Chapter 590 Movement Disorders
Once the category of movement disorder is recognized, etiology can be considered. Clinical history, including birth history, medication/toxin exposure, trauma, infections, family history, progression of the involuntary movements, developmental progress, and behavior should be explored as the underlying cause is established. Hyperkinetic movement disorders are more common than akinetic-rigid syndromes in childhood (Table 590-1).
590.1 Ataxias
Ataxia is the inability to make smooth, accurate, and coordinated movements, usually due to a dysfunction of the cerebellum, its inputs or outputs, sensory pathways in the posterior columns of the spinal cord, or a combination of these. Ataxias may be generalized or primarily affect gait or the hands and arms; they may be acute (Table 590-2) or chronic (Table 590-3). Congenital anomalies of the posterior fossa, including the Dandy-Walker syndrome, Chiari malformation, and encephalocele, are prominently associated with ataxia because of their destruction or replacement of the cerebellum (Chapters 585.9 and 585.11). Agenesis of the cerebellar vermis presents in infancy with generalized hypotonia and decreased deep tendon reflexes. Delayed motor milestones and truncal ataxia are typical. Joubert syndrome is an autosomal recessive disorder marked by agenesis of the cerebellar vermis, ataxia, hypotonia, oculomotor apraxia, neonatal breathing problems, and mental retardation. Mutations have been identified in the AHI1 gene on chromosome 6, encoding the Jouberin protein. This gene is strongly expressed in embryonic hindbrain, especially in neurons that give rise to the axons of the corticospinal tract and superior cerebellar peduncles, which fail to cross properly in Joubert syndrome. MRI is the method of choice for investigating congenital abnormalities of the cerebellum, vermis, and related structures. In Joubert syndrome, MRI reveals enlargement of the 4th ventricle at the junction between the midbrain and medulla, creating the “molar tooth sign.”
Table 590-3 CHRONIC OR PROGRESSIVE ATAXIA
BRAIN TUMORS
CONGENITAL MALFORMATIONS
HEREDITARY ATAXIAS
From Fenichel GM: Clinical pediatric neurology, ed 5, Philadelphia, 2005, Elsevier, p 220.
The major infectious causes of ataxia include cerebellar abscess, acute labyrinthitis, and acute cerebellar ataxia. Acute cerebellar ataxia occurs primarily in children 1-3 yr of age and is a diagnosis of exclusion. The condition often follows a viral illness, such as varicella, coxsackievirus, or echovirus infection by 2-3 wk and is thought to represent an autoimmune response to the viral agent affecting the cerebellum (Chapters 242, 245, and 595). The onset is sudden, and the truncal ataxia can be so severe that the child is unable to stand or sit. Vomiting may occur initially, but fever and nuchal rigidity are absent. Horizontal nystagmus is evident in approximately 50% of cases and, if the child is able to speak, dysarthria may be impressive. Examination of the cerebrospinal fluid (CSF) is typically normal at the onset of ataxia; a pleocytosis of lymphocytes (10-30/mm3) is not unusual. Later in the course, the CSF protein undergoes a moderate elevation. The ataxia begins to improve in a few weeks but may persist for as long as 2 mo. The incidence of acute cerebellar ataxia appears to have declined with increased rates of vaccination against varicella. The prognosis for complete recovery is excellent; a small number have long-term sequelae, including behavioral and speech disorders as well as ataxia and incoordination. Acute labyrinthitis may be difficult to differentiate from acute cerebellar ataxia in a toddler. The condition is associated with middle-ear infections and intense vertigo, vomiting, and abnormalities in labyrinthine function, particularly ice water caloric testing.
Several metabolic disorders are characterized by ataxia, including abetalipoproteinemia, arginosuccinic aciduria, and Hartnup disease. Abetalipoproteinemia (Bassen-Kornzweig disease) begins in childhood with steatorrhea and failure to thrive (Chapter 592). A blood smear shows acanthocytosis and decreased serum levels of cholesterol and triglycerides, and the serum β-lipoproteins are absent. Neurologic signs become evident by late childhood and consist of ataxia, retinitis pigmentosa, peripheral neuritis, abnormalities in position and vibration sense, muscle weakness, and mental retardation. Vitamin E is undetectable in the serum of patients with neurologic symptoms.
Degenerative diseases of the central nervous system (CNS) represent an important group of ataxic disorders of childhood because of the genetic consequences and poor prognosis. Ataxia-telangiectasia, an autosomal recessive condition, is the most common of the degenerative ataxias and is heralded by ataxia beginning at about age 2 yr and progressing to loss of ambulation by adolescence (Chapter 589). Ataxia-telangiectasia is caused by mutations in the ATM gene located at 11q22-q23. ATM is a phosphytidylinositol-3 kinase that phosphorylates proteins involved in DNA repair and cell cycle control. Oculomotor apraxia of horizontal gaze, defined as having difficulty fixating smoothly on an object and therefore overshooting the target with lateral movement of the head, followed by refixating the eyes, is a frequent finding, as is strabismus, hypometric saccade pursuit abnormalities, and nystagmus. Ataxia-telangiectasia may present with chorea rather than ataxia. The telangiectasia becomes evident by mid-childhood and is found on the bulbar conjunctiva, over the bridge of the nose, and on the ears and exposed surfaces of the extremities. Examination of the skin shows a loss of elasticity. Abnormalities of immunologic function that lead to frequent sinopulmonary infections include decreased serum and secretory IgA as well as diminished IgG2, IgG4, and IgE levels in more than 50% of patients. Children with ataxia-telangiectasia have a 50- to 100-fold greater chance over the normal population of developing lymphoreticular tumors (lymphoma, leukemia, and Hodgkin disease) as well as brain tumors. Additional laboratory abnormalities include an increased incidence of chromosome breaks, particularly of chromosome 14, and elevated levels of α-fetoprotein. Death results from infection or tumor dissemination.
Additional degenerative ataxias include Pelizaeus-Merzbacher disease, neuronal ceroid lipofuscinoses, and late-onset GM2 gangliosidosis (Chapter 592). Rare forms of progressive cerebellar ataxia have been described in association with vitamin E deficiency. A number of autosomal dominant progressive spinocerebellar ataxias have been defined at the molecular level, including those caused by unstable trinucleotide repeat expansions.
Albin RL. Dominant ataxias and Friedreich ataxia: an update. Curr Opin Neurol. 2003;16:507-514.
Beau-Salinas F, Guitteny MA, Donadieu J, et al. High doses of deferiprone may be associated with cerebellar syndrome. BMJ. 2009;338:653.
Farr AK, Shalev B, Crawford TO, et al. Ocular manifestations of ataxia-telangiectasis. Am J Ophthalmol. 2002;134:891-896.
Ferland RJ, Eyaid W, Collura RV, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1126.
Hart PE, Lodi R, Rajagopalan B, et al. Antioxidant treatment of patients with Friedreich ataxia: four-year followup. Arch Neurol. 2005;62:621-626.
Pandolfo M. Friedreich ataxia. Semin Pediatr Neurol. 2003;10:163-172.
Paulson H, Ammache Z. Ataxia and hereditary disorders. Neurol Clin. 2001;19:759-782.
Schols L, Bauer P, Schmidt T, et al. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291-304.
590.2 Chorea, Athetosis, Tremor
Chorea, meaning “dance-like” in Greek, refers to rapid, chaotic movements that seem to flow from one body part to another. Affected individuals exhibit motor impersistence, with difficulty keeping the tongue protruded (“darting tongue”) or maintaining grip (“milkmaid grip”). Chorea tends to occur both at rest and with action. Patients often attempt to incorporate the involuntary movements into more purposeful movements, making them appear fidgety. Chorea increases with stress and disappears in sleep. Chorea can be divided into primary (i.e., disorders in which chorea is the dominant symptom and the etiology is presumed to be genetic) and secondary forms, with the vast majority of pediatric cases falling into the latter category (Tables 590-4 and 590-5).
Table 590-4 ETIOLOGICAL CLASSIFICATION OF CHOREIC SYNDROMES
GENETIC CHOREAS
STRUCTURAL BASAL-GANGLIA LESIONS
PARAINFECTIOUS AND AUTOIMMUNE DISORDERS
INFECTIOUS CHOREA
METABOLIC OR TOXIC ENCEPHALOPATHIES
DRUG-INDUCED CHOREA (SEE TABLE 590-6)
From Cardoso F, Seppi K, Mair KJ, et al: Seminar on choreas, Lancet 5:589–602, 2006.
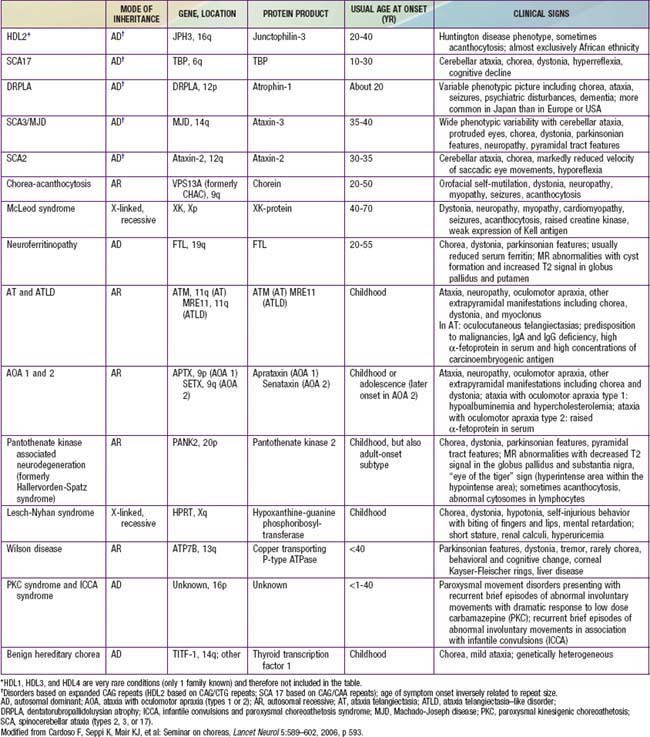
Sydenham chorea (SC, St. Vitus dance) is the most common acquired chorea of childhood. It occurs in 10% to 20% of patients with acute rheumatic fever, typically weeks to months after a group A β-hemolytic streptococcal infection (Chapter 176.1). Peak incidence is at age 8 to 9 yr, with a female predominance of 2 : 1.
Additional causes of secondary chorea include metabolic (hyperthyroidism, hypoparathyroidism), infectious (Lyme disease), immune-mediated (systemic lupus erythematosus), vascular (stroke, moyamoya disease), heredodegenerative disorders (Wilson disease), and drugs (Table 590-6). Although chorea is a hallmark of Huntington disease in adults, children who develop Huntington disease tend to present with rigidity and bradykinesia (Westphal variant) or dystonia rather than chorea.
Table 590-6 DRUGS THAT CAN INDUCE CHOREA
DOPAMINE RECEPTOR BLOCKING AGENTS
ANTIPARKINSONIAN DRUGS
ANTIEPILEPTIC DRUGS
PSYCHOSTIMULANTS
CALCIUM CHANNEL BLOCKERS
OTHERS
From Cardoso F, Seppi K, Mair KJ, et al: Seminar on choreas, Lancet 5:589–602, 2006.
While ET is the most common primary etiology of tremor in children, there are numerous secondary etiologies (Table 590-7). Holmes tremor, previously referred to as midbrain or rubral tremor, is characterized by a slow frequency, high amplitude tremor that is present at rest and with intention. It is a symptomatic tremor, which usually results from lesions of the brainstem, cerebellum, or thalamus. Psychogenic tremor is distinguished by its variable appearance, abrupt onset and remission, nonprogressive course, and association with selective but not task-specific disabilities. In some cases, tremor may even occur as a manifestation of another movement disorder, as is seen with position- or task-specific tremor (e.g., writing tremor), dystonic tremor, and myoclonic tremor.
Table 590-7 SELECTED CAUSES OF TREMOR IN CHILDREN
BENIGN
STATIC INJURY/STRUCTURAL
HEREDITARY/DEGENERATIVE
METABOLIC
DRUGS/TOXINS
PERIPHERAL NEUROPATHIES
PSYCHOGENIC
Avcin T, Benseler SM, Tyrrell PN, et al. A followup study of antiphospholipid antibodies and associated neuropsychiatric manifestations in 137 children with systemic lupus erythematosus. Arthritis Rheum. 2008;59(2):206-213.
Cardoso F, Seppi K, Mair KJ, et al. Seminar on choreas. Lancet Neurol. 2006;5:589-602.
Demiroren K, Yavuz H, Cam L, et al. Sydenham’s chorea: a clinical follow-up of 65 patients. J Child Neurol. 2007;22:550-554.
Keller S, Dure LS. Tremor in childhood. Semin Pediatr Neurol. 2009;16:60-70.
Kirvan CA, Cox CJ, Swedo SE, et al. Tubulin is a neuronal target of autoantibodies in Sydenham’s chorea. J Immunol. 2007;178:7412-7421.
Lehman R, Mink JW. Movement disorders in adolescents. In Fisher M, Alderman E, Kreipe R, et al, editors: Textbook of adolescent health care, (in press).
Paz JA, Silva CAA, Marques-Dias MJ. Randomized double-blind study with prednisone in Sydenham’s chorea. Pediatr Neurol. 2006;34(4):264-269.
590.3 Dystonia
Metabolic Disorders
Wilson disease is an autosomal recessive inborn error of copper transport characterized by cirrhosis of the liver and degenerative changes in the CNS, particularly the basal ganglia (Chapter 349.2). It has been determined that there are multiple mutations in the Wilson disease gene (WND), accounting for the variability in presentation of the condition. The neurologic manifestations of Wilson disease rarely appear before age 10 yr, and the initial sign is often progressive dystonia. Tremors of the extremities develop, unilaterally at first, but they eventually become coarse, generalized, and incapacitating. Other neurologic signs of Wilson disease relate to progressive basal ganglia disease, such as parkinsonism, dysarthria, dysphonia, and choreoathetosis. Less frequent are ataxia, and pyramidal signs. The MRI or CT scan shows ventricular dilatation in advanced cases with atrophy of the cerebrum, cerebellum, and/or brainstem along with signal intensity change in the basal ganglia, thalamus, and/or brainstem, particularly the midbrain.
Although dystonia may present in isolation as the 1st sign of a metabolic or neurodegenerative disorder, this group of diseases should be considered mainly in those who demonstrate signs of systemic disease, (e.g., organomegaly, short stature, hearing loss, vision impairment, epilepsy), those with episodes of severe illness, evidence of regression, or cognitive impairment. Additional features suggestive of specific disorders are outlined in Table 590-8.
Table 590-8 SELECTED CAUSES OF PRIMARY AND SECONDARY DYSTONIA IN CHILDHOOD
| DIAGNOSIS | ADDITIONAL CLINICAL FEATURES |
|---|---|
| Aicardi-Goutieres syndrome |
Agrawal SK, Rittey CD, Harrower NA, et al. Movement disorders associated with complex regional pain syndrome in children. Dev Med Child Neurol. 2009;51(7):557-562.
Fasano A, Nardocci N, Elia AE, et al. Non-DYT1 early-onset primary torsion dystonia: comparison with DYT1 phenotype and review of the literature. Mov Disord. 2006;21(9):1411-1418.
Heiman GA, Ottman R, Saunders-Pullman RJ, et al. Increased risk for recurrent major depression in DYT1 dystonia mutation carriers.[see comment]. Neurology. 2004;63(4):631-637.
Hilaire M-HS, Burke RE, Bressman SB, et al. Delayed-onset dystonia due to perinatal or early childhood asphyxia. Neurology. 1991;41(2 Part 1):216.
Nardocci N, Zorzi G, Barzaghi C, et al. Myoclonus-dystonia syndrome: clinical presentation, disease course, and genetic features in 11 families. Mov Disord. 2008;23(1):28-34.
Natascarona C, Petrovi I, Klein C, et al. Delayed-onset dystonia due to perinatal asphyxia: a prospective study. Mov Disord. 2007;22(16):2426-2429.
Pearl PL, Taylor JL, Trzcinski S, et al. The pediatric neurotransmitter disorders. J Child Neurol. 2007;22(5):606-616.
Rosman NP, Douglass LM, Sharif UM, et al. The neurology of benign paroxysmal torticollis of infancy: report of 10 new cases and review of the literature. J Child Neurol. 2009;24(2):155-160.
Sweney MT, Silver K, Gerard-Blanluet M, et al. Alternating hemiplegia of childhood: early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics. 2009;123(3):e534-e541.
Taly AB, Meenakshi-Sundaram S, Sinha S, et al. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine. 2007;82(2):112-121.
Van Hove JL, Steyaert J, Matthijs G, et al. Expanded motor and psychiatric phenotype in autosomal dominant Segawa syndrome due to GTP cyclohydrolase deficiency. J Neurol Neurosurg Psychiatry. 2006;77(1):18-23.





