[level-membership-for-pediatrics-category]
Chapter 346 Morphogenesis of the Liver and Biliary System
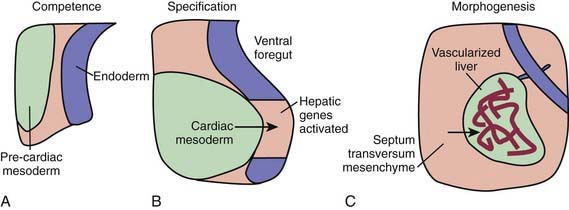
During the early embryonic process of gastrulation, the 3 embryonic germ layers (endoderm, mesoderm, ectoderm) are formed. The liver and biliary system arise from cells of the ventral foregut endoderm; their development can be divided into 3 distinct processes (Fig. 346-1). First, through unknown mechanisms, the ventral foregut endoderm acquires competence to receive signals arising from the cardiac mesoderm. These mesodermal signals, in the form of various fibroblast growth factors (FGFs) and bone morphogenetic proteins (BMPs), lead to specification of cells that will form the liver and activation of liver-specific genes. During this period of hepatic fate decision, “pioneer” transcription factors, including Foxa and Gata4, bind to specific binding sites in compacted chromatin, open the local chromatin structure, and mark genes as competent. But these will only be expressed if they are correctly induced by additional transcription factors. Newly specified cells then migrate in a cranial ventral direction into the septum transversum in the 4th wk of human gestation to initiate liver morphogenesis.
The growth and development of the newly budded liver require interactions with endothelial cells. Certain proteins are important for liver development in animal models (Table 346-1). In addition to these proteins, microRNA, which consists of small noncoding, single-stranded RNA, have a functional role in the regulation of gene expression and hepatobiliary development in a zebrafish model.
Table 346-1 SELECTED GROWTH FACTORS, RECEPTORS, PROTEIN KINASES AND TRANSCRIPTION FACTORS REQUIRED FOR NORMAL LIVER DEVELOPMENT IN ANIMAL MODELS
INDUCTION OF HEPATOCYTE FATE THROUGH CARDIAC MESODERM
INDUCTION OF HEPATOCYTE FATE THROUGH SEPTUM TRANSVERSUM
Bone morphogenetic proteins 2, 4, 7
STIMULATION OF HEPATOBLAST GROWTH AND PROLIFERATION
SPECIFICATION OF HEPATOCYTE LINEAGE
SPECIFICATION OF CHOLANGIOCYTE LINEAGE
Within the ventral mesentery, proliferation of migrating cells forms anastomosing hepatic cords, with the network of primitive liver cells, sinusoids, and septal mesenchyme establishing the basic architectural pattern of liver lobule (Fig. 346-2). The solid cranial portion of the hepatic diverticulum (pars hepatis) eventually forms the hepatic parenchyma and the intrahepatic bile ducts. The hepatic lobules are identifiable in the 6th week of human gestation. The bile canalicular structures, including microvilli and junctional complexes, are specialized loci of the liver cell membrane; these appear very early in gestation, and large canaliculi bounded by several hepatocytes are seen by 6-7 wk.
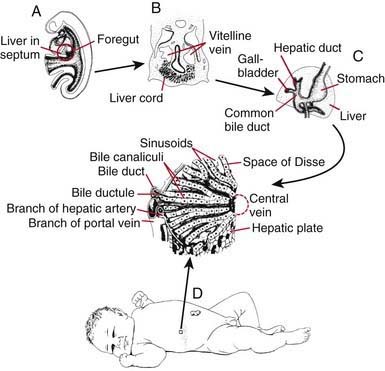
The caudal part (pars cystica) of the hepatic diverticulum becomes the gallbladder, cystic duct, and common bile duct. The distal portions of the right and left hepatic ducts develop from the extrahepatic ducts, whereas the proximal portions develop from the first intrahepatic ductal plates. The extrahepatic bile ducts and the developing intrahepatic biliary tree maintain luminal continuity and patency from the beginning of organogenesis (see Fig. 346-2C).
The transport and metabolic activities of the liver are facilitated by the structural arrangement of liver cell cords, which are formed by rows of hepatocytes, separated by sinusoids that converge toward the tributaries of the hepatic vein (the central vein) located in the center of the lobule (see Fig. 346-2D). This establishes the pathways and patterns of flow for substances to and from the liver. In addition to arterial input from the systemic circulation, the liver also receives venous input from the gastrointestinal tract via the portal system. The products of the hepatobiliary system are released by 2 different paths: through the hepatic vein and through the biliary system back into the intestine. Plasma proteins and other plasma components are secreted by the liver. Absorbed and circulating nutrients arrive through the portal vein or the hepatic artery and pass through the sinusoids and past the hepatocytes to the systemic circulation at the central vein. Biliary components are transported via the series of enlarging channels from the bile canaliculi through the bile ductule to the common bile duct.
Hepatic Ultrastructure
Hepatocytes exhibit various ultrastructural features that reflect their biologic functions (Fig. 346-3). Hepatocytes, like other epithelial cells, are polarized, meaning that their structure and function are directionally oriented. One result of this polarity is that various regions of the hepatocyte plasma membrane exhibit specialized functions. Bidirectional transport occurs at the sinusoidal surface, where materials reaching the liver via the portal system enter and compounds secreted by the liver leave the hepatocyte. Canalicular membranes of adjacent hepatocytes form bile canaliculi, which are bounded by tight junctions, preventing transfer of secreted compounds back into the sinusoid. Within hepatocytes, metabolic and synthetic activities are contained within a number of different cell organelles. The oxidation and metabolism of heterogeneous classes of substrates, fatty acid oxidation, key processes in gluconeogenesis, and the storage and release of energy occur in the abundant mitochondria.
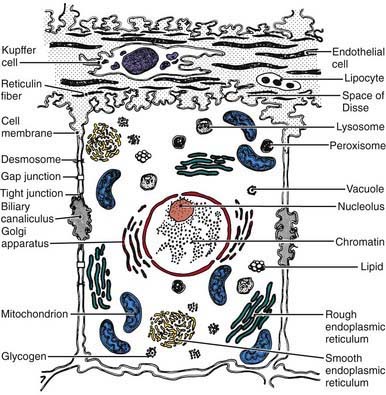
Metabolic Functions of the Liver
Hepatic Excretory Function
Many of the processes related to immaturity of the newborn in liver morphogenesis and function as discussed earlier are implied in the increased susceptibility of infants to liver disease associated with parenteral nutrition. The reduced bile salt pool, hepatic glutathione depletion, and deficient sulfation contribute to production of toxic lithocholic bile acids and cholestasis, whereas deficiencies of essential amino acids including taurine and cysteine can cause hepatic steatosis in these infants. Beyond the neonatal period, disturbances in bile acid metabolism may be responsible for diverse effects on hepatobiliary and intestinal function (Table 346-2).
Table 346-2 CAUSES OF IMPAIRED BILE ACID METABOLISM AND ENTEROHEPATIC CIRCULATION
DEFECTIVE BILE ACID SYNTHESIS OR TRANSPORT
ABNORMALITIES OF BILE ACID DELIVERY TO THE BOWEL
LOSS OF ENTEROHEPATIC CIRCULATION OF BILE ACIDS
BILE ACID MALABSORPTION
DEFECTIVE UPTAKE OR ALTERED INTRACELLULAR METABOLISM
Bates MD, Balistreri WF. The gastrointestinal tract: development of the human digestive system. In: Fanaroff AA, Martin RJ, editors. Neonatal-perinatal medicine: diseases of the fetus and infant. ed 7. St Louis: Mosby; 2002:1255-1263.
Beath SV. Hepatic function and physiology in the newborn. Semin Neonatol. 2003;8:337-346.
Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation.”. Hepatology. 1992;16:1069-1083.
Hofmann A. Bile acids: trying to understand their chemistry and biology with the hope of helping patients. Hepatology. 2009;49:1403-1418.
Lemaigre F, Zaret KS. Liver development update: new embryo models, cell lineage control, and morphogenesis. Curr Opin Genet Dev. 2004;14:582-590.
Meier PJ, Stieger B. Molecular mechanisms of bile formation. News Physiol Sci. 2000;15:89-93.
Roskams T, Desmet V. Embryology of extra- and intrahepatic bile ducts, the ductal plate. Anat Rec. 2008;291:628-635.
Rudolph AM. Hepatic and ductus venosus blood flows during fetal life. Hepatology. 1983;3:254-258.
Wuestefeld T, Zaret K. Liver development: from endoderm to hepatocyte. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver disease in children. ed 3. New York: Cambridge University Press; 2007:3-13.
Zaret KS. Regulatory phases of early liver development: paradigms of organogenesis. Nat Rev Genet. 2002;3:499-512.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 346 Morphogenesis of the Liver and Biliary System
During the early embryonic process of gastrulation, the 3 embryonic germ layers (endoderm, mesoderm, ectoderm) are formed. The liver and biliary system arise from cells of the ventral foregut endoderm; their development can be divided into 3 distinct processes (Fig. 346-1). First, through unknown mechanisms, the ventral foregut endoderm acquires competence to receive signals arising from the cardiac mesoderm. These mesodermal signals, in the form of various fibroblast growth factors (FGFs) and bone morphogenetic proteins (BMPs), lead to specification of cells that will form the liver and activation of liver-specific genes. During this period of hepatic fate decision, “pioneer” transcription factors, including Foxa and Gata4, bind to specific binding sites in compacted chromatin, open the local chromatin structure, and mark genes as competent. But these will only be expressed if they are correctly induced by additional transcription factors. Newly specified cells then migrate in a cranial ventral direction into the septum transversum in the 4th wk of human gestation to initiate liver morphogenesis.
The growth and development of the newly budded liver require interactions with endothelial cells. Certain proteins are important for liver development in animal models (Table 346-1). In addition to these proteins, microRNA, which consists of small noncoding, single-stranded RNA, have a functional role in the regulation of gene expression and hepatobiliary development in a zebrafish model.
Table 346-1 SELECTED GROWTH FACTORS, RECEPTORS, PROTEIN KINASES AND TRANSCRIPTION FACTORS REQUIRED FOR NORMAL LIVER DEVELOPMENT IN ANIMAL MODELS
INDUCTION OF HEPATOCYTE FATE THROUGH CARDIAC MESODERM
INDUCTION OF HEPATOCYTE FATE THROUGH SEPTUM TRANSVERSUM
Bone morphogenetic proteins 2, 4, 7
STIMULATION OF HEPATOBLAST GROWTH AND PROLIFERATION
SPECIFICATION OF HEPATOCYTE LINEAGE
SPECIFICATION OF CHOLANGIOCYTE LINEAGE
Within the ventral mesentery, proliferation of migrating cells forms anastomosing hepatic cords, with the network of primitive liver cells, sinusoids, and septal mesenchyme establishing the basic architectural pattern of liver lobule (Fig. 346-2). The solid cranial portion of the hepatic diverticulum (pars hepatis) eventually forms the hepatic parenchyma and the intrahepatic bile ducts. The hepatic lobules are identifiable in the 6th week of human gestation. The bile canalicular structures, including microvilli and junctional complexes, are specialized loci of the liver cell membrane; these appear very early in gestation, and large canaliculi bounded by several hepatocytes are seen by 6-7 wk.
The caudal part (pars cystica) of the hepatic diverticulum becomes the gallbladder, cystic duct, and common bile duct. The distal portions of the right and left hepatic ducts develop from the extrahepatic ducts, whereas the proximal portions develop from the first intrahepatic ductal plates. The extrahepatic bile ducts and the developing intrahepatic biliary tree maintain luminal continuity and patency from the beginning of organogenesis (see Fig. 346-2C).
The transport and metabolic activities of the liver are facilitated by the structural arrangement of liver cell cords, which are formed by rows of hepatocytes, separated by sinusoids that converge toward the tributaries of the hepatic vein (the central vein) located in the center of the lobule (see Fig. 346-2D
[/not-level-membership-for-pediatrics-category]
