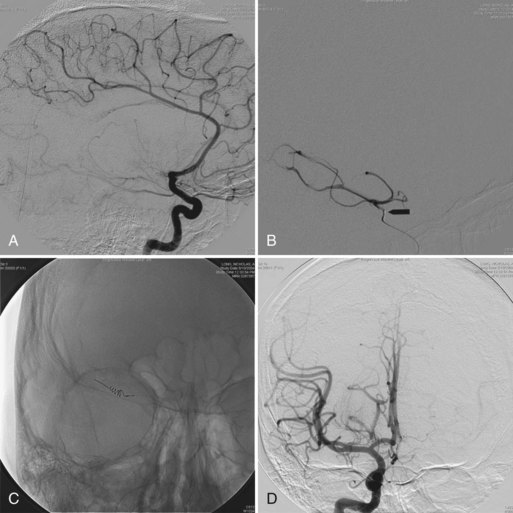
Chapter 71 Movement Disorders
Movement Disorders and the Basal Ganglia
Basal Ganglia Anatomy
There is no clear consensus on which structures should be included in the basal ganglia. For the purposes of this discussion, we consider those structures in the striatopallidal circuits involved in modulation of the thalamocortical projection: the caudate nucleus, the putamen, the external segment of the GP (GPe), and the internal segment of the GP (GPi). In addition, the SNc, the substantia nigra pars reticulata (SNr), and the STN are included in the basal ganglia. The substantia nigra (SN), a melanin-containing (pigmented) nucleus, normally contains about 500,000 dopaminergic neurons. Several transcriptional regulators including Nurr1, Lmx1a, Lmx1b, Msx1, and Pitx3 are responsible for the development and maintenance of midbrain dopaminergic neurons (Le et al., 2009).
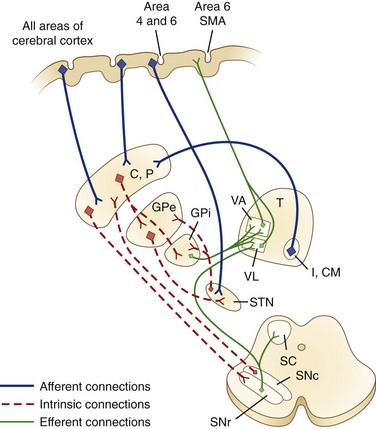
The caudate nucleus is a curved structure that traverses the deep hemisphere at the lateral edge of each lateral ventricle. Its diameter is largest at its head, tapering to a small tail. It is continuous with the putamen at the head and tail. The caudate and putamen together are called the striatum, and they form the major target for projections from the cerebral cortex and the SN. The putamen and the GP together form a wedge-shaped structure called the lenticular nucleus. The GP is divided into two parts, the GPe and the GPi. The GPi is structurally and functionally homologous with the SNr. The SNr and SNc extend the length of the midbrain ventral to the red nucleus and dorsal to the cerebral peduncles. The STN is a small lens-shaped structure at the border between the cerebrum and the brainstem. The basal ganglia and its relation to the thalamus and overlying cortex are illustrated in Fig. 71.1.
Functional Organization of the Basal Ganglia and Other Pathways
Afferent projections to the striatum arise from all areas of the cerebral cortex, the intralaminar nuclei of the thalamus, mesencephalic SN, and from the brainstem locus ceruleus and raphe nuclei. There is also a projection from the cerebral cortex to the STN. The major efferent projections are from the GPi and SNr to the thalamus and brainstem nuclei such as the pedunculopontine nucleus (PPN). The GPi and SNr project to ventral anterior and ventrolateral thalamic nuclei. The GPi also projects to the centromedian thalamic nuclei, and the SNr projects to the mediodorsal thalamic nuclei and superior colliculi. The ventral anterior and ventrolateral thalamic nuclei then project to the motor and premotor cortex. Throughout, these projections are somatotopically organized (Rodriguez-Oroz et al., 2009).
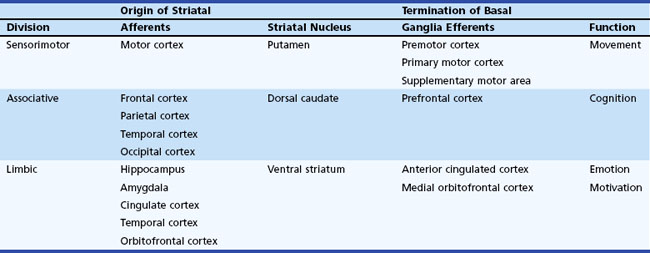
The basal ganglia has dense internuclear connections (see Fig. 71.1). Five parallel and separate closed circuits through the basal ganglia have been proposed. These are the motor, oculomotor, dorsolateral prefrontal, lateral orbitofrontal, and limbic loops (Rodriguez-Oroz et al., 2009). It is now generally agreed that these loops form three major divisions—sensorimotor, associative, and limbic—that are related to motor, cognitive, and emotional functions, respectively (Table 71.1). The functions of the sensorimotor striatum are subserved mainly by the putamen, which derives its afferent cortical inputs from both motor cortices. Sensorimotor pathways are somatotopically organized, and the pathway ultimately terminates in the premotor and primary motor cortices and the supplementary motor area. Cognitive functions are largely mediated by the associative striatum, particularly the dorsal caudate nucleus, which receives afferent input from the homolateral frontal, parietal, temporal, and occipital cortices. Projections from this pathway ultimately terminate in the prefrontal cortex. The limbic striatum subserves emotional and motivational functions. Its input derives from the cingulate, temporal, and orbitofrontal cortices, the hippocampus, and the amygdala. It mainly comprises the ventral striatum, with ultimate projections to the anterior cingulate and medial orbitofrontal cortices (Rodriguez-Oroz et al., 2009). Whether these divisions are interconnected or organized in parallel remains a topic of debate.
Within each basal ganglia circuit lies an additional level of complexity. Each circuit contains two pathways by which striatal activity is translated into pallidal output. These two pathways are named the direct and indirect pathways, depending on whether striatal outflow connects directly with the GPi or first traverses the GPe and STN before terminating in the GPi. The direct and indirect pathways have opposite effects on outflow neurons of the GPi and SNr (Fig. 71.2, A).

Fig. 71.2 Schematic drawing of internuclear connections of basal ganglia, including (A) direct and indirect pathways and (B) direct pathway. (See Fig. 71.3 for depiction of indirect pathway.) Excitatory pathways in solid lines, inhibitory pathways in dotted lines. D1, Dopamine D1 receptor; D2, dopamine D2 receptor; GABA, γ-aminobutyric acid; glu, glutamate; GPe, external segment of the globus pallidus; GPi, internal segment of the globus pallidus; MC, motor cortex; PMC, premotor cortex; SMA, supplementary motor area; SN, substantia nigra; STN, subthalamic nucleus; VA/VL, ventral anterior/ventrolateral thalamic nuclei.
In the motor direct pathway, excitatory neurons from the cerebral cortex synapse on putamenal neurons, which in turn send inhibitory projections to the GPi and its homolog, the SNr. The GPi/SNr sends an inhibitory outflow to the thalamus (see Fig. 71.2, B). Activity in the direct pathway disinhibits the thalamus, facilitating the excitatory thalamocortical pathway and enhancing activity in its target, the motor cortices. Thus, the direct pathway constitutes part of an excitatory cortical-cortical circuit that likely functions to maintain ongoing motor activity. In the indirect pathway, excitatory axons from the cerebral cortex synapse on putaminal neurons. These neurons send inhibitory projections to the GPe, and the GPe sends an inhibitory projection to the STN. The net effect of these projections is disinhibition of the STN. The STN in turn has an excitatory projection to the GPi (see Fig. 71.2, C). Activity in the indirect pathway thus excites the GPi/SNr, which in turn inhibits the thalamocortical pathway. Thus, the net effect of increased activity in the indirect pathway is cortical inhibition.
The striatum also receives robust afferent input from the SNc. This projection from the SNc, an important modifier of striatal activity, facilitates activity in the direct pathway, mediated via D1 dopamine receptors, and inhibits activity in the indirect pathway via D2 dopamine receptors (see Fig. 71.2, A).
Disorders of the basal ganglia result in prominent motor dysfunction, though not generally in frank weakness. The absence of direct primary or secondary sensory input and lack of a major descending pathway below the level of the brainstem suggest that the basal ganglia moderates rather than controls movement. The direct pathway is important in initiation and maintenance of movement, and the indirect pathway apparently plays a role in the suppression of extraneous movement. From this model of basal ganglia connectivity, hypotheses about the motor function of the basal ganglia have been proposed. One hypothesis is that the relative activities of the direct and indirect pathways serve to balance the facilitation and inhibition of the same population of thalamocortical neurons, thus controlling the scale of movement. A second hypothesis proposes that direct pathway-mediated facilitation and indirect pathway-mediated inhibition of different populations of thalamocortical neurons serve to focus movement in an organization reminiscent of center-surround inhibition. These hypotheses relate activity in the direct and indirect pathways mainly to rates of firing in the STN and GPi. Thus, death of neurons in the SNc, as in nigrostriatal degeneration associated with PD, decreases activity in the direct pathway and increases activity in the indirect pathway. These changes cause an increased rate of firing of subthalamic and GPi neurons, with excessive inhibition of thalamocortical pathways, and produce the behavioral manifestations of bradykinesia in PD (Fig. 71.3, A) (Hallett, 2011).
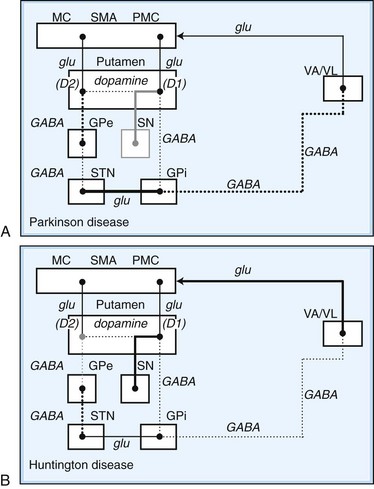
Fig. 71.3 Schematic drawing of functional activities in the direct and indirect pathways in Parkinson disease (PD) and Huntington disease (HD). A, In PD, reduced dopaminergic facilitation of direct pathway and inhibition of indirect pathway due to death of dopaminergic neurons causes increased firing and increased inhibition of thalamocortical pathways, producing bradykinesia. B, In HD, loss of striatal neurons leads to reduced activity in indirect pathway, causing reduced inhibition of thalamocortical pathways, with production of excessive or involuntary movements. (See Fig. 71.2 for explanations to abbreviations.)
On the other hand, predominant loss of indirect pathway neurons, as in HD, interferes with suppression of involuntary movements. Choreic involuntary movements are the usual result (see Fig. 71.3, B). Direct electrophysiological recordings of the STN and GP during stereotactic functional neurosurgical procedures confirm that the GPi and STN are overly active in patients with PD. The activity of these nuclei returns toward normal with effective pharmacotherapy, and chorea is associated with lower firing rates of neurons in these nuclei. Unfortunately, this model does not completely explain some important features of movement disorders. For example, bradykinesia and chorea coexist in HD and in patients with PD treated with levodopa (LD). Thalamic lesions that might be expected to worsen parkinsonism by reducing excitatory thalamocortical activity do not do so. Pallidal lesions that might be expected to worsen chorea by decreasing inhibition of thalamocortical pathways instead are dramatically effective at reducing chorea. The model is even more problematic when applied to dystonia. It has been suggested that in dystonia, there is overactivity of both the direct and indirect pathways. Yet, intraoperative recordings in dystonia have shown low rates and abnormal patterns of neuronal firing in the GPi. A simple change in firing rate of the STN or GPi is thus insufficient to explain the underlying physiology of dystonia. It is likely that disordered patterns and synchrony of pallidal firing, as well as changes in sensorimotor integration and the control of spinal and brainstem reflexes, are important. These factors are under investigation, but current models remain useful for understanding the rationale of pharmacological and ablative surgical procedures for certain movement disorders.
Biochemistry
The major neurotransmitters of the basal ganglia are outlined in Table 71.2 (see also Fig. 71.2). Most excitatory synapses of the basal ganglia and its connections—including those from the cerebral cortex to the striatum, the STN to the GPi, and the thalamocortical projections—use glutamate. Projections from the striatum to the GPe and GPi, from the GPe to the STN, and from the GPi to the thalamus are inhibitory and employ GABA. Medium spiny GABAergic neurons in the direct pathway co-localize substance P and dynorphin. GABAergic neurons in the indirect pathway co-localize enkephalin. Dopamine is the major neurotransmitter in the nigrostriatal dopamine system; it has excitatory or inhibitory actions depending on the properties of the stimulated receptor. Acetylcholine is found in large aspiny striatal interneurons and the PPN. Norepinephrine, important in the autonomic nervous system, is most concentrated in the lateral tegmentum and locus ceruleus. Serotonin is found in the dorsal raphe nucleus of the brainstem, hippocampus, cerebellum, and spinal cord.
| Pathway | Transmitter |
|---|---|
| STRIATAL AFFERENTS | |
| Cerebral cortex → striatum | Glutamate |
| Cerebral cortex → STN | Glutamate |
| Locus ceruleus → striatum | Norepinephrine |
| Locus ceruleus → SN | Norepinephrine |
| Raphe nuclei → striatum | Serotonin |
| Raphe nuclei → SN | Serotonin |
| Thalamus → striatum | Acetylcholine? |
| SNc → striatum | Glutamate? |
| Dopamine, cholecystokinin | |
| INTRINSIC CONNECTIONS | |
| Striatal interneurons | GABA, acetylcholine |
| Striatum → GPi | Somatostatin, neuropeptide Y |
| Striatum → SNr | Nitric acid, calretinin |
| Striatum → GPe | GABA, substance P |
| GPe → STN | GABA, dynorphin, substance P |
| STN → GPi, SNr, GPe | GABA, enkephalin, glutamate |
| STRIATAL EFFERENTS | |
| GPi → thalamus | GABA |
| SNr → thalamus | GABA |
GABA, γ-Aminobutyric acid; GPe, external segment of the globus pallidus; GPi, internal segment of the globus pallidus; SN, substantia nigra; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; STN, subthalamic nucleus.
Mechanisms of Neurodegeneration
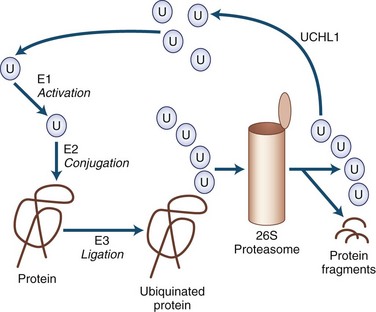
Many of the neurodegenerative movement disorders share the property of neuronal damage caused by the accumulation of aggregation-prone proteins that have toxic effects (Table 71.3). For a protein to function normally, it must be properly synthesized and folded into its normal three-dimensional structure. Nascent proteins are aided in folding by molecular chaperones. Proteins that are not properly folded, are otherwise damaged, or are beyond their useful lives are degraded by the ubiquitin-dependent proteasome protein degradation system (Gupta et al., 2008). In the ubiquitin-dependent proteasome system, proteins are first labeled for degradation by attachment of a polyubiquitin chain (Fig. 71.4). This three-step process involves activation, conjugation, and ligation steps catalyzed by three types of enzymes—E1, E2, and E3, respectively. Polyubiquinated protein enters the 26S proteasome, a cylindrical complex of peptidases. The end products of proteasome action are protein fragments and polyubiquitin. The polyubiquitin is then degraded and recycled to the cellular ubiquitin pool, a process requiring enzymatic action by ubiquitin carboxyterminal hydrolase 1.
Table 71.3 Toxic Proteins and Neurodegenerative Movement Disorders
| α-Synuclein | Tau | Polyglutamine Tract |
|---|---|---|
| Parkinson disease | Four-repeat tau | Huntington disease |
| Diffuse Lewy body disease | Progressive supranuclear palsy | Spinocerebellar ataxias |
| Multiple system atrophy | Corticobasal degeneration Frontotemporal dementia with parkinsonism (chromosome 17) Parkinsonism dementia complex of Guam Postencephalitic parkinsonism |
Dentatorubral-pallidoluysian atrophy |
The cascade of pathogenic events linking abnormal protein aggregation to cell death is the subject of intense investigation. Although aggregates are the most striking physical change in surviving cells, the actual role of the aggregate remains a mystery. Indeed, many now believe that the formation of aggregates may be a protective mechanism sequestering the wayward protein from vulnerable cell processes. Misfolded proteins may produce the most mischief as they form protofibrils. A number of mechanisms have been described. In some cases, these are specifically related to the type of protein, but in many other cases, they are nonspecific mechanisms shared by all the misfolded protein diseases. Some potential mechanisms of neurodegeneration related to misfolded protein stress are listed in Box 71.1. The mutant protein may be unable to perform a vital function or may interfere with the function of the wild-type protein. Mutant protein, protofibrils, or aggregates might interfere with other proteins. Interference with transcription factors may be particularly important in this regard. Mutant proteins may activate caspases or in other ways activate the apoptotic cascade. They may interfere with intracellular transport or other vital processes. They may suppress activity of the proteasome, enhancing protein aggregation. They may interfere with mitochondrial function, making cells more vulnerable to excitotoxicity. In addition to the ubiquitin-proteasome system, lysosomes play an important role in degrading intracellular proteins by a process termed autophagy (Pan et al., 2008). When the function of the ubiquitin proteosome system is not sufficient to clear the accumulating cellular proteins, the autophagy lysosome pathway becomes the other important route for degradation of aggregated/misfolded proteins as well as sick or abnormal mitochondria. Accumulation of iron, increased oxidative stress, and microglial activation have also been thought to play important roles in the pathogenesis of various neurodegenerative disorders (Zhu et al., 2010).
Recent ultrastructural work has elucidated that many degenerative movement disorders can be linked to abnormal synthesis, folding, or degradation of specific proteins or protein families. Thus, a number of parkinsonian disorders are thought to result from neuronal accumulation of α-synuclein or tau proteins. Certain other disorders are linked to abnormalities in pathways involving proteins with expanded polyglutamine tracts (polyQ). Among these disorders, clinical and pathological differences depend on the distribution of protein aggregates or in the abnormal configuration they assume when they aggregate. The synucleinopathies include PD, Lewy body disease (LBD), and multiple system atrophy (MSA). The tauopathies include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), familial frontotemporal dementia with parkinsonism (FTDP), postencephalitic parkinsonism (PEP), posttraumatic parkinsonism, and amyotrophic lateral sclerosis (ALS)-PD of Guam. The polyQ disorders include HD, dentatorubral-pallidoluysian atrophy (DRPLA), and many spinocerebellar ataxias (Ling et al., 2010; Williams and Lees, 2009).
Parkinsonian Syndromes
Parkinson Disease
Although the disease was described by others before him, James Parkinson is attributed with rendering the first cogent description of PD. In his monograph, The Shaking Palsy (1817), he identified the hallmark features of the illness through descriptions of cases observed in the streets of London as well as in his own patients. Over time, Parkinson disease or idiopathic PD has replaced the original term paralysis agitans as the name for the clinical syndrome of asymmetrical parkinsonism, usually with rest tremor, in association with the specific pathological findings of depigmentation of the SN due to loss of melanin-laden dopaminergic neurons containing eosinophilic cytoplasmic inclusions (Lewy bodies) (see Chapter 21). Dopamine deficiency in parkinsonian brain homogenates was described by Hornykiewicz in 1959, a discovery that ultimately led to highly effective pharmacotherapy with LD and direct-acting dopamine agonists (DAs). Recently, genetic forms of parkinsonism that are clinically indistinguishable from PD have been linked to mutations in several genes (Shulman et al., 2011). The discovery of different genetic forms of parkinsonism with variable penetrance has led to the current concept of PD as a syndrome with genetic and environmental etiologies, but overall, gene mutations are a rare cause of parkinsonism, particularly in those patients with late-onset disease (Gandhi and Wood, 2010).
Epidemiology and Clinical Features
In community-based series, PD accounted for more than 80% of all parkinsonism, with a prevalence of approximately 360 per 100,000 and an incidence of 18 per 100,000 per year (de Lau and Breteler, 2006). PD is an age-related disease, showing a gradual increase in prevalence beginning after age 50, with a steep increase in prevalence after age 60 (Driver et al., 2009). Disease before 30 years of age is rare and often suggests a hereditary form of parkinsonism. Prevalence rates in the United States are higher than those in Africa and China, but the role of race remains unclear. Within the United States, race-specific prevalence rates vary, with some studies suggesting a similar prevalence among whites and blacks. Unfortunately, blacks make up only a small fraction of most specialty clinic populations and thus are underrepresented in clinic-based studies and clinical trials. One study showed the world’s highest prevalence of PD may be among the Amish in the U.S. Northeast—nearly 6% of those 60 years of age or older, more than 3 times the prevalence for the rest of the country (Racette et al., 2009).
Typically, the onset and progression of PD are gradual. The most common presentation is with rest tremor in one hand, often associated with decreased arm swing and shoulder pain (Jankovic, 2008). Bradykinesia and rigidity are often detectable on the symptomatic side, and midline signs such as reduced facial expression or mild contralateral bradykinesia and rigidity may already be present. The presentation may be delayed if bradykinesia is the earliest symptom, particularly when the onset is on the nondominant side. The disorder usually remains asymmetrical throughout much of its course. With progression of the illness, generalized bradykinesia may cause difficulty arising from a chair or turning in bed. The gait and balance are progressively affected, and falls may occur. Sudden arrests in movement, also called freezing or motor blocks, soon follow, first with gait initiation, turning and traversing narrow or crowded environments, and then during walking. Bulbar functions deteriorate, impairing communication and nutrition. The tremor-dominant form of PD generally has a more favorable clinical course than PD dominated by gait disorder and postural instability (van Rooden et al., 2011).
The Unified Parkinson’s Disease Rating Scale (UPDRS) has been used to quantitate the various motor symptoms and signs of PD and to chart the course of the disease. This traditional scale, now known as the Movement Disorder Society (MDS)-UPDRS, has been revised to clarify some ambiguities in the original version and to capture early motor and also nonmotor symptoms associated with PD (www.movementdisorders.org). The Hoehn and Yahr stage, first described before effective dopaminergic treatment became available, outlines the milestones in progression of the illness from mild unilateral symptoms through the end-stage nonambulatory state. A modified version of the Hoehn and Yahr stage is commonly used in contemporary clinical trials (Table 71.4).
| ORIGINAL SCALE | |
| Stage | Disease State |
| I | Unilateral involvement only, minimal or no functional impairment |
| II | Bilateral or midline involvement, without impairment of balance |
| III | First sign of impaired righting reflex, mild to moderate disability |
| IV | Fully developed, severely disabling disease; patient still able to walk and stand unassisted |
| V | Confinement to bed or wheelchair unless aided |
| MODIFIED SCALE | |
| Stage | Disease State |
| 0 | No signs of disease |
| 1 | Unilateral disease |
| 1.5 | Unilateral plus axial involvement |
| 2.0 | Bilateral disease without impairment of balance |
| 2.5 | Mild bilateral disease with recovery on pull test |
| 3.0 | Mild to moderate bilateral disease, some postural instability, physically independent |
| 4.0 | Severe disability, still able to walk or stand unassisted |
| 5.0 | Wheelchair bound or bedridden unless aided |
Nonmotor symptoms are increasingly recognized as a major cause of disability in PD and contribute prominently to declining quality of life, particularly in the more advanced stages of the disease (Lim et al., 2009). Autonomic symptoms include reduced gastrointestinal transit time with postprandial bloating and constipation, urinary frequency and urgency (sometimes with urge incontinence), impotence, disordered sweating, and orthostatic hypotension (Mostile and Jankovic, 2009). Cognitive and behavioral changes are also very common. Attention and concentration wane. Executive dysfunction with diminished working memory, planning, and organization is common. Global dementia occurs in approximately 30% of patients, increasing in frequency with the age of the patient. Those with prominent early executive dysfunction and more severe motor signs seem particularly at risk. Anxiety, depression, and other mood disorders are common in PD. Sleep disturbance is nearly universal in PD and is multifactorial.
Disordered sleep onset and maintenance lead to fragmentation of nocturnal sleep. A variety of motor movements including restless legs and periodic leg movements of sleep may be seen, and many patients have rapid eye movement (REM) sleep behavior disorder (RBD) with active motor movements during REM sleep (Postuma et al., 2010). Some patients have sleep apnea. Vivid dreams and nightmares are very common, particularly in treated patients. Sleep disorders in PD variably relate to the pathological changes of the disease itself, arousals due to immobility, comorbid primary sleep disorders, and side effects of antiparkinsonian medications. Many patients with PD are excessively sleepy during the day, sometimes with serious consequences such as unintended sleep episodes while driving. In most cases, this excessive daytime drowsiness is related to dopaminergic drugs. Fatigue is a common and complex symptom of PD. The differentiation of fatigue from excessive daytime sleepiness, depression, apathy, and other conditions can be difficult, and there is not yet a useful body of literature on its assessment and treatment.
Clinicopathological studies have found that the clinical syndrome that best reflects the typical pathological changes of PD, in the absence of other diagnoses known to cause parkinsonism, is an asymmetrical illness with rest tremor along with rigidity or bradykinesia and marked improvement with LD. Misdiagnosed cases generally are found to have MSA, PSP, or subcortical vascular disease. When making the diagnosis of early PD, the clinician should be aware of a number of red flags (Box 71.2) (see Chapter 21). Cognitive impairment within the first year should raise the possibility of Alzheimer disease (AD), dementia with Lewy bodies (DLB), CBD, PSP, or FTDP. Symmetrical or prominent midline or bulbar signs suggest MSA or PSP. Early gait disorder with falls points to the diagnosis of PSP or to subcortical vascular disease. Dependence on a wheelchair within 5 years of onset is suggestive of PSP or MSA. Early orthostatic hypotension or incontinence points to the autonomic dysfunction of MSA. Severe sleep apnea, inspiratory stridor, or involuntary sighing also suggests MSA. Apraxia, alien limb, or cortical sensory loss is typically seen in CBD.
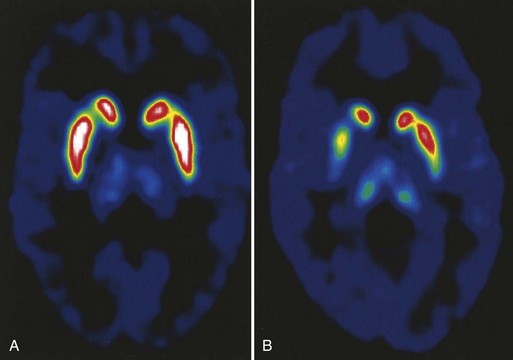
Neuroimaging studies such as computed tomography (CT) and magnetic resonance imaging (MRI) are also usually unhelpful in making a diagnosis of PD, because they are generally normal or show only incidental abnormalities. Sometimes neuroimaging abnormalities can be useful in suggesting alternative diagnoses such as PSP or MSA. The radiopharmaceutical 6-[18F]-fluorodopa (F-dopa) is taken up by dopaminergic neurons in the SN and metabolized to 6-[18F]-fluorodopamine. Positron emission tomography (PET) scans using this radiopharmaceutical agent show reduced F-dopa uptake in dopaminergic nerve terminals in the putamen and caudate proportional to the severity of degeneration in the ipsilateral SN and symptoms in the contralateral hemibody (Fig. 71.5). Although these tests are used in PD research, they are not readily clinically available at this time. Single-photon emission CT (SPECT) with radioligands that label the dopamine transporter on nerve terminals in the striatum is another research tool, and its clinical relevance is now being studied. There is no role for routine electrophysiological testing in the diagnosis of PD. Because identified genetic causes are rare and their interpretation for genetic counseling is difficult, deoxyribonucleic acid (DNA) tests should not be used routinely in the diagnosis of PD (see Chapter 21).
Pathology
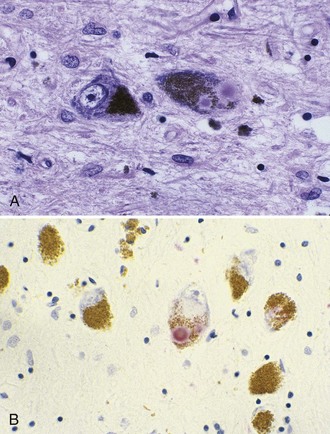
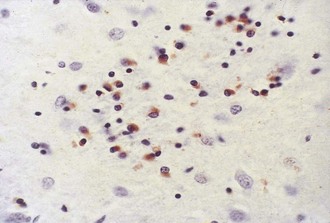
The most striking pathological changes in PD occur in the SNc. The SN appears pale to the naked eye. Microscopic changes include neuronal loss, gliosis, and the presence of extracellular pigment. Surviving neurons may show characteristic cytoplasmic inclusions (Fig. 71.6). These inclusions, called Lewy bodies, have a dense eosinophilic core and a pale halo. They contain hyperphosphorylated neurofilament proteins, lipids, iron, ubiquitin, and α-synuclein (Lees et al., 2009). Pigmented nuclei elsewhere in the brainstem, including the locus ceruleus, dorsal motor nucleus of the vagus, and others, may also show Lewy bodies and characteristic degenerative changes. The substantia innominata and intermediolateral cell column in the spinal cord also are affected. Patients with PD and dementia show more diffuse Lewy body pathology or comorbid AD. Even the myenteric intestinal and cardiac plexus of patients with PD may contain Lewy bodies, showing that PD is not just a CNS disease.
A staging system introduced by Braak (Braak and Del Tredici, 2008) has been developed to characterize the progression of neuropathological changes associated with PD. According to Braak staging, during the presymptomatic stages (1 and 2), the PD-related inclusion body pathology remains confined to the medulla oblongata and olfactory bulb. In stages 3 and 4, the substantia nigra and other nuclear grays of the midbrain and basal forebrain are the focus of initially subtle and then severe changes, and the illness reaches its symptomatic phase. In the end stages (5 and 6), the pathological process encroaches upon the telencephalic cortex. Although the Braak hypothesis is supported by early olfactory, sleep, and autonomic involvement in patients with PD, the staging proposal has been challenged for many reasons and inconsistencies, such as absence of cell counts to correlate with the described synuclein pathology, absence of immuno-histochemistry to identify neuronal types, absence of observed asymmetry in the pathological findings that would correlate with the well-recognized asymmetry of clinical findings, absence of bulbar symptoms as early features of PD, and the observation that brain synucleinopathy consistent with Braak stages 4 and 6 has been found in individuals without any neurological signs. There are several other concerns about the Braak hypothesis (Halliday et al., 2008).
Etiology
Studies of large numbers of patients with PD have suggested that PD is a multifactorial illness with likely genetic and environmental determinants (Shulman et al., 2011). Although strongly hereditary PD is rare, there is a tendency for familial aggregation of PD. Twin studies suggest that heredity plays a relatively small role in the population at large, but the hereditary component is greater if one twin has disease onset at younger than age 50. Moreover, PET studies of twins suggest that most monozygotic twins of patients with PD show subclinical declines in dopamine innervation, strengthening the evidence for a significant hereditary contribution irrespective of age at onset.
Although the majority of cases of PD appear to be sporadic, it is becoming increasingly evident that genetic factors play an important role in the pathogenesis of PD, particularly if onset is earlier than age 50 (see Chapter 21, Table 21.1). Some 20% to 25% of patients have at least one first-degree relative with PD, and first-degree relatives are 2 to 3 times as likely as relatives of controls to develop PD. The most cogent evidence for genetic contribution to the pathogenesis of PD has been provided by reports of large multicase kindreds with dominantly inherited autopsy-proven PD. A genome scan in the Contursi kindred of Greek-Italian origin found a genetic marker on chromosome 4q21-q23 linked to the PD phenotype. Subsequent studies identified at least three different mutations in the α-synuclein gene (SNCA), the first monogenetic form of PD, designated PARK1 (see Table 21.1). In addition to the typical PD features, this family exhibited dementia, severe central hypoventilation, orthostatic hypotension, prominent myoclonus, urinary incontinence, and pathological involvement of the brainstem pigmented nuclei, hippocampus, and temporal neocortex. Later, the application of quantitative real-time PCR amplification of the SNCA gene showed that some families with a PD phenotype, originally designated as PARK 4, had duplication and triplication of the gene, with marked increase in the amount of α-synuclein protein. Thus, an overexpression of α-synuclein may lead to neurodegenerative disease with features overlapping with PD, DLB, and MSA. Based on screening, the entire coding region of the gene in a large number of PD patients shows that mutation in the SNCA gene is a rare cause of PD.
A growing number of novel genes have been implicated in the pathogenesis of PD (see Table 21.1). One locus for PD has been found on chromosome 2p13 (PARK3) in rare European families with a mean age at onset of 61 years. Although some members of the kindreds had prominent dementia, the autopsy in some cases confirmed the typical pathological features of PD, including Lewy bodies. Another piece of evidence that an impairment of normal protein degradation is an important mechanism of neurodegeneration in PD is the finding of a mutation in the de-ubiquinating enzyme, ubiquitin carboxyterminal hydrolase L1 (UCHL1) gene, on 4p14 in a German family with autosomal dominant PD (PARK5). Mutations in the PTEN-induced putative kinase 1 (PINK1) gene on chromosome 1p36 were identified in autosomal recessive families with early-onset parkinsonism (PARK6). The PINK1 gene codes for a putative serine-threonine kinase located in the mitochondria, thus providing further support for the role of oxidative stress in the pathogenesis of PD. The mean age at onset is in the fourth decade, and the course is quite benign, associated with LD-induced dyskinesias. These clinical features are similar to those of another autosomal recessive form of PD (PARK7) localized to the same chromosomal region in the DJ-1 gene. Besides slow progression and good, prolonged response to LD, patients with a DJ-1 mutation may exhibit blepharospasm, leg dystonia, anxiety, and parkinsonism-dementia-ALS complex. In contrast to parkin mutations that may account for up to 50% of young-onset PD, the DJ-1 mutations account for approximately 1% of all young-onset PD cases.
Another locus mapped to 12p11.23-q13.11 (PARK8) was initially identified in a Japanese family with typical PD inherited in an autosomal dominant pattern with incomplete penetrance and has been subsequently found to be the most common form of familial adult-onset PD (Marras et al., 2011). The course of the disease is relatively benign, usually presenting with unilateral hand or leg tremor without cognitive deficit; the patients respond well to LD. Other clinical phenotypes have included parkinsonism with dementia, hallucinations, dysautonomia, amyotrophy, or both and otherwise typical essential tremor. Autopsy studies demonstrate variable pathology ranging from Lewy body and tau neurofibrillary tangle pathology to no pathological changes. The gene responsible for PARK8 on 12p11.2-q13.1, called LRRK2 (leucine-rich repeat kinase 2), belongs to the ROCO protein family and includes a protein kinase domain of the MAPKKK class and several other major functional domains. The gene product, a protein called dardarin (from Basque word dardara, meaning “tremor”), is a novel protein that probably functions as a cytoplasmic kinase involved in phosphorylation of proteins such as α-synuclein and tau. LRRK2 is closely associated with a variety of membrane and vesicular structures, membrane-bound organelles, and microtubules, suggesting its role in vesicular transport and membrane and protein turnover, including the lysosomal degradation pathway. This mutation has been found to be particularly frequent in PD patients of North African origin and in Ashkenazi Jewish patients. The penetrance is quite variable, and many elderly individuals with the mutation but no signs of PD have been reported. The variable penetrance and growing number of causative and susceptibility genes, coupled with a growing number of commercially available DNA tests, has obvious implications for genetic counseling (Gandhi and Wood, 2010).
Epidemiological studies suggest that exposure to environmental metals or organic toxins may be associated with an increased risk of PD or an earlier age at onset (Tanner et al., 2009). Case-controlled studies have suggested that the risk of PD is increased in persons who have worked in the agricultural industry, have been exposed to pesticides, or have sustained significant head injury. Whether exposure to welding predisposes to earlier onset of PD, possibly as a result of manganese poisoning, is controversial (Jankovic, 2005). On the other hand, the risk of PD seems lower in those with a high dietary intake of antioxidant-rich foods, as well as caffeine drinkers and those who have smoked cigarettes (Chen et al., 2010). These epidemiological studies have failed to find a “smoking gun” in the etiopathogenesis of PD.
Although PD and cancer are two distinct diseases that result from either degeneration or over-proliferation, respectively, several recent studies have provided evidence that while PD provides some type of biological protection against most types of cancers, the disease confers increased risk for other cancers such as melanoma (Inzelberg and Jankovic, 2007). The relationship between PD and melanoma is being explored, but the higher frequency of melanoma does not appear to be due to LD. It is possible that high concentrations of α-synuclein in the skin of patients with PD may increase the risk of melanoma in PD patients by inhibiting tyrosine hydroxylase, an enzyme for dopamine and melanin, or by some other mechanism (Pan et al., 2011).
Treatment
Before discussing specific treatment strategies for PD, it is important to recognize that the quantitative assessment of clinical symptoms and progression of the course is an essential component of any therapeutic trial (Jankovic, 2008). In addition to sensitive clinical rating scales (e.g., MDS-UPDRS), reliable diagnostic, presymptomatic, and progression biomarkers are needed (Wu et al., 2011) (Table 71.5).
Table 71.5 Candidate Neuroprotective Approaches in Parkinson Disease
| Mechanism | Potential Neuroprotective Approaches |
|---|---|
| Oxidative stress | Antioxidants (e.g., monoamine oxidase inhibitors, coenzyme Q10) |
| Mitochondrial dysfunction | Coenzyme Q10, creatine |
| Excitotoxicity | Glutamate antagonists (e.g., riluzole) |
| Caspase activation | Caspase inhibitors (e.g., minocycline) |
| Apoptosis | Antiapoptotic agents (e.g., mixed-lineage kinase inhibitors) |
| Inflammation | Antiinflammatory drugs |
| Trophin deficiency | Neurotrophins |
Neuroprotective Or Disease-Modifying Therapies For Parkinson Disease
A preclinical period lasting years, its slow progression rate, and our increasing understanding of disease etiopathogenesis make PD an ideal candidate for neuroprotective therapeutic strategies. However, double-blind placebo-controlled trials designed to explore therapies that may have favorable disease-modifying effects and slow disease progression have been thus far disappointing. The first “neuroprotective” trial, DATATOP (Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism), randomized patients with early PD to treatment with placebo, tocopherol, selegiline (Deprenyl), or both, using the time until the patients needed potent symptomatic dopaminergic therapy, LD, as a proxy endpoint for disease progression. The selective monoamine oxidase (MAO-B) inhibitor, selegiline, successfully delayed this endpoint, but interpretation of the study was contaminated by the drug’s mild antiparkinsonian and antidepressant properties, as well as the potential effects of its amphetamine metabolites. Although disease-modifying effects of selegiline have been suggested by some clinical trials (Palhagen et al., 2006), further studies are needed before it can be concluded that selegiline is neuroprotective in PD. Another MAO-B inhibitor, rasagiline, has been shown to have modest symptomatic benefit, but its effects on disease progression are also still being debated. In a randomized multicenter, double-blind, placebo-controlled, parallel-group study prospectively examining rasagiline’s potential disease-modifying effects (ADAGIO [Attenuation of Disease Progression with Azilect Given Once-Daily]), delayed-start design was used to assess the potential disease-modifying effects of rasagiline (Olanow et al., 2009). A total of 1176 patients with early untreated PD (mean time from diagnosis, 4.5 months) from 129 centers in 14 countries were randomized into 4 treatment groups (either 1 or 2 mg/day, early-start versus delayed treatment, 9 months each). Early-start treatment consisted of 72 weeks of rasagiline (either 1 or 2 mg once daily), and delayed-start treatment consisted of 36 weeks of placebo followed by 36 weeks of rasagiline (either 1 or 2 mg once daily [active treatment phase]). The primary analyses of the trial were based on change in total UPDRS score and included slope superiority of rasagiline over placebo in the placebo-controlled phase, change from baseline to week 72, and noninferiority of early-start versus delayed-start slopes during weeks 48 through 72 of the active phase. The 1-mg dose group met all three endpoints, but there was no observable benefit with the higher 2-mg dose, although when analyzing the upper quartile group, the 2-mg dose group met all the primary endpoints. Some possible explanations for the seemingly confusing outcome include early symptomatic treatment helping some compensatory mechanism, cumulative symptomatic effect, and other possibilities.
Many studies have concluded that that DAs such as pramipexole, ropinirole, and rotigotine, when introduced early in the course of PD treatment, may delay LD-related complications such as motor fluctuations and dyskinesias (Jankovic and Aguilar, 2008). But evidence is lacking to support the hypothesis that early introduction of DAs slows progression of the disease. The PROUD study (Pramipexole on Underlying Disease), which assessed early versus delayed pramipexole treatment in early PD, involved 535 untreated PD patients who were randomized to double-blind placebo or pramipexole (1.5 mg/day) for 6 to 9 months and continued with pramipexole for up to 15 months. The researchers found no difference in UPDRS (−0.4 UPDRS units) or PDQ-39 scores in the 411 patients who completed the 15-month study, but at the end of the placebo-controlled phase, the difference in adjusted means was −4.8 UPDRS units (95% CI, −6.3, −3.2; P < 0.0001), and there was a significant difference in PDQ-39 (P = 0.0001), both in favor of pramipexole (Schapira et al., 2010). Furthermore, many studies have shown that LD is more effective than DAs in reducing motor symptoms in early as well as advanced stages of PD (Parkinson Study Group, 2009).
Symptomatic Treatment Of Parkinson Disease
Many types of medications are available for symptomatic treatment of PD: anticholinergics, amantadine, LD, MAO inhibitors (MAOIs), catechol-O-methyltransferase inhibitors (COMTIs), and DAs (Table 71.6). Anticholinergics such as trihexyphenidyl and benztropine antagonize the effects of acetylcholine at muscarinic receptors postsynaptic to striatal interneurons. They reduce tremor and rigidity but have no effects on bradykinesia. Toxicity relates to antagonism of acetylcholine at central receptors, causing confusion, and peripheral receptors, causing blurred vision, dry mouth, constipation, and urine retention. Although amantadine has been available for nearly 4 decades (it was originally marketed as an antiviral agent), its antiparkinsonian mechanisms have been poorly understood. It has been thought to stimulate release of endogenous dopamine stores, block reuptake of dopamine from the synaptic cleft, and have anticholinergic properties. However, amantadine has been found to have antiglutamatergic properties and as such is the only antiparkinsonian drug that improves levodopa-induced dyskinesia.
| Drug | Usual Starting Dose | Usual Daily Dose |
|---|---|---|
| ANTICHOLINERGICS | ||
| Trihexyphenidyl | 1 mg | 2-12 mg |
| Benztropine | 0.5 mg | 0.5-6 mg |
| Biperiden | 1 mg | 2-16 mg |
| AMANTADINE | 100 mg | 100-300 mg |
| LEVODOPA (WITH CARBIDOPA) | ||
| Immediate-release | 100 mg | 150-800 mg |
| Controlled-release | 100 mg | 200-1000 mg |
| DOPAMINE AGONISTS | ||
| Bromocriptine | 1.25 mg | 15-40 mg |
| Pergolide | 0.05 mg | 2-4 mg |
| Pramipexole | 0.375 mg | 1.5-4.5 mg |
| Ropinirole | 0.75 mg | 8-24 mg |
| Cabergoline | 0.25 mg | 0.25-4 mg |
| CATECHOL-O-METHYL TRANSFERASE INHIBITORS | ||
| Entacapone | 200 mg | 200 mg |
| Tolcapone | 300 mg | 600 mg |
A number of direct-acting DAs are available, including bromocriptine, pergolide, pramipexole, and ropinirole. Because of various adverse effects related to their ergot structure, particularly fibroproliferative lesions of heart valves, lung, and other tissues, bromocriptine, pergolide, and cabergoline are only rarely used today. Cabergoline, lisuride, piribedil, and rotigotine are not available in the United States, but these DAs are available in other countries. Apomorphine, a nonergoline DA, is water soluble and lipophilic and is therefore suitable for intravenous, subcutaneous, sublingual, intranasal, or transdermal administration. Apomorphine is available as an acute intermittent subcutaneous injection, as a rapid rescue from hypomobility off episodes (end-of-dose wearing off and unpredictable on/off episodes) associated with advanced PD. DAs cause side effects similar to those of LD, although orthostatic hypotension, sleepiness, and hallucinations are more common or severe. One major concern with DAs is the emergence of a variety of behavioral problems that include pathological gambling, compulsive shopping and eating, hypersexuality, and other impulse-control disorders (Voon et al., 2009).
Selegiline and rasagiline block MAO-B–dependent dopamine degradation and have modest effects in potentiating the action of LD. A novel formulation of selegiline was approved by the U.S. Food and Drug Administration (FDA) in 2006 as an adjunctive therapy to LD in the management of PD. Zydis (Zelapar) is formulated in a freeze-dried tablet that contains 1.25 mg of fast-dissolving selegiline that results in a Tmax of only 15 minutes—4 times shorter than conventional selegiline. This is particularly useful for patients who have difficulty swallowing. Furthermore, the pregastric absorption avoids extensive first-pass metabolism in the liver, and the concentration of amphetamine-like metabolites is therefore much lower. In a 3-month placebo-controlled study of patients with PD who were experiencing levodopa-related motor fluctuations, Zydis selegiline was associated with a 2.2-hour reduction in the total number of off-hours compared to placebo, and the number of dyskinesia-free on-hours increased by 1.8 hours (Waters et al., 2004).
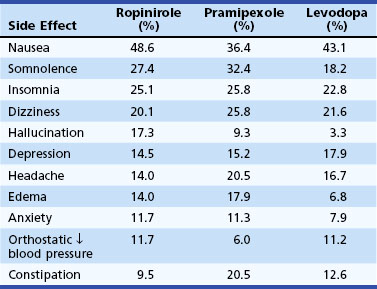
Clinical experience with LD treatment of PD suggests that there is a progressive increase in the prevalence of drug-related motor fluctuations (wearing off, dyskinesia) over time, and that about half of patients experience wearing off, and a third experience dyskinesias within 2 years after initiation of LD therapy. Wearing off results from loss of DA storage in the striatum as a result of loss of nigrostriatal terminals. Experiments in animal models relate the development of dyskinesia to changes in striatal glutamate receptor sensitivity consequent to pulsatile stimulation of striatal dopamine receptors. Continuous dopamine receptor stimulation with LD or with long-acting DAs prevents or reverses this phenomenon. Several clinical trials were designed to address the question of whether the more sustained dopamine receptor stimulation afforded by DA therapy (pergolide, pramipexole, ropinirole) was less likely to induce motor fluctuations and dyskinesia than therapy with the shorter half-life agent, LD. The studies showed that the efficacy of LD for the primary symptoms of PD was superior to that of DAs. Also, LD was better tolerated than DAs, which caused more edema, sleepiness, and hallucinations than LD (Parkinson Study Group, 2009) (Table 71.7). The risk of dyskinesia or motor fluctuations, however, was substantially reduced in the DA monotherapy group and even in subjects who ended the study on LD and a DA. At this time, it remains unclear whether the delay in development of early dyskinesias and fluctuations is worth the poorer efficacy and tolerability and greater expense of agonists. Clearly, by the second decade of therapy, essentially all patients are on LD, often in combination with a DA, and it is not yet clear whether initial treatment influences overall long-term function or quality of life. In many specialty centers, otherwise healthy and cognitively intact patients younger than age 70 are usually treated first with DAs, and those who are cognitively impaired or elderly are started on LD.
Irrespective of initial treatment, virtually all patients with PD experience an evolution of their response to LD that includes wearing off and other motor fluctuations. In eliciting a description of the patient’s response to medication, it is important to understand the severity of symptoms in the morning on arising and the magnitude and duration of benefit from each dose of antiparkinsonian drug. The usefulness of historical information may be augmented by careful patient education on symptom recognition and the development of a shared vocabulary. Completing motor diaries (Fig. 71.7) helps both the patient and the treating physician recognize patterns of motor response and adjust the medications accordingly.
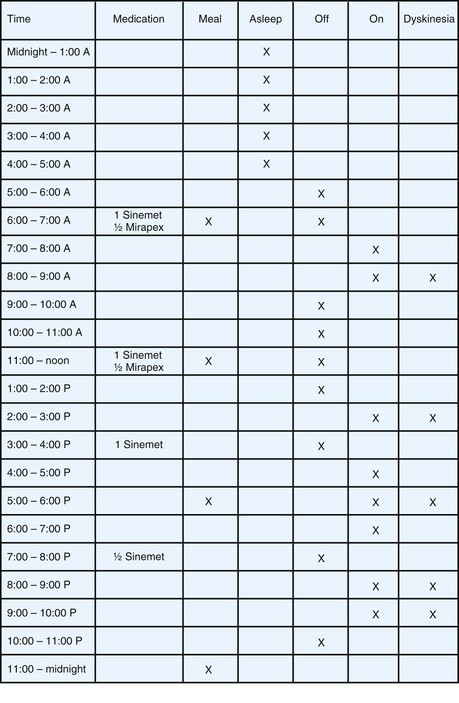
Armed with a few basic principles and a commonsense approach, the clinician can usually smooth out fluctuations for most patients with appropriate selection of drugs and dose (Box 71.3). Delay to onset of therapeutic benefit can be hastened by avoiding or reducing protein ingestion or by crushing the LD tablet and mixing it with a carbonated beverage. The duration of benefit increases when the incremental dose is increased or dopamine metabolism is blocked with an MAO-B or COMT inhibitor. The brittle patient with motor fluctuations and dyskinesia may do better on smaller, more frequent LD doses.
Surgical Treatment Of Parkinson Disease
Thalamic DBS reduces or eliminates contralateral rest tremor in 75% to 85% of patients, but there is less effect on rigidity and no effect on bradykinesia. The chief advantage of this procedure over ablative lesioning is that the stimulation parameters can be customized to the needs of the patient to optimize the benefits and control adverse effects such as paresthesia, dysarthria, and balance difficulties—the latter two more frequent with bilateral stimulation. Thalamic DBS is most frequently used to control high-amplitude tremor, but STN or GPi are the most frequent targets for DBS treatment of patients with PD with disabling LD-related complications. To address the question of whether optimal medical therapy or DBS provides more robust improvement, 255 patients at 7 Veterans Affairs and 6 university hospitals were enrolled in a randomized controlled trial designed to compare the effects of DBS (STN, n = 60; or GPi, n = 61) and “best medical therapy” (n = 134) after 6 months of treatment (Weaver et al., 2009). Patients treated with DBS gained a mean of 4.6 hours/day of on time without troubling dyskinesia, compared to 0 hours/day for patients who received best medical therapy (P<0.001). Furthermore, motor function improved by 5 or more points on the motor UPDRS in 71% of DBS and 32% of medical therapy patients. This was accompanied by improvements in the majority of PD-related HRQOL (health-related quality of life) measures and only minimal decrement in neurocognitive testing. The overall risk of experiencing a serious adverse event, however, was 3.8 times higher in the DBS than in the medical therapy group (40% versus 11%). In a follow-up analysis of the Veterans Affairs Cooperative Studies Program outcomes, STN and GPi DBS were analyzed after 24 months in 299 patients, and there were no differences in mean changes in the motor (part III) UPDRS between the two targets (Follett et al., 2010). Patients undergoing STN required a lower dose of DAs than those undergoing pallidal stimulation (P = 0.02), and visuomotor processing speed declined more after STN than after GPi stimulation (P = 0.03). On the other hand, there was worsening of depression after STN DBS, but mood improved after GPi DBS (P = 0.02). Slightly more than half of the patients experienced serious adverse events, but there was no difference in the frequency of these events between the two groups. Based on these and other studies, there is emerging evidence that GPi DBS may be particularly suitable for patients who may have troublesome dyskinesias as well as mild cognitive or behavioral impairment, whereas bilateral STN DBS may be the surgical choice for patients who are cognitively intact but in whom reduction in levodopa dosage is the primary goal. While DBS is a proven effective therapeutic strategy its success depends on the appropriate selection of patients and the experience and skill of the stereotactic surgeon in order to optimize the results and minimize complications (Baizabal Carvallo et al., 2011).
Pharmacological suppression of excessive STN firing may be possible with a gene therapy approach that uses adeno-associated viral vector delivery of glutamic acid decarboxylase (GAD), the enzyme that synthesizes GABA. This technique is now being investigated in human trials. Glial cell line–derived neurotrophic factor–like molecules such as neurturin are being explored as possible trophic factors with the potential for clinical use. Surgical gene delivery of adeno-associated virus-based vector encoding human neurturin (AAV2-NTN, also called CERE-120), has been tested in parkinsonian animals and is currently undergoing further clinical trials (Marks et al., 2008). Human fetal brain transplantation is not likely to be a useful intervention for PD, partly because of lack of efficacy demonstrated by controlled trials and because of the possibility that the disease can propagate from host to graft cells, as suggested by synuclein pathology found at autopsy in some graft cells.
Multiple System Atrophy
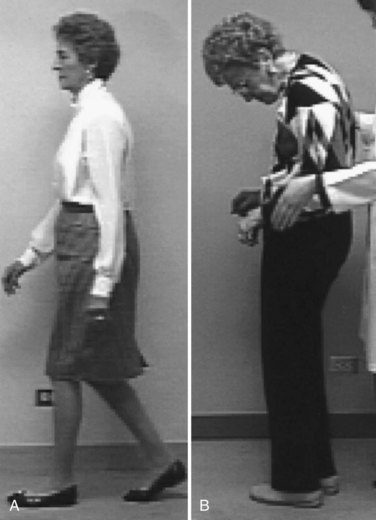
Several clinical studies have addressed differentiating between MSA-P and parkinsonism, and a collection of “red flags” has been generated and recently validated as having high diagnostic specificity (Stefanova et al., 2009). The red flags were grouped into 6 categories: (1) early instability, (2) rapid progression, (3) abnormal postures (includes Pisa syndrome, disproportionate anterocollis, and/or contractures of hands or feet) (Fig. 71.8), (4) bulbar dysfunction (includes severe dysphonia, dysarthria, and/or dysphagia), (5) respiratory dysfunction (includes diurnal or nocturnal inspiratory stridor and/or inspiratory sighs) (Mehanna and Jankovic, 2010), and (6) emotional incontinence (includes inappropriate crying and/or laughing). They proposed that a combination of 2 out of these 6 red-flag categories be used as additional criteria for the diagnosis of probable MSA-P. Other characteristic features of MSA include early hypokinetic dysarthria, distal myoclonus, and a variety of autonomic symptoms and signs such as cold hands and feet with bluish discoloration of the distal extremities, orthostatic hypotension, incontinence, and impotence. MSA patients also have more autonomic symptoms at baseline and more progression to global anhidrosis than patients with PD (Lipp et al., 2009). The autonomic symptoms (particularly sexual dysfunction) and RBD may precede the onset of motor symptoms by years or even decades. Patients with MSA-C have parkinsonism with prominent cerebellar signs, especially wide-based ataxic gait. Although there may be a positive response to LD, this is generally relatively short lived and often associated with facial and oromandibular dyskinesia.
Clinical tests of autonomic dysfunction may be helpful in diagnosis or treatment (Mostile and Jankovic, 2009). Testing of cardiovascular reflexes such as heart rate variability at rest and during forced respiration, as well as blood pressure changes during head-up tilt, may help establish a clinical diagnosis of MSA. A lack of responsiveness of growth hormone to clonidine challenges and denervation on rectal sphincter electromyography (EMG) are also characteristic findings. T2-weighted MRI brain scans may show a hyperintense rim at the lateral edge of the dorsolateral putamen, with decreased signal within the putamen. Cruciform hyperintensity within the pons, the so-called hot-cross-bun sign, may also be a helpful marker (Brooks et al., 2009). PET scan with [11C]PMP that images subcortical AChE activity was significantly more decreased in MSA-P and PSP than in PD, possibly reflecting greater impairment in the pontine cholinergic group (PPN); this may account for the greater gait disturbances in the early stages of these two disorders compared to PD (Gilman et al., 2010).
At autopsy, MSA brains show neuronal loss and gliosis in the striatum, SN, locus ceruleus, inferior olive, pontine nuclei, Purkinje cells, intermediolateral cell column, and the Onuf nucleus in the sacral spinal cord. Glial cytoplasmic inclusions containing α-synuclein (Fig. 71.9) are the most characteristic histological feature linking the different types of MSA. There is a severe depletion of cholinergic neurons in the PPN and laterodorsal tegmental nucleus. The etiology of MSA remains unknown, but genetic factors do not seem to play an important role.
Progressive Supranuclear Palsy
First described in 1964 by Steele, Richardson, and Olszewski as a progressive illness characterized by vertical supranuclear ophthalmoplegia, axial rigidity, pseudobulbar palsy, and mild dementia, PSP is the second most common form of neurodegenerative atypical parkinsonism (Azher and Jankovic, 2008). After vascular parkinsonism, PSP represents the third most common cause of parkinsonism, but it is still a relatively rare disease, with prevalence estimates ranging from 1.39 to 6.4 per 100,000. Men are affected more often than women. PSP typically begins with gait disorder and falling in the sixth to seventh decades of life. Patients develop an akinetic rigid state with symmetrical signs and prominent axial rigidity. In contrast to the flexed posture of patients with PD, those with PSP may have an extended trunk or retrocolic neck posture. A characteristic facial appearance features a wide-eyed stare, furrowing of the forehead, and deepening of other facial creases, allowing experienced clinicians to make an instant diagnosis (Fig. 71.10). Pseudobulbar palsy with dysarthria and dysphagia lend the patient a characteristic dysarthria with spasticity, hypokinesia, and ataxia and often “silent” aspiration. Frontal lobe features are common. There is striking executive dysfunction early in the disease course; concrete thought, difficulty shifting set, decreased verbal fluency, and personality changes such as impulsivity and poor judgment are nearly universal. A progressive apathetic state ensues, but true dementia may not be prominent until the advanced stages of the disease. Although considered a clinical hallmark of PSP, supranuclear vertical gaze palsy may not appear until later in the disease course, and some patients may never develop gaze palsy. Atypical presentations are often seen, especially pure akinesia manifested by severe motor blocks while walking (freezing). PSP is rapidly progressive; by the fourth year of illness, half of patients need assistance for walking and have troublesome dysarthria and visual symptoms. Dysphagia becomes prominent shortly thereafter. The diagnosis of PSP is usually made by clinical criteria. Abnormal vertical saccades, best demonstrated by examination for opticokinetic nystagmus, compared to horizontal saccades, is one of the earliest ophthalmological signs of PSP. Electro-oculographic recordings in PSP show decreased amplitude and normal latency of horizontal saccadic eye movements. Typical MRI signs of PSP include midbrain atrophy, increased signal in the midbrain and GP, atrophy or increased signal in the red nucleus, third ventricle dilation, and atrophy of the frontal or temporal lobes. On the midsagittal view of the MRI, as a result of atrophy of the rostral midbrain tegmentum, the most rostral midbrain, the midbrain tegmentum, the pontine base, and the cerebellum appear to correspond to the bill, head, body, and wing, respectively, of a hummingbird or a penguin.

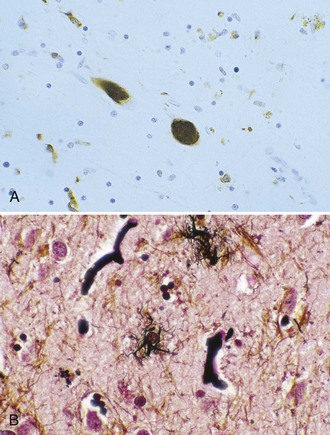
At autopsy, the midbrain in PSP is atrophied, and the sylvian aqueduct is dilated. The SN is depigmented and appears orange and shrunken. The locus ceruleus may also show some depigmentation, but this is less prominent than in idiopathic PD. Other structures may also show atrophy, most notably the frontal lobe, STN, and superior cerebellar peduncle. Histopathologically, the degenerative process involves mainly the basal ganglia, diencephalons, and brainstem. Pathological findings include neuronal loss, gliosis, neurofibrillary tangles, and granulovacuolar degeneration in neurons of the brainstem. There are tufted astrocytes in the motor cortex and the striatum, and the typical neuronal lesion is the globose neurofibrillary tangle, made up of hyperphosphorylated four-repeat tau protein filaments (Fig. 71.11).
On the basis of an analysis of 103 pathologically confirmed consecutive cases of PSP, PSP was divided into two categories: Richardson syndrome, characterized by the typical features described in the original report, and PSP-P, in which the clinical features overlap with PD and the course is more benign (Williams and Lees, 2009). The latter group, representing about a quarter of all patients with PSP, has less tau pathology than the classic Richardson syndrome. The mean 4R-tau/3R-tau ratio of the isoform composition of insoluble tangle-tau isolated from the pons was significantly higher in Richardson syndrome (2.84) than in PSP-P syndrome (1.63). Further studies are needed to confirm or refute this classification. Another subgroup of PSP that has been identified is the so-called frontal PSP, representing about 20% of all PSP patients. These patients initially present with behavioral and cognitive symptoms, with or without ophthalmoparesis, and then evolve into typical PSP.
PSP almost always occurs sporadically, yet an increasing number of familial cases suggests a genetic etiology in some cases. Pedigrees with apparent dominant and recessive inheritance have been described. Affected families may show phenotypical heterogeneity, with some affected persons showing dementia, dystonia, gait disorder, or tics. Mutations in the tau gene have been reported in patients with a familial PSP-like illness, but these have been quite rare, and mutations are not believed responsible for most PSP cases. However, patients with PSP are homozygous for a common haplotype that contains a normally occurring polymorphism in the tau intron immediately preceding exon 10. There is growing support for the notion of altered regulation of tau gene expression in PSP (Höglinger et al., 2011). No toxic, viral, or other environmental risk factors have been described.
Corticobasal Degeneration
In 1967, Rebeiz and colleagues described three patients with akinetic rigidity, apraxia, dystonia, tremor, and aphasia, who at autopsy had pale achromatic ballooned neurons similar to those seen in Pick disease. The condition was named corticodentatonigral degeneration with neuronal achromasia in 1989 but has since become known simply as corticobasal degeneration. Although CBD generally brings to mind a particular motor syndrome of asymmetrical rigidity, apraxia, and cortical sensory dysfunction, its underlying pathological features may be seen in other clinical syndromes including PSP, progressive aphasia, and FTDP. One study based on 35 cases from the Queen Square Brain Bank, in which there were 21 clinically diagnosed cases of corticobasal syndrome (CBS) and 19 pathologically diagnosed with CBD, was designed to address the clinical and pathological overlap between CBD and PSP (Ling et al., 2010). Of 19 pathologically confirmed CBD cases, only 5 had been diagnosed correctly in life (sensitivity = 26.3%). All had a unilateral presentation, a clumsy useless limb, limb apraxia, and myoclonus; 4 had cortical sensory impairment and focal limb dystonia, and 3 had an alien limb. Eight cases of CBD had been clinically diagnosed as PSP, all of whom had vertical supranuclear palsy, and 7 had falls within the first 2 years. Of 21 cases with CBS, only 5 had CBD (positive predictive value = 23.8%); 6 others had PSP pathology, 5 had AD, and the remaining 5 had other non-tau pathologies. Forty-two percent of CBD cases presented clinically with a PSP phenotype, and 29% of CBS cases had underlying PSP pathology. The authors suggested the CBD-Richardson syndrome for the overlap cases and concluded that CBD “is a discrete clinico-pathological entity but with a broader clinical spectrum than was originally proposed” (Ling et al., 2010).
CBD is one of the least common and most asymmetrical forms of atypical parkinsonism. The mean age at onset is 60 to 64 years. In its most recognizable form, it is predominantly a motor disease, but its presentation is clinically heterogeneous. In addition to parkinsonism with strikingly asymmetrical rigidity, CBD patients often exhibit asymmetrical dystonia, myoclonus, apraxia, alien limb, and cortical sensory loss. They may also present with primary progressive aphasia and may evolve into global dementia (Lee et al., 2011).
At autopsy, patients with a clinical syndrome consistent with CBD have gross brain atrophy. Typical microscopic changes are tau-positive neuronal and glial lesions, especially gray and white matter astrocytic plaques and threadlike lesions, and neuronal loss in the cortex and SN. The inclusions are formed of hyperphosphorylated four-repeat tau. Overlap with other conditions including AD, PSP, PD, FTDP, and hippocampal sclerosis is common (Ling et al., 2010).
Dementia with Lewy Bodies
PD dementia (PDD) is defined as cognitive impairment that includes cognitive and motor slowing, executive dysfunction, and impaired memory retrieval. The relationship of PDD to AD and other dementing disorders such as DLB has not yet been well defined. Although some investigators suggest that clear clinicopathological separation is possible between the three disorders, the differences in neuropathological and neurochemical characteristics suggest that there is a continuum (Lippa et al., 2007).
Frontotemporal Degeneration with Parkinsonism Linked to Chromosome 17
Frontotemporal degeneration is a group of disorders characterized by behavioral changes and neuropsychological evidence of frontal lobe dysfunction. They include PSP, CBD, Pick disease, pallidopontonigral degeneration, disinhibition-dementia-parkinsonism-amyotrophy, familial multiple system tauopathy with presenile dementia, familial subcortical gliosis, frontotemporal dementia (FTD), FTD with ALS, FTD with inclusion body myopathy, and FTDP-17 (Josephs, 2008). In up to 60% of patients with FTD, there is a positive family history. Genetic loci on chromosomes 17 (FTDP-17), 9 (FTD with ALS; FTD with inclusion body myositis), and 3 (FTD) have been described. The prototype of FTDP is an inherited parkinsonism-dementia disorder, initially described as Wilhelmsen-Lynch disease (disinhibition-dementia-parkinsonism-amyotrophy complex) and subsequently found to be due to mutations in the tau gene on chromosome 17q21. Although tau mutations account for many of these diseases, similar phenotypes have been attributed to mutations in other genes such as p97 (also known as valosin-containing protein) on chromosome 9p21-p12, CHMP2B (charged multivesicular body protein 2B) on the pericentromeric region of chromosome 3, and progranulin (PGRN) on chromosome 17q21 (1.5 Mb centromeric of tau). Plasma and cerebrospinal fluid (CSF) levels of progranulin have been found to be reduced nearly fourfold in affected and unaffected subjects with PGRN mutations, and low (75% reduction) plasma progranulin levels may be used as a screening tool for PGRN mutations (Finch et al., 2009).
Guadeloupean Parkinsonism
Over the past 5 years, a focus of atypical parkinsonism has been described in the French West Indies. So-called Guadeloupean parkinsonism shows clinical features of LD-unresponsive parkinsonism, postural instability with early falls, and pseudobulbar palsy. More than 25% of these patients have a phenotype like that of PSP. The etiology of this form of parkinsonism is unknown, but exposure to dietary or other environmental toxins is suspected. The case-control survey (Lannuzel et al., 2007) found that the disease is associated with the use of indigenous plants (Annona muricata [synonyms: soursop, corossol, guanbana, graviola, and sweetsop]) that contain the mitochondrial complex I and dopaminergic neuronal toxins, reticuline and coreximine.
Vascular Parkinsonism
In many as 3% to 6% of patients with parkinsonism, the disease has a vascular etiology, but the true frequency is unknown. Vascular changes on imaging studies are common, but the cause and effect are not always clearly established. Among stroke patients, parkinsonism is more common in patients with lacunar stroke. Vascular parkinsonism usually presents as “lower body parkinsonism” with a broad-based shuffling gait and prominent start and terminal hesitation, as well as freezing. Postural instability and a history of falls are common. Many patients have dementia and corticospinal findings of incontinence. In a systematic review of 25 articles, patients with vascular parkinsonism were older, had a shorter duration of illness, presented with symmetrical gait difficulties, were less responsive to levodopa, and were more prone to postural instability, falls, and dementia (Kalra et al., 2010). Pyramidal signs, pseudobulbar palsy, and incontinence were more common in vascular parkinsonism, but tremor was not a main feature. Structural neuroimaging was abnormal in 90% to 100% of vascular cases, compared to 12% to 43% of PD cases. In contrast to PD, there is usually no abnormality in presynaptic striatal dopamine transporters as measured by SPECT in vascular parkinsonism.
Drug-Induced Parkinsonism
Dopamine receptor–blocking drugs reproduce the major clinical features of PD, although signs are usually symmetrical, and the tremor is more often present during posture holding than at rest. The most common causes of drug-induced parkinsonism (DIP) are the typical neuroleptic antipsychotic drugs, antidopaminergic antiemetics, and drugs that deplete presynaptic nerve terminals of dopamine, such as reserpine and tetrabenazine. Among the newer, or atypical, antipsychotics, the relative propensity to cause DIP is as follows: risperidone = ziprasidone > olanzapine > quetiapine > clozapine. This ranking reflects their respective affinity for the D2 receptor. A number of other drugs have been associated with DIP: selective serotonin reuptake inhibitors, lithium, phenytoin, methyldopa, valproic acid, and the calcium channel antagonists, flunarizine and cinnarizine, which are not marketed in the United States. DIP generally appears subacutely after weeks to months of therapy. Although it is reversible, DIP may resolve very slowly over a period of up to 6 months, and symptomatic treatment with anticholinergics, amantadine, or LD may be required. Occasionally, parkinsonism does not resolve, suggesting that the offending drug likely has unmasked an underlying parkinsonism. The use of antipsychotic medications is a strong predictor of subsequent PD; in one study, patients taking neuroleptics were found to be 5.4 times more likely to begin antiparkinsonian medications than nonusers (Noyes et al., 2006).
Toxin-Induced Parkinsonism
Manganese toxicity is associated with LD-unresponsive symmetrical parkinsonism with dystonic features such as oculogyric crisis. The disorder may progress for years after cessation of exposure (Jankovic, 2005). Striatal MRI T2-weighted hyperintensity may be present during the acute phase of poisoning. F-dopa PET scans in subjects with manganism show normal presynaptic dopamine function, suggesting postsynaptic pathology. The fungicide, maneb (manganese ethylene-bis-dithiocarbamate), has also been shown to induce a toxic parkinsonism.