. However, stimulation with ACTH 1-24 best combines high accuracy with practicality and is felt to best correlate with the results of an insulin tolerance test.19,20 There is debate on whether one should conduct this test with 1 or 250 μg of ACTH 1-24. One caveat of the ACTH 1-24 stimulation test is its accuracy is limited until about 6 weeks following a pituitary insult; despite the limitations of the cosyntropin-stimulation tests, there is little evidence that clinically significant adrenal insufficiency will be missed by either test.21
Oral regimens for adrenal hormone replacement in a non-stressed patient include hydrocortisone 15 to 30 mg daily, usually in divided doses, dexamethasone 0.25 mg at bedtime, or prednisone 5 mg daily.22 Adjustments in dosing are based on clinical symptoms and signs of either glucocorticoid excess or deficiency, as there is, at this point, no reliable, reproducible laboratory test to assess the adequacy of treatment. The HPA axis is usually reassessed 6 to 8 weeks after surgery to determine if ongoing chronic treatment is necessary. Many patients will experience improvement in pituitary function after surgery and their glucocorticoid replacement may be abruptly discontinued. Patients with Cushing’s disease may experience a period of temporary adrenal insufficiency of 6 to 18 months due to suppression of normal corticotroph cells by pre-existing hypercortisolism. They must be reassessed at regular intervals to determine the need for ongoing steroid replacement.
Treatment with replacement doses of glucocorticoids is indicated in all patients with documented central adrenal insufficiency. Empiric treatment prior to surgery is acceptable in the setting of pituitary apoplexy, especially in the setting of large space occupying lesions, and when an urgent or emergent procedure is indicated even in the absence of documented adrenal dysfunction. A reasonable therapeutic regimen when the patient is under significant physiological stress is 25 mg of intravenous hydrocortisone every 8 hours. In a crisis, one may want to initiate intravenous glucocorticoid replacement with 100 mg initially and 50 mg every 8 hours thereafter. Mineralocorticoid replacement is not necessary in secondary adrenal insufficiency.23
Pituitary Hypersecretion
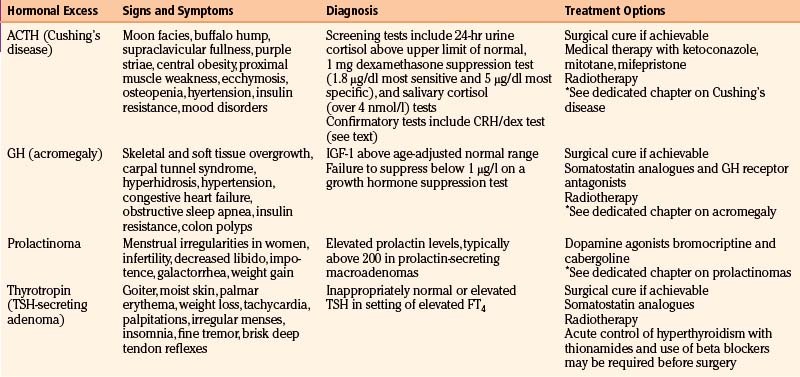
Pituitary adenomas can secrete hormones in excess resulting in particular clinical syndromes. Other sellar and parasellar tumors will not present in this fashion, although mild prolactin elevations can be seen with many large tumors due to compression of the infundibulum. The major hypersecretory states are discussed individually in other chapters, but all should be considered in the initial evaluation of a pituitary mass. Table 16-2 summarizes some of the possible signs and symptoms associated with various hypersecretory states.
Prolactinoma
Prolactinomas represent approximately 40% of pituitary tumors.1 They are more common in women with a gender ratio of 10:1.24 Women usually present when their tumors are microadenomas. Men are more likely to present with macroadenomas. Whether this is function of delayed presentation (women may present quickly with menstrual irregularities) or of unique biological properties of tumors in men is unclear.25
A serum prolactin should be obtained in all patients with pituitary tumors, as this information will affect treatment decisions. Prolactin levels of greater than 200 μg/l are almost always associated with a prolactin-secreting macroadenoma. Prolactin levels less than 200 μg/l can be caused by microprolactinomas and also a variety of parasellar lesions (pseudoprolactinoma), infiltrative processes, as well as physiologic and pharmacologic causes.24 Any tumor or infiltrative process that interferes with the delivery of dopamine, which inhibits prolactin production, to the normal lactotrophs can cause hyperprolactinemia that is often referred to as “stalk-effect hyperprolactinemia.” Pregnancy should be excluded in any woman with hyperprolactinemia and amenorrhea. A variety of drugs can cause hyperprolactinemia. If medications are suspected as a cause of hyperprolactinemia, they should be discontinued if possible, and the prolactin level reevaluated 1 month thereafter. Severe primary hypothyroidism can cause hyperprolactinemia. Thus, a serum TSH level is valuable during an initial evaluation of hyperprolactinemic patients.
The clinician must be aware that the differential diagnosis for hyperprolactinemia is large encompassing pathological and physiological causes. Defining the cause of hyperprolactinemia is crucial since true prolactinomas are treated medically with dopamine agonists, pseudo-prolactinomas are surgically removed and other causes have their own unique treatments. Table 16-3 presents a summary of the differential diagnosis of hyperprolactinemia.
| Physiologic |
Source: Adapted from Mancini T, Casanueva FF, Giustina A. Hyperprolactinemia and prolactinomas. Endocrinol Metab Clin North Am. 2008;37:67-99, viii.
A small subset of extremely large prolactinomas can produce enormous amounts of prolactin that overwhelm antibodies used in the assay resulting in a false lowering of prolactin levels.26 This is known as the “hook effect.” In the setting of a macroadenoma, when a prolactinoma is suspected, prolactin levels should be performed on diluted serum samples to avoid this error in laboratory diagnosis. Most modern radioimmunoassays are able to detect prolactin levels as great as 4000 μg/l without being subject to the “hook effect.” We recommend that treating physicians being aware of the prolactin test performance in the laboratories they employ to evaluate their patients.27
On occasion a patient will be found to have an elevated prolactin but without symptoms of hyperprolactinemia. This is often due to a condition known as macroprolactinemia. In this disorder, prolactin aggregates with circulating IgG antibodies resulting in decreased clearance of the complex and thus elevated prolactin levels.28–30 However, the prolactin-IgG complex is devoid of biological activity and thus the absence of symptoms. This condition does not require treatment and needs to be distinguished from true hyperprolactinemia. Incubating the serum with polyethylene glycol, which removes the prolactin-IgG complex prior to performing the assay, can identify the phenomenon.
Cushing’s Disease
Recent guidelines review proposed diagnostic approaches to the evaluation of patients with suspected Cushing’s syndrome.31 The inclusion of an endocrinologist experienced with this disorder to establish and confirm the diagnosis is strongly advised. Generally speaking, and depending on the clinical situation, one should start with screening tests, then proceed to diagnostic or confirmatory tests, and finally, to differential diagnostic tests.
There are at least three reliable screening tests to evaluate patients with suspected hypercortisolism. Keep in mind that screening tests are designed to be sensitive but not specific. Thus, there will be false positive tests and not everyone with a positive screening test will be ultimately diagnosed with pathologic hypercortisolism. Appropriate screening tests include the overnight 1 mg dexamethasone suppression test, the 24-hour urine collection for free cortisol and creatinine, and the midnight salivary cortisol collection.32–36 For most patients who require screening only, performance of one of these tests is indicated. The salivary cortisol and 24-hour urine free cortisol collection should be done at least twice to confirm abnormal initial findings. A positive dexamethasone suppression test is identified when an 8 a.m. cortisol is greater than 1.8 μg/dl following administration of 1 mg of dexamethasone at 11 p.m. the prior night. Using this cutoff gives the test a sensitivity of approximately 95% with specificity of 80%. To enhance specificity to over 95%, a cutoff of 5 μg/dl may be used, which will sacrifice sensitivity (falling to 85%).31 A 24-hour urine cortisol above the upper limits of normal may be considered a positive screening test, along with a late-night salivary cortisol level of 4 nmol/l or greater.31
Diagnostic tests are designed to balance sensitivity and specificity. They are used to confirm the diagnosis of pathologic hypercortisolism. Diagnostic tests for hypercortisolism include the 24-hour urine cortisol, the dexamethasone suppressed CRH stimulation test, (CRH/Dex test) and the formal low-dose dexamethasone suppression test.37,38 A 24-hour urine cortisol excretion rates greater than two to three times the upper limit of normal is generally considered to be a positive test. The CRH/Dex test is performed by administering dexamethasone, 0.5 mg every 6 hours for 2 days followed by CRH stimulation on the morning of the third day. A cortisol value greater than 1.4 μg/dl 15 minutes after CRH administration indicates an abnormal result.39 Treating physicians are encouraged to review the performance of tests employed by their laboratory and draw upon their own experiences to determine what constitutes an abnormal response to a formal dexamethasone suppression test.
Inferior Petrosal Sinus Sampling
Inferior petrosal sinus sampling is a procedure that may be required to confirm the source of ACTH-dependent Cushing’s syndrome. Lab studies can often differentiate ectopic from pituitary ACTH-driven Cushing’s syndrome, but it is clear that the levels, even on dynamic testing, may have some overlap. IPSS may be particularly useful when the lab evaluation suggests a pituitary source for ACTH but none can be identified by MRI scanning. Studies have varied in their estimation of the sensitivity and specificity of the test. Earlier studies have shown it to be very sensitive (96%) and specific (100%), but there have been recent studies questioning these estimates.40,41 It is clear that the experience of the practitioner has an effect on the utility of the test, and even some large medical centers may not be able to perform the procedure reliably.
The technique is performed by cannulation of the bilateral femoral veins, and advancing catheters into the inferior petrosal sinuses via the internal jugular veins. Baseline ACTH values are obtained from the periphery and the inferior petrosal sinuses both before and after CRH stimulation (1 μg/kg of body weight). A ratio of 2.0 from the petrosal sinus to the periphery before CRH stimulation, or 3.0 afterward is felt to be consistent with pituitary Cushing’s disease. In ectopic disease, the ratio is typically less than 2.0 both before and after CRH stimulation.42
False negative tests have been described in several situations. Ectopic pituitary tumors can render it inaccurate, as can hypoplastic or anomalous inferior petrosal sinuses. While IPSS results can lateralize, those findings may be misleading and are not highly specific. One study suggested that a gradient of 1.4 across sides was able to predict tumor location 78% of the time.42
Acromegaly
Growth hormone–secreting pituitary tumors account for 95% of acromegaly. Rarely, ectopic GH- or GH-releasing-hormone (GHRH)–secreting tumors are a cause. Most pituitary tumors are macroadenomas at the time of diagnosis. Approximately one third of tumors secrete additional pituitary hormones such as prolactin or TSH.43
In most patients, screening with an IGF-1 level is sufficient to establish the diagnosis. IGF-1 levels are reported as age-adjusted and attention should be paid to where the level falls within the normal range. When the IGF-1 levels are slightly elevated, an oral glucose suppression test is indicated. This test is performed by the oral administration of 75 grams of glucose and the measurement of serum GH levels every 30 minutes for 2 hours. Using the modern ultrasensitive assay, a normal individual will suppress GH levels to below 1 μg/l.44
Hyperthyroidism
TSH-secreting adenomas are uncommon and account for less than 1% of all pituitary adenomas. These aggressive tumors usually present as macroadenomas with a goiter and clinical features of hyperthyroidism. Generally, the free T4 and T3 levels are elevated in the setting of an inappropriately normal or elevated serum TSH concentration. TSH-secreting tumors often co-secrete other hormones; therefore measurement of PRL, GH and alpha subunit should be performed.45,46 The alpha subunit can be useful in distinguishing TSH-secreting pituitary adenomas from thyroid hormone resistance (either pituitary, peripheral or generalized types). A suggested method to confirm the presence of a TSH-secreting tumor is to calculate the molar ratio of alpha subunit to TSH: (alpha subunit in nmol/l)/(serum TSH in μIU/l) × 10. A ratio exceeding 1.0 is found in 80% of patients with TSH-secreting adenomas.45 Preoperative control of the hyperthyroidism is necessary and can be achieved with somatostatin analogs or the brief use of antithyroid medication, possibly in combination with beta-blockade.
Radiologic Studies
The differential diagnosis of sellar and parasellar neoplasms is quite large (Table 16-4). In many cases, an accurate diagnosis may be arrived at based on clinical symptoms and signs, and specific radiographic features. Occasionally, however, sellar lesions mimic one another and the actual diagnosis may only be resolved by histologic examination of resected tissue. Magnetic resonance imaging (MRI) is the primary modality employed in imaging of sellar and parasellar neoplasms. A typical MRI protocol calls for sagittal and coronal imaging, with thin-sectioned (2-3 mm) T1-weighted images with and without contrast. Some disease processes have very well defined characteristics on MRI that permit their identification prior to surgery. Computed tomography has a role in the evaluation for calcifications within the lesion and in defining the integrity of the parasellar bony anatomy. It may also be acutely useful in suspected apoplexy to identify hemorrhage.
| Type of Lesion | MRI/CT Findings | Clinical Clues |
|---|---|---|
| Physiologic enlargement of the pituitary | Larger than average gland without discrete tumor seen |
Source: Adapted from Kaltsas GA, Nomikos P, Kontogeorgos G, Buchfelder M, Grossman AB. Clinical review: diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. 2005;90:3089-3099.
Pituitary Hyperplasia
The normal pituitary gland is 9 to 10 mm in greatest dimension. It is rectangular on coronal section and somewhat semilunar on sagittal section. Physiologic enlargement of the pituitary gland, as can be seen in young menstruant women, and hyperplasia that can be seen during pregnancy, in the setting of primary hypothyroidism, primary adrenal insufficiency, or hypogonadism, can lead to a trapezoidal shape of the gland on coronal sections and a globular shape on sagittal sections. These entities should be considered prior to assuming that pituitary enlargement represents a pituitary tumor.4,47
Pituitary Adenomas
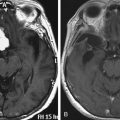
Microadenomas of the pituitary gland typically show diminished enhancement on postcontrast T1 images through the sella. The normal pituitary will appear hyperintense in relationship to the tumor. The pituitary stalk will often deviate laterally towards the contralateral side from the tumor on coronal imaging. Occasionally, however, postcontrasted images fail to delineate the tumor. In this setting, and when there is obvious evidence for a syndrome of hormone hypersecretion, serial dynamic contrast-enhanced images often delineate a microadenoma. Macroadenomas of the gland are obvious. On occasion, these lesions are hypodense. The normal pituitary is often displaced laterally or superiorly and will enhance. Occasionally, tumors invade the cavernous sinuses or extend inferiorly into the sphenoid sinuses, inferiorly and laterally into the pterygopalatine fossa and related spaces, or extend superiorly into the suprasellar cistern and elevate the optic chiasm. Magnetic resonance imaging provides for excellent definition of the limits of growth of a pituitary macroadenoma in most cases.
The syndrome of pituitary apoplexy can result from either hemorrhage or infarction of a pre-existing pituitary tumor. It has some unique presenting characteristics that are quite different from other pathology in the region. Early during the initial event, CT may be helpful, but if not directed at the sella details may be missed. MRI of the hemorrhage in the first few days will be hyperintense on T1 weighted imaging and hypointense on T2. As time progresses, hemorrhage will be hyperintense on T1 and T2 weighted imaging as hemoglobin degrades to methemoglobin, and fluid levels may begin to be seen as sedimentation begins. In the case of visual loss, more rapid surgical intervention may lead to better outcomes.48
Pituitary Cystic Lesions
Some pituitary adenomas undergo degenerative change and may present as predominately cystic or solid and cystic lesions within the sella. The cyst fluid is usually hypointense on T1 images with or without contrast. Cystic adenomas may be hyperintense on T2 imaging. Occasionally, hemorrhage is seen within pituitary tumors. Blood products are usually hyperintense on T1 images. Primary pituitary cysts, such as pars intermedia cysts and Rathke’s cleft cysts are usually hypointense on T1 imaging. The latter lesions, however, may mimic a pituitary tumor. These lesions may not be hyperintense on T2 imaging if the fluid is very proteinaceous. Arachnoid cysts are most commonly suprasellar, but can be intrasellar and are often hypointense and distort the sellar contents and displace the pituitary stalk posteriorly.47,49 Patients who have primary or secondary empty sella often have a small gland, usually situated in the floor of the sella, and the pituitary stalk traverses the empty sella as it passes to the residual or remnant gland. In many patients, the stalk is straight and tapered and has the appearance of a sword leading to the well known “sword sign” taken to confirm the presence of an empty sella and exclude the diagnosis of an intrasellar cyst.
Craniopharyngiomas
Craniopharyngiomas are epithelial tumors that arise along the path of the craniopharyngeal duct. They are the most common lesions of the parasellar area in children. There is a bimodal age distribution, with peak prevalence seen in children 5 to 14 years old and adults 50 to 74 years old.50 They can have imaging characteristics in common with cystic pituitary lesions. CT scanning may be useful when a craniopharyngioma is suspected, and the presence of calcium within the lesion on CT is highly suggestive, but is only seen in 50% of such tumors. The tumors may be solid, cystic, or both. The solid component will often enhance with contrast on T1 weighted MRI imaging and the cystic component will usually be hyperintense on T2 weighted imaging.51 Craniopharyngiomas may also be located in the region of the pituitary stalk or situated with an epicenter in the hypothalamus. The majority will be between 2 and 4 cm in diameter, but they can be bigger.50 Suprasellar craniopharyngiomas may result in mass effect on the ventricular system and are often accompanied by a high rate of hypopituitarism and diabetes insipidus.
Meningiomas
Meningiomas are usually benign neoplasms of the dura mater. The majority are WHO grade I, but atypical (WHO II) and anaplastic (WHO III) lesions are described.52 Peak incidence is in the 4th to 7th decade.53 In adults, they are the second most common lesion encountered in the parasellar area after pituitary adenomas.54 Parasellar meningiomas include those that arise from the diaphragma sella, tuberculum sella, or the dura overlying the medial walls of the cavernous sinuses. They are often diagnosed based on imaging characteristics. They tend to have very uniform enhancement with gadolinium contrast and their appearance is similar to gray matter both on CT and MRI.47 A gnarled appearance is often seen in cavernous sinus meningiomas. Tuberculum sellae meningiomas often have a dural tail that extends over the planum sphenoidale, which distinguishes them from pituitary adenomas.49 On coronal sections, this tail often gives the appearance of a bird’s beak. Adjacent bone may be thickened or sclerotic. Meningiomas often encase arterial vessels and narrow their lumen, which is again atypical of pituitary adenomas.51
Chordomas and Chondrosarcomas
Chordomas and chondrosarcomas are cartilaginous tumors arising from the notochord remnant in the skull base. Chordomas are low-grade tumors, but can be quite destructive and have a significant mortality risk (5-year survival 65%).55 Intracranial chondrosarcomas are far less common than chordomas and fewer than 200 are described in the literature.56 These lesions grow in all directions and can efface normal anatomic structures, compress the brainstem, and obliterate the sella. They tend to invade and destroy the sella rather causing the expansion one may see with a typical pituitary adenoma. They often cause bony destruction and may contain calcifications best seen on CT. They tend to be hyperintense on T2 weighted imaging and enhance with contrast. Like craniopharyngiomas, these lesions may be calcified on CT scanning.
Initial presenting complaints are likely to be related to visual symptoms or headaches. Diplopia is more likely than visual field defects, reflecting their propensity to affect the cranial nerves within the cavernous sinuses.51 They are not likely to be associated with pituitary insufficiency, but it has been described, particularly in association with chondrosarcoma.56
Other Malignant Lesions of the Sella
Parasellar gliomas, malignant lesions that can range from low to high grade, arise from glial tissue located in the hypothalamus, optic chiasm, optic nerves, or the optic tracts. Examples include pilocytic astrocytomas, a low-grade (WHO I) lesion, that is often associated with neurofibromatosis type 1 (NF-1). Higher-grade gliomas in the parasellar region include astrocytomas (WHO II) and anaplastic astrocytomas. Occasionally astrocytomas will progress to gliobastomas (WHO grades III-VI).51 These lesions tend to be large, potentially infiltrative, suprasellar masses. They tend to be hypointense to brain on T1- weighted images and hyperintense on T2 weighted images. They may demonstrate heterogeneous enhancement with contrast.5
Germ-cell tumors (germinomas, choriocarcinomas, teratomas) are most commonly seen in childhood and adolescence. There are often synchronous lesions located in the suprasellar region, the vicinity of the third ventricle, and in the area of the pineal gland. These lesions often are suspected based on the radiographic characteristics in conjunction with the clinical presentation characterized by profound hypopituitarism and diabetes insipidus. These lesions tend to enhance with CT contrast and be isointense to brain on T1-weighted images and hyperintense on T2-weighted images.
Primary CNS lymphomas in the parasellar region have been described in both immunocompromised and immunocompetent patients. Presenting features include including diabetes insipidus, pituitary stalk thickening, and fever.57 Synchronous lesions are not uncommon and there may be evidence of disseminated lymphoma. Contrast-enhancing lesions in contact with the subarachnoid space and without necrosis are characteristic of parasellar lymphomas.
Metastases to the parasellar region may mimic a lesion of the pituitary stalk or of the sella. In most cases, there is additional evidence for intracranial metastases or distant metastases to other sites including the skeletal system and the liver. Melanoma and carcinomas of the breast and lung account for most cases of metastases to the pituitary stalk. Imaging characteristics may mimic a primary stalk tumor such as a glioma or choristoma or an infiltrative process causing thickening of the pituitary stalk.5 These tumors tend to have irregular borders.
Inflammatory Lesions
Sarcoidosis may present as a sellar mass, as thickening of the pituitary stalk, or even as a hypothalamic mass. Some patients may have communicating hydrocephalus and leptomeningeal enhancement. The disorder may be limited to the central nervous system or may be seen in association with the typical pulmonary or skin findings of sarcoidosis. Hyperprolactinemia due to stalk effect, DI, and hypopituitarism are not uncommon. An elevated serum or CSF ACE level may be helpful in securing the diagnosis but biopsy is often required. The pituitary “bright spot” on T1 weighted imaging may not be seen in patients who have resultant diabetes insipidus. Sarcoid granulomata tend to be isointense on T1 weighted imaging and will often enhance with gadolinium.58
Lymphocytic hypophysitis is characterized by either focal or diffuse infiltration of the pituitary by lymphocytes with accompanying inflammation and is felt to be an autoimmune disorder. Hypophysitis often mimics a pituitary tumor. The inflammatory mass is often diffuse and hyperintense on postcontrast T1 images. Patients usually have profound degrees of hypopituitarism that are not usually seen when similar degrees of enlargement of the sellar contents are seen in other disorders such as meningiomas, pituitary adenomas, and so on.49 Lymphocytic neuroinfundibulohypophysitis is associated with diabetes insipidus and loss of the pituitary bright spot on MRI. Xanthomatous hypophysitis is described as a reaction to ruptured cyst components.58 The appearance on MRI may be indistinguishable from sacrcoidosis, and clinical manifestations are similar.
Wegener’s granulomatosis may affect the pituitary and mimic an inflammatory condition. Most patients have evidence of coexisting renal and pulmonary disease. MRI often reveals a thickened infundibulum, but will not reliably distinguish this lesion from other inflammatory processes.58
Langerhan’s cell histiocytosis is characterized by proliferation of specific dendritic cells of the reticuloendothelial tissue. This disorder is more common in children (70%) than it is in adults (30%) and is quite rare. Diabetes insipidus is one of the more common early manifestations of the disorder. A thickened pituitary stalk and lack of a posterior pituitary bright spot on MRI may be seen.58 Occasionally patients also have lesions involving the skin, liver, lungs, spleen, and bone marrow.
Infectious Disorders
Pituitary abscesses may be seen in the setting of a pituitary tumor and are more common in the setting of a pre-existing Rathke’s cleft cyst. These infections may also be seen in patients with sphenoid sinusitis. On MRI, a pituitary abscess is typically cystic, hypo- or iso-intense on T1 weighted imaging and hyper or iso-intense on T2 imaging. Ring enhancement is often seen. Fever is not reliably present. Pituitary abscesses are often first diagnosed at the time of pituitary surgery when the surgeon encounters pus. Cultures should be obtained to permit appropriate identification of the infectious agent and directed antibiotic therapy.59,60
Intracranial tuberculosis is uncommon. The prevalence depends upon the underlying prevalence of tuberculosis in particular patient populations. Affected patients usually have systemic manifestation of the disease process but pituitary and suprasellar lesions have been described as the sole or presenting feature of the infection. A recent report illustrates a case where a tuberculoma was misdiagnosed as idiopathic granulomatous hypophysitis for which steroid treatment was initiated. The patient developed tuberculous meningitis, illustrating the need to maintain a degree of suspicion and to ensure antibiotic therapy is provided when indicated.61
Vascular Disorders
Intrasellar aneurysms arise from the carotid arteries but can arise from the anterior communicating arteries. These lesions often mimic pituitary adenomas but can usually be readily distinguished by appropriate imaging techniques. They may appear black on conventional MRI and show homogenous blush on contrasted CT images.58 MRA or conventional angiogram may confirm the diagnosis, and should be considered if an aneurysm is suspected.
Ophthalmologic Studies
The ophthalmologic exam and associated testing is a critical component of the evaluation of the patient with a sellar or parasellar lesion. In many cases these patients may present with visual complaints that ultimately lead to the discovery of their lesion. An experienced neuro-opthalmologist can often discern a great deal about a tumor’s location and some clues about its etiology from a detailed exam. Alternatively, the lesion may be discovered by other means, and the ophthalmologic exam may reveal previously unknown visual symptomatology that may influence the treatment plan, particularly in regards to surgery.
The optic chiasm is typically few millimeters above the diaphragma sellae separated by the suprasellar cistern. However, the chiasm can occupy both a “prefixed” or “postfixed” relationship to the diaphragma sellae (each in about 5% of the population). In the prefixed position, the chiasm is placed more anteriorly, with a shorter length of the optic nerves traveling over the pituitary gland and any potential associated pathology. In the postfixed position, the chiasm is more posterior, and a longer section of optic nerve is above the gland. The position of the chiasm will produce differing clinical manifestations when compression occurs from below. Lateral to the pituitary gland cranial nerves III, IV, V1, V2, and VI traverse the cavernous sinus.62
A thorough ophthalmologic examination is extremely important. This is performed preferably with the patient’s own corrective lenses, or if unavailable, with manifest refraction or correction by pinhole. Usually, in early chiasmal compression from a pituitary tumor, visual acuity and visual fields will be normal or near normal. Loss of visual acuity may suggest a very large pituitary tumor or pathology other than a typical pituitary lesion. Visual testing should also include color vision testing with pseudoisochromatic plates and formal visual field testing. Desaturation of color vision may be one of the earliest signs of a chiasmal lesion, preceding actual field deficits.63
All patients suspected of having a visual problem should have confrontational field testing. However, in the ophthalmologist’s office, automated visual fields, usually by Humphrey automated perimetry, should be performed. This testing is critical, as it may be used as a baseline against which to compare future examinations and is often the first clue to the examining physician that the patient may have a pituitary tumor large enough to cause a visual field deficit. The most typical initial visual field defect associated with a pituitary adenoma is a bitemporal superior quadrantic defect, produced due to pressure from below on the chiasm. A bitemporal inferior quadrantic defect is suggestive of pressure from above, as might be seen from a craniopharyngioma growing downward onto the chiasm. More severe pressure from either direction can produce a dense bitemporal hemianopsia.62,63
Formal testing of ocular motility, including ductions, saccades, pursuit, and alternate cover tests may reveal pathology related to the compression of cavernous sinuses and CN III, IV, and VI. Pathology in this area should certainly lead to consideration of cavernous sinus extension of a pituitary tumor, but also to alternative pathology. Involvement of CN III may result in a unilateral dilated pupil, ptosis, and a globe that is deviated inferiorly and laterally. Involvement of CN IV will result in vertical diplopia and upward deviation. Involvement of CN VI will result in horizontal diplopia due to the inability to abduct the involved eye. It is likely that deficits will be seen in combination. Horner’s syndrome may develop due to compression of the sympathetic neurons in the cavernous sinus.62,63
Coordination of Care
The diseases discussed in this chapter are uncommon and complex. Several studies illustrate that experienced pituitary neurosurgeons obtain greater likelihoods of surgical success and have lower rates of surgical morbidity and mortality.64 Neuroradiologists play a key role in providing expert interpretation of radiographic images and in establishing a differential diagnosis. When indicated, neuro-ophthalmologists conduct in-depth evaluations to document whether pituitary and parasellar lesions have affected the visual pathways. Radiation oncologists are essential in the management of patients with residual tumors, and in particular those with residual hormone-secreting neoplasms.
Bertherat J., Chanson P., Montminy M. The cyclic adenosine 3’,5’-monophosphate-responsive factor CREB is constitutively activated in human somatotroph adenomas. Mol Endocrinol. 1995;9:777-783.
Buchfelder M., Schlaffer S. Surgical treatment of pituitary tumours. Best Pract Res Clin Endocrinol Metab. Oct 2009;23(5):677-692.
Buurman H., Saeger W. Subclinical adenomas in postmortem pituitaries: classification and correlations to clinical data. Eur J Endocrinol. May 2006;154(5):753-758.
Castro M., Elias P.C., Quidute A.R., et al. Out-patient screening for Cushing’s syndrome: the sensitivity of the combination of circadian rhythm and overnight dexamethasone suppression salivary cortisol tests. J Clin Endocrinol Metab. Mar 1999;84(3):878-882.
DiGiovanni R., Serra S., Ezzat S., Asa S.L. .A.I.P. Mutations are not identified in patients with sporadic pituitary adenomas. Endocr Pathol. Summer 2007;18(2):76-78.
Elston M., McDonald K., Clifton-Bligh R., Robinson B. Familial pituitary tumor syndromes. Nat Rev Endocrinol. Aug 2009;5(8):453-461.
Ezzat S., Asa S.L., Couldwell W.T., et al. The prevalence of pituitary adenomas: a systematic review. Cancer. Aug 1 2004;101(3):613-619.
Fernandez A., Karavitaki N., Wass J.A. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72:377-382.
FitzPatrick M., Tartaglino L.M., Hollander M.D., et al. Imaging of sellar and parasellar pathology. Radiol Clin North Am. Jan 1999;37(1):101-121. x
Georgitsi M., Heliovaara E., Paschke R., et al. Large genomic deletions in AIP in pituitary adenoma predisposition. J Clin Endocrinol Metab. Oct 2008;93(10):4146-4151.
Glezer A., Paraiba D.B., Bronstein M.D. Rare sellar lesions. Endocrinol Metab Clin North Am. Mar 2008;37(1):195-211. x
Hamacher C., Brocker M., Adams E.F., et al. Overexpression of stimulatory G protein alpha-subunit is a hallmark of most human somatotrophic pituitary tumours and is associated with resistance to GH-releasing hormone. Pituitary. Apr 1998;1(1):13-23.
Heliovaara E., Raitila A., Launonen V., et al. The expression of AIP-related molecules in elucidation of cellular pathways in pituitary adenomas. Am J Pathol. 2009;175:2501-2507.
Kaltsas G.A., Evanson J., Chrisoulidou A., Grossman A.B. The diagnosis and management of parasellar tumours of the pituitary. Endocr Relat Cancer. Dec 2008;15(4):885-903.
Kaltsas G.A., Nomikos P., Kontogeorgos G., et al. Clinical review: diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. May 2005;90(5):3089-3099.
King J.T.Jr., Justice A.C., Aron D.C. Management of incidental pituitary microadenomas: a cost-effectiveness analysis. J Clin Endocrinol Metab. Nov 1997;82(11):3625-3632.
Krikorian A., Aron D. Evaluation and management of pituitary incidentalomas—revisiting an acquaintance. Nat Clin Pract Endocrinol Metab. Mar 2006;2(3):138-145.
Lanzino G., Dumont A., Lopes M., Laws E.J. Skull base chordomas: overview of disease, management options, and outcome. Neurosurg Focus. 2001;10(3):E12.
Melmed S., Colao A., Barkan A., et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. May 2009;94(5):1509-1517.
Molitch M.E. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. Mar 2008;37(1):151-171. xi
Powell M., Lightman S.L., Laws E.R. Management of Pituitary Tumours: the Clinician’s Practical Guide, 2nd ed. Totowa, NJ: Humana Press; 2003.
Vance M.L. Perioperative management of patients undergoing pituitary surgery. Endocrinol Metab Clin North Am. Jun 2003;32(2):355-365.
Vierimaa O., Georgitsi M., Lehtonen R., et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312(5777):1228-1230. May 26
Yanoff M., Duker J.S., Augsburger J.J. Ophthalmology, 3rd ed. Edinburgh: Mosby Elsevier; 2009.
Zee C.S., Go J.L., Kim P.E., Mitchell D., Ahmadi J. Imaging of the pituitary and parasellar region. Neurosurg Clin North Am. Jan 2003;14(1):55-80. vi
1. Ezzat S., Asa S.L., Couldwell W.T., et al. The prevalence of pituitary adenomas: a systematic review. Cancer. Aug 1 2004;101(3):613-619.
2. Fernandez A., Karavitaki N., Wass J.A. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72:377-382.
3. Buurman H., Saeger W. Subclinical adenomas in postmortem pituitaries: classification and correlations to clinical data. Eur J Endocrinol. May 2006;154(5):753-758.
4. Molitch M.E. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. Mar 2008;37(1):151-171. xi
5. Kaltsas G.A., Nomikos P., Kontogeorgos G., et al. Clinical review: diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. May 2005;90(5):3089-3099.
6. Elston M., McDonald K., Clifton-Bligh R., Robinson B. Familial pituitary tumor syndromes. Nat Rev Endocrinol. Aug 2009;5(8):453-461.
7. Vierimaa O., Georgitsi M., Lehtonen R., et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. May 26 2006;312(5777):1228-1230.
8. DiGiovanni R., Serra S., Ezzat S., Asa Slaip. Mutations are not identified in patients with sporadic pituitary adenomas. Endocr Pathol. 2007;18(2):76-78.
9. Georgitsi M., Heliovaara E., Paschke R., et al. Large genomic deletions in AIP in pituitary adenoma predisposition. J Clin Endocrinol Metab. Oct 2008;93(10):4146-4151.
10. Heliovaara E., Raitila A., Launonen V., et al. The expression of AIP-related molecules in elucidation of cellular pathways in pituitary adenomas. Am J Pathol. 2009;175:2501-2507.
11. Bertherat J., Chanson P., Montminy M. The cyclic adenosine 3’,5’-monophosphate-responsive factor CREB is constitutively activated in human somatotroph adenomas. Mol Endocrinol. Jul 1995;9(7):777-783.
12. Hamacher C., Brocker M., Adams E.F., et al. Overexpression of stimulatory G protein alpha-subunit is a hallmark of most human somatotrophic pituitary tumours and is associated with resistance to GH-releasing hormone. Pituitary. Apr 1998;1(1):13-23.
13. Spada A., Arosio M., Bochicchio D., et al. Clinical, biochemical, and morphological correlates in patients bearing growth hormone-secreting pituitary tumors with or without constitutively active adenylyl cyclase. J Clin Endocrinol Metab. Dec 1990;71(6):1421-1426.
14. Peri A., Conforti B., Baglioni-Peri S., et al. Expression of cyclic adenosine 3’,5’-monophosphate (cAMP)-responsive element binding protein and inducible-cAMP early repressor genes in growth hormone-secreting pituitary adenomas with or without mutations of the Gsalpha gene. J Clin Endocrinol Metab. May 2001;86(5):2111-2117.
15. King J.T.Jr., Justice A.C., Aron D.C. Management of incidental pituitary microadenomas: a cost-effectiveness analysis. J Clin Endocrinol Metab. Nov 1997;82(11):3625-3632.
16. Krikorian A., Aron D. Evaluation and management of pituitary incidentalomas—revisiting an acquaintance. Nat Clin Pract Endocrinol Metab. Mar 2006;2(3):138-145.
17. Howlett T.A., Como J., Aron D.C. Management of pituitary incidentalomas. A survey of British and American endocrinologists. Endocrinol Metab Clin North Am. Mar 2000;29(1):223-230. xi
18. Singer P.A., Sevilla L.J. Postoperative endocrine management of pituitary tumors. Neurosurg Clin North Am. Jan 2003;14(1):123-138.
19. Thaler L.M. Blevins LS Jr. The low dose (1-microg) adrenocorticotropin stimulation test in the evaluation of patients with suspected central adrenal insufficiency. J Clin Endocrinol Metab. Aug 1998;83(8):2726-2729.
20. Kazlauskaite R., Evans A.T., Villabona C.V., et al. Corticotropin tests for hypothalamic-pituitary- adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab. Nov 2008;93(11):4245-4253.
21. Agha A., Tomlinson J.W., Clark P.M., et al. The long-term predictive accuracy of the short synacthen (corticotropin) stimulation test for assessment of the hypothalamic-pituitary-adrenal axis. J Clin Endocrinol Metab. Jan 2006;91(1):43-47.
22. Vance M.L. Perioperative management of patients undergoing pituitary surgery. Endocrinol Metab Clin North Am. Jun 2003;32(2):355-365.
23. Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J Clin Endocrinol Metab. Apr 2009;94(4):1059-1067.
24. Mancini T., Casanueva F.F., Giustina A. Hyperprolactinemia and prolactinomas. Endocrinol Metab Clin North Am. Mar 2008;37(1):67-99. viii
25. Colao A., Sarno A.D., Cappabianca P., et al. Gender differences in the prevalence, clinical features and response to cabergoline in hyperprolactinemia. Eur J Endocrinol. Mar 2003;148(3):325-331.
26. Delgrange E., de Hertogh R., Vankrieken L., Maiter D. Potential hook effect in prolactin assay in patients with giant prolactinoma. Clin Endocrinol (Oxf). Oct 1996;45(4):506-507.
27. Frieze T.W., Mong D.P., Koops M.K. “Hook effect” in prolactinomas: case report and review of literature. Endocr Pract. Jul-Aug 2002;8(4):296-303.
28. Alfonso A., Rieniets K.I., Vigersky R.A. Incidence and clinical significance of elevated macroprolactin levels in patients with hyperprolactinemia. Endocr Pract. 2006;12(3):275-280.
29. McKenna T.J. Should macroprolactin be measured in all hyperprolactinaemic sera? Clin Endocrinol (Oxf). Oct 2009;71(4):466-469.
30. Ellis M.J., Livesey J.H., Soule S.G. Macroprolactin, big-prolactin and potential effects on the misdiagnosis of hyperprolactinemia using the Beckman Coulter Access Prolactin assay. Clin Biochem. Oct 2006;39(10):1028-1034.
31. Nieman L.K., Biller B.M., Findling J.W., et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. May 2008;93(5):1526-1540.
32. Meikle A.W. Dexamethasone suppression tests: usefulness of simultaneous measurement of plasma cortisol and dexamethasone. Clin Endocrinol (Oxf). Apr 1982;16(4):401-408.
33. Pecori Giraldi F., Ambrogio A.G., De Martin M., et al. Specificity of first-line tests for the diagnosis of Cushing’s syndrome: assessment in a large series. J Clin Endocrinol Metab. Nov 2007;92(11):4123-4129.
34. Kidambi S., Raff H., Findling J.W. Limitations of nocturnal salivary cortisol and urine free cortisol in the diagnosis of mild Cushing’s syndrome. Eur J Endocrinol. Dec 2007;157(6):725-731.
35. Castro M., Elias P.C., Quidute A.R., et al. Out-patient screening for Cushing’s syndrome: the sensitivity of the combination of circadian rhythm and overnight dexamethasone suppression salivary cortisol tests. J Clin Endocrinol Metab. Mar 1999;84(3):878-882.
36. Papanicolaou D.A., Mullen N., Kyrou I., Nieman L.K. Nighttime salivary cortisol: a useful test for the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. Oct 2002;87(10):4515-4521.
37. Kennedy L., Atkinson A.B., Johnston H., et al. Serum cortisol concentrations during low dose dexamethasone suppression test to screen for Cushing’s syndrome. Br Med J (Clin Res Ed). Nov 3 1984;289(6453):1188-1191.
38. Gatta B., Chabre O., Cortet C., et al. Reevaluation of the combined dexamethasone suppression-corticotropin-releasing hormone test for differentiation of mild cushing’s disease from pseudo-Cushing’s syndrome. J Clin Endocrinol Metab. Nov 2007;92(11):4290-4293.
39. Yanovski J.A., Cutler G.B.Jr., Chrousos G.P., Nieman L.K. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test differentiates mild Cushing’s disease from normal physiology. J Clin Endocrinol Metab. Feb 1998;83(2):348-352.
40. Newell-Price J., Trainer P., Besser M., Grossman A. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. Oct 1998;19(5):647-672.
41. Swearingen B., Katznelson L., Miller K., et al. Diagnostic errors after inferior petrosal sinus sampling. J Clin Endocrinol Metab. Aug 2004;89(8):3752-3763.
42. Lad S.P., Patil C.G., Laws E.R.Jr., Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus. 2007;23(3):E2.
43. Simard M.F. Pituitary tumor endocrinopathies and their endocrine evaluation. Neurosurg Clin North Am. Jan 2003;14(1):41-54. vi
44. Melmed S., Colao A., Barkan A., et al. Guidelines for acromegaly management: An update. J Clin Endocrinol Metab. May 2009;94(5):1509-1517.
45. Losa M., Fortunato M., Molteni L., et al. Thyrotropin-secreting pituitary adenomas: biological and molecular features, diagnosis and therapy. Minerva Endocrinol. Dec 2008;33(4):329-340.
46. Sanno N., Teramoto A., Osamura R.Y. Thyrotropin-secreting pituitary adenomas. Clinical and biological heterogeneity and current treatment. J Neurooncol. Sep 2001;54(2):179-186.
47. Chandler W.F., Barkan A.L. Treatment of pituitary tumors: a surgical perspective. Endocrinol Metab Clin North Am. Mar 2008;37(1):51-66. viii
48. Murad-Kejbou S., Eggenberger E. Pituitary apoplexy: evaluation, management, and prognosis. Curr Opin Ophthalmol. Nov 2009;20(6):456-461.
49. Rennert J., Doerfler A. Imaging of sellar and parasellar lesions. Clin Neurol Neurosurg. Feb 2007;109(2):111-124.
50. Karavitaki N., Wass J.A. Craniopharyngiomas. Endocrinol Metab Clin North Am. Mar 2008;37(1):173-193. ix-x
51. Kaltsas G.A., Evanson J., Chrisoulidou A., Grossman A.B. The diagnosis and management of parasellar tumours of the pituitary. Endocr Relat Cancer. Dec 2008;15(4):885-903.
52. Hartmann C., Boström J., Simon M. Diagnostic and molecular pathology of meningiomas. Expert Rev Neurother. Nov 2006;6(11):1671-1683.
53. Zee C.S., Go J.L., Kim P.E., et al. Imaging of the pituitary and parasellar region. Neurosurg Clin North Am. Jan 2003;14(1):55-80. vi
54. FitzPatrick M., Tartaglino L.M., Hollander M.D., et al. Imaging of sellar and parasellar pathology. Radiol Clin North Am. Jan 1999;37(1):101-121. x
55. Lanzino G., Dumont A., Lopes M., Laws E.J. Skull base chordomas: overview of disease, management options, and outcome. Neurosurg Focus. 2001;10(3):E12.
56. Allan C., Kaltsas G., Evanson J., et al. Pituitary chondrosarcoma: an unusual cause of a sellar mass presenting as a pituitary adenoma. J Clin Endocrinol Metab. Jan 2001;86(1):386-391.
57. Liu J.K., Sayama C., Chin S.S., Couldwell W.T. Extranodal NK/T-cell lymphoma presenting as a pituitary mass. Case report and review of the literature. J Neurosurg. Sep 2007;107(3):660-665.
58. Glezer A., Paraiba D.B., Bronstein M.D. Rare sellar lesions. Endocrinol Metab Clin North Am. Mar 2008;37(1):195-211. x
59. Sabbah P., Bonardel G., Herve R., et al. CT and MRI findings in primitive pituitary abscess: a case report and review of literature. J Neuroradiol. Oct 1999;26(3):196-199.
60. Somali M.H., Anastasiou A.L., Goulis D.G., et al. Pituitary abscess presenting with cranial nerve paresis. Case report and review of literature. J Endocrinol Invest. Jan 2001;24(1):45-50.
61. Husain N., Husain M., Rao P. Pituitary tuberculosis mimicking idiopathic granulomatous hypophysitis. Pituitary. 2008;11(3):313-315.
62. Yanoff M., Duker J.S., Augsburger J.J. Ophthalmology, 3rd ed. [Edinburgh]: Mosby Elsevier; 2009.
63. Powell M., Lightman S.L., Laws E.R. Management of pituitary tumours: the clinician’s practical guide, 2nd ed. Totowa, NJ: Humana Press; 2003.
64. Buchfelder M., Schlaffer S. Surgical treatment of pituitary tumours. Best Pract Res Clin Endocrinol Metab. Oct 2009;23(5):677-692.