Chapter 82 Mucopolysaccharidoses
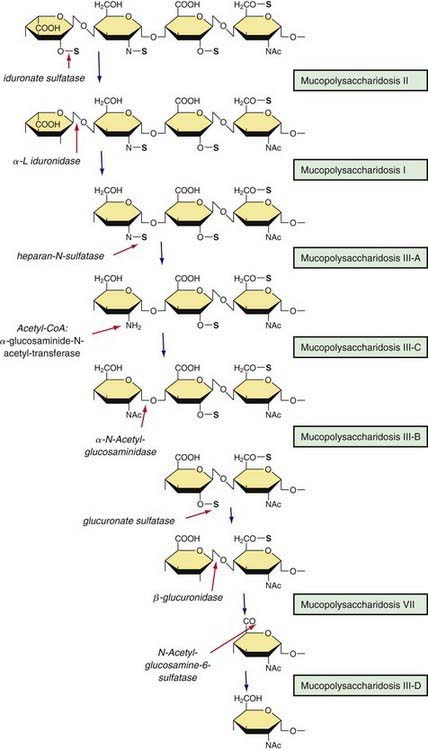
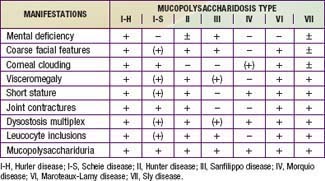
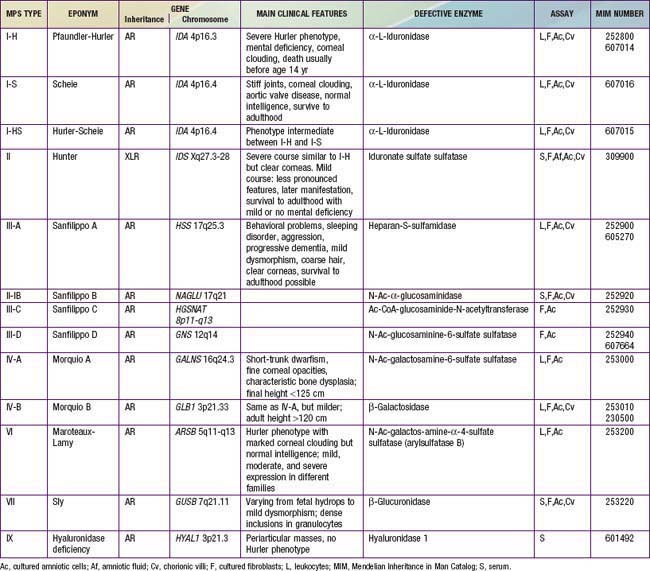
Mucopolysaccharidoses are hereditary, progressive diseases caused by mutations of genes coding for lysosomal enzymes needed to degrade glycosaminoglycans (acid mucopolysaccharides). Glycosaminoglycan (GAG) is a long-chain complex carbohydrate composed of uronic acids, amino sugars, and neutral sugars. The major GAGs are chondroitin-4-sulfate, chondroitin-6-sulfate, heparan sulfate, dermatan sulfate, keratan sulfate, and hyaluronan. These substances are synthesized and, with the exception of hyaluronan, linked to proteins to form proteoglycans, major constituents of the ground substance of connective tissue, as well as nuclear and cell membranes. Degradation of proteoglycans starts with the proteolytic removal of the protein core followed by the stepwise degradation of the GAG moiety. Failure of this degradation due to absent or grossly reduced activity of mutated lysosomal enzymes results in the intralysosomal accumulation of GAG fragments (Fig. 82-1). Distended lysosomes accumulate in the cell, interfere with cell function, and lead to a characteristic pattern of clinical, radiologic, and biochemical abnormalities (Table 82-1, Fig. 82-2). Within this pattern, specific diseases can be recognized that evolve from the intracellular accumulation of different degradation products (Table 82-2). As a general rule, the impaired degradation of heparan sulfate is more closely associated with mental deficiency and the impaired degradation of dermatan sulfate, chondroitin sulfates, and keratan sulfate with mesenchymal abnormalities. Variable expression within a given entity results from allelic mutations and varying residual activity of mutated enzymes. Allelic mutations of the gene encoding L-iduronidase may result in severe Hurler disease with early death or in mild Scheie disease manifesting only with limited joint mobility, mild skeletal abnormalities, and corneal opacities. Mucopolysaccharidoses are autosomal recessive disorders, with the exception of Hunter disease, which is X-linked recessive. Their overall frequency is between 3.5/100,000 and 4.5/100,000. The most common subtype is MPS-III, followed by MPS-I and MPS-II.
Clinical Entities
Mucopolysaccharidosis I
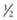 the MPS-I alleles in the white population. The mutations introduce stop codons with ensuing absence of functional enzyme (null alleles); homozygosity or compound heterozygosity gives rise to Hurler disease. Other mutations occur in only one or a few individuals.
the MPS-I alleles in the white population. The mutations introduce stop codons with ensuing absence of functional enzyme (null alleles); homozygosity or compound heterozygosity gives rise to Hurler disease. Other mutations occur in only one or a few individuals.Hurler Disease
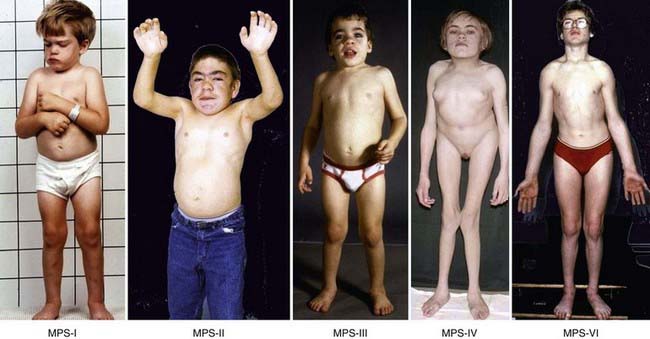
This form of MPS I (MPS I-H) is a severe, progressive disorder with multiple organ and tissue involvement that results in premature death, usually by 10 yr of age. An infant with Hurler syndrome appears normal at birth, but inguinal hernias are often present. Diagnosis is usually made between 6 and 24 mo of age with evidence of hepatosplenomegaly, coarse facial features, corneal clouding, large tongue, prominent forehead, joint stiffness, short stature, and skeletal dysplasia (see Fig. 82-2). Acute cardiomyopathy has been found in some infants <1 yr of age. Most patients have recurrent upper respiratory tract and ear infections, noisy breathing, and persistent copious nasal discharge. Valvular heart disease with incompetence, notably of the mitral and aortic valves, regularly develops, as does coronary artery narrowing. Obstructive airway disease, notably during sleep, may necessitate tracheotomy. Obstructive airway disease, respiratory infection, and cardiac complications are the common causes of death.
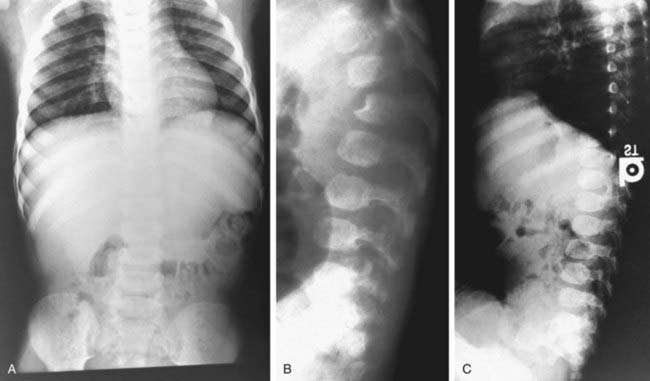
Most children with Hurler syndrome acquire social but only limited language skills because of developmental delay, combined conductive and neurosensory hearing loss, and an enlarged tongue. Progressive ventricular enlargement with increased intracranial pressure caused by communicating hydrocephalus may be responsible for headache and sleep disturbance. Corneal clouding, glaucoma, and retinal degeneration are common. Radiographs show a characteristic skeletal dysplasia known as dysostosis multiplex (Figs. 82-3 and 82-4). The earliest radiographic signs are thick ribs and ovoid vertebral bodies. Skeletal abnormalities in addition to those shown in the figures include enlarged, coarsely trabeculated diaphyses of the long bones with irregular metaphyses and epiphyses. With progression of the disease, macrocephaly develops, with thickened calvarium, premature closure of lambdoid and sagittal sutures, shallow orbits, enlarged J-shaped sella, and abnormal spacing of teeth with dentigenous cysts.
Mucopolysaccharidosis II
Hunter disease (MPS II) is an X-linked disorder caused by the deficiency of iduronate-2-sulfatase (IDS). The gene encoding IDS is mapped to Xq28. Point mutations of the IDS gene have been detected in about 80% of patients with MPS II. Major deletions or rearrangements of the IDS gene have been found in the rest; these are usually associated with a more severe clinical phenotype (see Fig. 82-2). Hunter disease manifests almost exclusively in males; it has been observed in a few females and this is explained by skewed inactivation of the X chromosome carrying the normal gene.
Mucopolysaccharidosis III
Sanfilippo disease (MPS III) makes up a genetically heterogeneous but clinically similar group of 4 recognized types. Each type is caused by a different enzyme deficiency involved in the degradation of heparan sulfate (see Fig. 82-1). Mutations have been found in all the MPS III disorders for which the genes have been isolated.
Phenotypic variation exists in MPS III patients but to a lesser degree than in other MPS disorders. Patients with Sanfilippo disease are characterized by slowly progressive, severe CNS involvement with mild somatic disease. Such disproportionate involvement of the CNS is unique to MPS III. Onset of clinical features usually occurs between 2 and 6 yr in a child who previously appeared normal. Presenting features include delayed development, hyperactivity with aggressive behavior, coarse hair, hirsutism, sleep disorders, and mild hepatosplenomegaly (see Fig. 82-2). Delays in diagnosis of MPS III are common due to the mild physical features, hyperactivity, and slowly progressive neurologic disease. Severe neurologic deterioration occurs in most patients by 6-10 yr of age, accompanied by rapid deterioration of social and adaptive skills. Severe behavior problems such as sleep disturbance, uncontrolled hyperactivity, temper tantrums, destructive behavior, and physical aggression are common. Profound mental retardation and behavior problems often occur in patients with normal physical strength, making management particularly difficult.
Mucopolysaccharidosis IV
Both types of Morquio disease are characterized by short-trunk dwarfism, fine corneal deposits, a skeletal dysplasia that is distinct from other mucopolysaccharidoses, and preservation of intelligence. MPS IV-A is usually more severe than MPS IV-B, with adult heights of <125 cm in the former and >150 cm in the latter. There is considerable variability of expression in both subtypes, however. The appearance of genua valga, kyphosis, growth retardation with short trunk and neck, and waddling gait with a tendency to fall are early symptoms of MPS IV (see Fig. 82-2). Extra skeletal manifestations include mild corneal clouding, small teeth with abnormally thin enamel, frequent caries formation, and, occasionally, hepatomegaly, and cardiac valvular lesions. Instability of the odontoid process and ligamentous laxity are regularly present and can result in life-threatening atlantoaxial instability and dislocation. Surgery to stabilize the upper cervical spine, usually by posterior spinal fusion before the development of cervical myelopathy, can be lifesaving.
Mucopolysaccharidosis VI
Maroteaux-Lamy disease (MPS VI) is caused by mutations of the ARSB gene on chromosome 5q11-13 encoding N-acetylgalactosamine-4-sulfatase (arylsulfatase B). It is characterized by severe to mild somatic involvement, as seen in MPS I, but with preservation of intelligence. The somatic involvement of the severe form of MPS VI is characterized by corneal clouding, coarse facial features, joint stiffness, valvular heart disease, communicating hydrocephalus, and dysostosis multiplex (see Fig. 82-2). In the severe form, growth can be normal for the 1st few years of life but seems virtually to stop after age 6-8 yr. The mild to intermediate forms of Maroteaux-Lamy disease can be easily confused with Scheie syndrome. Spinal cord compression from thickening of the dura in the upper cervical canal with resultant myelopathy is a frequent occurrence in patients with MPS VI.
Diagnosis and Differential Diagnosis
Radiographs of chest, spine, pelvis, and hands are useful to detect early signs of dysostosis multiplex (see Figs. 82-3 and 82-4). Semiquantitative spot tests for increased urinary GAG excretion are quick, inexpensive, and useful for initial evaluation but are subject to both false-positive and false-negative results. Chemical quantification of uronic acid–containing substances is required to assess the total urinary excretion of GAG. Quantitative analysis of single GAG by various methods, or of oligosaccharides by tandem mass spectrometry, reveals type-specific profiles. Morquio disease is often missed in urinary assays but can reliably be diagnosed in serum using monoclonal antibodies to keratan sulfate. Any individual who is suspected of an MPS disorder based on clinical features, radiographic results, or urinary GAG screening tests should have a definitive diagnosis established by enzyme assay. Serum, leukocytes, or cultured fibroblasts are used as the tissue source for measuring lysosomal enzymes (see Table 82-2). Prenatal diagnosis is available for all mucopolysaccharidoses and is carried out on cultured cells from amniotic fluid or chorionic villus biopsy. Measurement of GAGs in amniotic fluid is unreliable. Carrier testing in Hunter syndrome, an X-linked disorder, requires analysis of the IDS gene once the specific mutation or chromosome arrangement in the family under consideration is known. Molecular analysis in patients with other mucopolysaccharidoses or in known carriers requires a specific rationale. Attempts are being made to develop methods for routine newborn screening.
Mucolipidoses and oligosaccharidoses manifest with the same clinical and radiographic features as mucopolysaccharidoses (Chapters 80.4 and 80.5). In these conditions, the urinary excretion of GAGs is not elevated. Hurler-like facial features, joint contractures, dysostosis multiplex, and elevated urinary GAG excretion differentiate the mucopolysaccharidoses from other neurodegenerative and dwarfing conditions.
Treatment
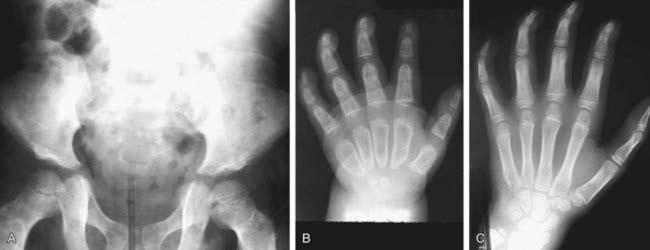
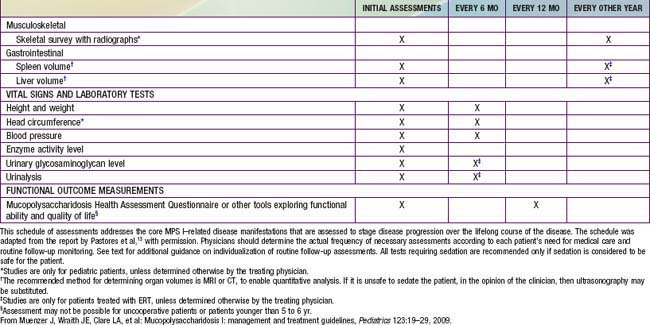
Primary prevention through genetic counseling and tertiary prevention to avoid or treat complications remains the mainstay of supportive pediatric care. Multidisciplinary attention to respiratory and cardiovascular complications, hearing loss, carpal tunnel syndrome, spinal cord compression, hydrocephalus, and other problems can greatly improve the quality of life for patients and their families (Table 82-3). The progressive nature of clinical involvement in MPS patients dictates the need for specialized and coordinated evaluation (Table 82-4).
| PROBLEM | PREDOMINANTLY IN | MANAGEMENT |
|---|---|---|
| NEUROLOGIC | ||
| Hydrocephalus | MPS I, II, VI, VII | Fundoscopy, CT scan, ventriculoperitoneal shunting |
| Chronic headaches | All | See behavioral disturbances |
| Behavioral disturbance | MPS III | Behavioral or medication therapy, sometimes CT scan, ventriculoperitoneal shunting |
| Disturbed sleep/wake circle | MPS III | Melatonin |
| Seizures | MPS I, II, III | EEG, anticonvulsants |
| Odontoid hypoplasia | MPS IV | Cervical MRI, upper cervical fusion |
| Spinal cord compression | All | Laminectomy, dural excision |
| OPHTHALMOLOGIC | ||
| Corneal opacity | MPS I, VI, VII | Corneal transplant |
| Glaucoma | MPS I, VI, VII | Medication, surgery |
| Retinal degeneration | MPS I, II | Night light |
| EARS, AIRWAYS | ||
| Recurrent otitis media | MPS I, II, VI, VII | Ventilating tubes |
| Impaired hearing | All except MPS IV | Audiometry, hearing aids |
| Obstruction | All except MPS III | Adenotomy, tonsillectomy, bronchodilator therapy, CPAP at night, laser excision of tracheal lesions, tracheotomy |
| CARDIAC | ||
| Cardiac valve disease | MPS I, II, VI, VII | Endocarditis prevention, valve replacement |
| Coronary insufficiency | MPS I, II, VI, VII | Medical therapy |
| Arrhythmias | MPS I, II, VI, VII | Antiarrhythmic medication, pacemaker |
| ORAL, GASTROINTESTINAL | ||
| Hypertrophic gums, poor teeth | MPS I, II, VI, VII | Dental care |
| Chronic diarrhea | MPS II | Diet modification, loperamide |
| MUSCULOSKELETAL | ||
| Joint stiffness | All except MPS IV | Physical therapy |
| Weakness | All | Physical therapy, wheelchair |
| Gross long bone malalignment | All | Corrective osteotomies |
| Carpal tunnel syndrome | MPS I, II, VI, VII | Electromyography, surgical decompression |
| ANESTHESIA | All except III | Avoid atlantoaxial dislocation, use angulated videointubation laryngoscope and small endotracheal tubes |
CPAP, continuous positive air pressure.
Boelens JJ, Wynn RF, O’Meara A, et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant. 2007;40:225-233.
Cheng Y, Verp MS, Knutel T, et al. Mucopolysaccharidosis type VII as a cause of recurrent non-immune hydrops fetalis. J Perinat Med. 2003;31:535-537.
Clarke LA, Wraith E, Beck M, et al. Long-term efficacy and safety of Laronidase in the treatment of mucopolysaccharidosis I. Pediatrics. 2009;123:229-240.
Fuller M, Rozaklis T, Ramsay SL, et al. Disease-specific markers for the mucopolysaccharidoses. Pediatr Res. 2004;56:733-738.
Guffon N, Bertrand Y, Forest I, et al. Bone marrow transplantation in children with Hunter syndrome: outcome after 7-17 years. J Pediatr. 2009;154:733-737.
Harmatz P, Giugliani R, Schwartz IV, et al. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI. Mol Genet Metab. 2008;94:469-475.
Harmatz P, Gugliani R, Schwartz I, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatese (recombinant human arylsulfatase B or RHASB) and follow-on, open-label extension study. J Pediatr. 2006;148:533-539.
Kamin W. Diagnosis and management of respiratory involvement in Hunter syndrome. Acta Paediat Suppl. 2008;97:57-60.
Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008:e377-e386.
Meikle PJ, Ranieri E, Skimonsen H, et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics. 2004;114:909-914.
Muenzer J, Wraith JE, Clare LA, et al. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19-29.
Orchard PJ, Blazar BR, Wagner J, et al. Hematopoietic cell therapy for metabolic disease. J Pediatr. 2007;151:340-346.
Robertson SP, Klug GL, Rogers JG. Cerebrospinal fluid shunts in the management of behavioural problems in Sanfilippo syndrome (MPS III). J Pediatr. 1998;157:653-655.
Schlander M, Beck M. Expensive drugs for rare disorders: to treat or not to treat. The case of enzyme replacement therapy for mucopolysaccharidosis VI. Curr Med Res Opin. 2009;25:1284-1293.
Wilcox WR. Lysosomal storage disorders: the need for better pediatric recognition and comprehensive care. J Pediatr. 2004;144:S3-S14.
Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome). A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediat. 2008;167:267-277.
Wynn RF, Mercer J, Page J, et al. Use of enzyme replacement therapy (Laronidase) before hematopoietic system cell transplantation for mucopolysaccharidosis I: experience in 18 patients. J Pediatr. 2009;154:135-139.
Wynn RF, Wraith JE, Mercer J, et al. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J Pediatr. 2009;154:609-611.
Yeung AH, Cowan MJ, Horn B, et al. Airway management in children with mucopolysaccharidoses. Arch Otolaryngol Head Neck Surg. 2009;135:73-79.





