Molecular Pathogenesis and Diagnostics of Hepatocellular Tumors
Prodromos Hytiroglou
Paulette Bioulac-Sage
Introduction
Identification of the molecular pathways altered in benign and malignant hepatocellular tumors has significantly increased our understanding of hepatocellular tumorigenesis. The immunohistochemical markers derived from these molecular alterations have been useful in clinical pathology practice, contributing to accurate diagnosis and classification of these tumors in resection and biopsy specimens.
Benign Hepatocellular Tumors
Hepatocellular adenoma (HCA) and focal nodular hyperplasia (FNH) are benign hepatocellular tumors. HCA is a hepatocytic neoplasm, whereas FNH is a hyperplastic hepatocytic response caused by localized hepatic vascular abnormalities.1
FNH and HCA occur most frequently in women of child-bearing age; these tumors are much less common in men, children, and postmenopausal women.2 Oral contraceptives are recognized as the main risk factor for HCA. These agents are probably not etiologically implicated in FNH. Most cases of HCA occurring in men are associated with the administration of anabolic steroids for Fanconi anemia, acquired aplastic anemia, or bodybuilding.3 HCAs also occur in the setting of metabolic disorders such as glycogenosis type 1 and 3,4 galactosemia, tyrosinemia, and familial diabetes mellitus.
The epidemiology of HCA has changed in recent years because low-dose contraceptive pills cause fewer cases of HCA than the pills used in the past. However, obesity and the metabolic syndrome are more frequently encountered in women and men with HCA, particularly the inflammatory subtype. FNH lesions, especially when multiple, may be associated with hepatic vascular disorders and tumors of the liver and other organs. FNH and HCA usually develop in livers that are otherwise histologically normal or sometimes steatotic.5
Focal Nodular Hyperplasia
Molecular Changes
The data regarding molecular aberrations in FNH are limited.6 Analysis of clonality using the human androgen receptor assay demonstrated the polyclonal nature of the hepatocytes in 60% to 100% of cases.7–11 Studies of chromosomal gains and losses using comparative genomic hybridization or allelotyping detected alterations in 14% to 50% of cases.11–13 However, genetic analysis failed to identify somatic gene mutations in CTNNB1 (which encodes β-catenin), TP53 (tumor protein 53), APC (adenomatous polyposis coli protein), and HNF1A (hepatocyte nuclear factor 1α).11,14,15
The mRNA expression levels of two angiopoietin genes, ANGPT1 and ANGPT2, involved in vascular maturation are altered in FNH, and the consistently increased ANGPT1/ANGPT2 ratio supports the role of a vascular trigger for this lesion.11,16 Studies have identified overexpression of genes encoding proteins of the extracellular matrix and cell–matrix adhesion,17 consistent with the finding of fibrosis in most cases of classic FNH. A twofold overexpression of the TGFB1 gene was detected, and other key genes involved in the fibrogenesis pathway, such as PDGFA and PDGFRB, were overexpressed in FNH.17 The finding of myofibroblasts expressing smooth muscle actin at the periphery and in the fibrotic areas of FNH is consistent with activation of the transforming growth factor-β (TGF-β) pathway in these cells.17 The NTS gene, which encodes neurotensin, was overexpressed in FNH, resulting in an increased ratio of NTS to HAL, a periportal area gene that encodes histidine ammonia lyase. This finding can discriminate FNH from other benign tumors.17
Alterations in the expression of physiologically zonated genes of the hepatic lobule have been detected in FNH.17 Thirteen genes of the periportal areas were found to be downregulated, whereas six genes of the perivenous areas were upregulated, indicating altered zonation in FNH. One of the perivenous area genes, GLUL, which encodes glutamate-ammonia ligase, also known as glutamine synthetase (GS), is upregulated by β-catenin.18 GLUL mRNA overexpression correlates with a slight but significant CTNNB1 mRNA overexpression in FNH compared with normal liver parenchyma,17 suggesting that upregulation of the β-catenin signaling pathway is restricted to areas surrounding the veins of FNH nodules. Upregulation causes overexpression of the active form of β-catenin in the absence of CTNNB1-activating mutations.
These molecular data indicate that the almost completely arterial inflow in FNH activates the β-catenin pathway, which can contribute to tumor formation by promoting hepatocyte proliferation and regeneration. However, the mechanism of β-catenin activation in FNH remains unresolved, because no alteration of the main known regulators of the WNT signaling pathway have been identified, including a lack of AXIN1 and APC inactivating mutations.14 Additional functional analyses of the interplay between the TGF-β and β-catenin pathways in hepatocytes will be required to understand the pathogenesis of FNH.
FNH-like nodules are lesions resembling FNH macroscopically and on imaging, but they occur in cirrhotic livers19,20 or in the setting of vascular hepatic disorders such as the Budd-Chiari syndrome.21,22 The gene expression profile of FNH-like nodules in cirrhosis is significantly different from that of classic FNH.17 The nodules do not show β-catenin activation, and the β-catenin–induced perivenous genes are significantly downregulated compared with those of nontumorous liver and classic FNH. The periportally zonated genes are not differentially expressed compared with nontumorous liver as determined by quantitative reverse transcription–polymerase chain reaction (RT-PCR), and the NTS/HAL expression ratio is not increased, which is observed in classic FNH.17
Molecular Diagnostics
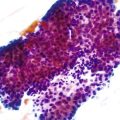
As a consequence of the deregulation of the β-catenin pathway, overexpression of GS is heterogeneously distributed in the hepatocytes of the FNH nodules. This can be demonstrated with immunohistochemical techniques as characteristic staining with relatively large areas anastomosed in maplike patterns (Fig. 44.1, A and B). The GS-positive areas are often centered on veins and are usually located at a distance from the fibrous bands.23 This specific pattern of GS staining in FNH contrasts with that of normal liver, in which staining is restricted to a few rows of hepatocytes surrounding the terminal hepatic venules. Due to the absence of β-catenin (CTNNB1) mutations, immunohistochemistry using a β-catenin antibody does not show abnormal cytoplasmic or nuclear β-catenin staining, even in hepatocytes overexpressing GS (see Fig. 44.1, B). The characteristic GS immunostaining and the lack of aberrant β-catenin expression provide a useful tool for the diagnosis of some FNH lesions that lack typical histologic features in surgical specimens or biopsies.23–25
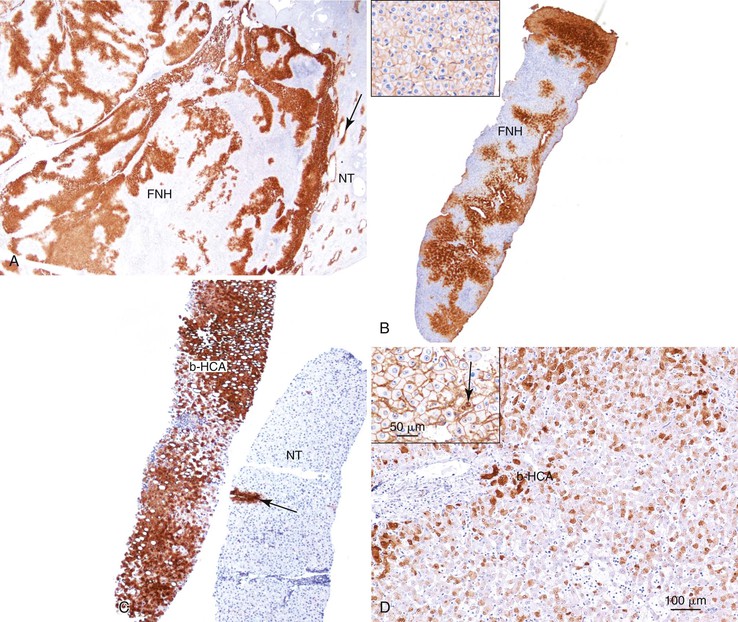
As expected from their molecular changes, FNH-like nodules show little or no parenchymal GS immunostaining.17 When present, GS staining is usually restricted to some persistent venous structures inside the nodules. This staining pattern is similar to that of cirrhotic nodules and quite different from that of the large, anastomosing, GS-positive areas of classic FNH.
Hepatocellular Adenoma
HCAs are a heterogenous group of monoclonal tumors in which several recurrent mutations have been identified, leading to a classification based on good genotypic-phenotypic correlations.26,27 Molecular changes and diagnostics are described for each HCA group.
Hepatocellular Adenoma with Inactivating Mutations of the HNF1A Gene
Molecular Changes
The HNF1A gene (formerly hepatic transcription factor 1 [TCF1]) is located on chromosome 12q24.2 and encodes the hepatocyte nuclear factor 1α protein (HNF1A), a 681–amino acid homeodomain transcription factor that is involved in hepatocyte differentiation.28 HNF1A controls the expression of liver-specific genes, such as those for β-fibrinogen (FGB), α1-antitrypsin (SERPINA1), and albumin (ALB).29 HNF1A is a human tumor suppressor gene involved in liver tumorigenesis.30
Biallelic inactivating HNF1A mutations have been detected in 35% to 40% of HCAs.31 In most cases, both mutations are of somatic origin, but in less than 10% of cases, one mutation is germline and the other somatic. Sporadic HNF1A-inactivated HCAs (H-HCAs) with somatic mutations occur almost exclusively in women and usually are associated with oral contraception. Patients with heterozygous germline HNF1A mutations are younger than those with somatic mutations and frequently have a family history of liver adenomatosis or diabetes (usually maturity-onset diabetes of the young type 3 [MODY3]), which is a consequence of heterozygous germline HNF1A mutations.32 The spectrum of HNF1A somatic mutations differs significantly from the germline changes in MODY3.33
Transcriptomic analysis showed that several metabolic pathways are altered in sporadic H-HCA, including neoglycogenesis repression, and glycolysis and fatty acid synthesis activation.29 The induction of glycolysis and lipogenesis in H-HCA is linked to HNF1A inactivation and is independent of the activation of sterol regulatory element–binding protein 1 (SREBP1) and MLX interacting protein-like (MLXIPL, formerly called carbohydrate response element–binding protein [ChREBP]), leading to the characteristic steatotic phenotype of these tumors.29 Genes for fatty acid binding protein 1 (FABP1) and UDP glucuronosyltransferase 2-B7 (UGT2B7) are positively regulated by HNF1A and are downregulated in H-HCA.27
Heterozygous, germline, inactivating mutations of CYP1B1 have been detected in 15% of women with H-HCA, suggesting that deregulation of the enzyme, which is responsible for the formation of genotoxic metabolites of estrogens, may confer a predisposition to sporadic H-HCA in women.34 CYP1B1 mutation modifies the penetrance of the liver adenomatosis phenotype in patients with HNF1A germline mutations.34
Molecular Diagnostics
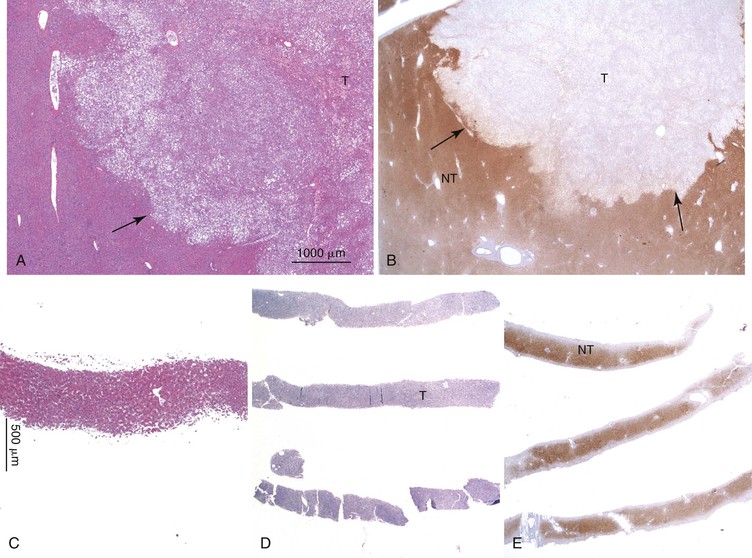
Downregulation of the FABP1 gene leads to a lack of liver-type fatty acid binding protein 1 (FABP1) expression in H-HCA, contrasting with normal expression in the surrounding nontumorous liver as demonstrated by immunohistochemistry.27 In addition to steatosis, which is common in H-HCA cases,26 the absence of liver-type FABP1 expression is an excellent marker for the diagnosis of this subtype of adenoma in resection specimens and biopsy samples (Fig. 44.2), irrespective of the degree of steatosis or size of the lesion. This feature also allows the diagnosis of HNF1A-inactivated adenomatosis to be made when multiple HCAs of various sizes are found throughout the liver. They are often associated with myriad steatotic microadenomas, which can be correctly identified by the lack of FABP1 staining.27,32
Hepatocellular Adenoma with Activating Mutations of the CTNNB1 Gene
Molecular Changes
Approximately 10% to 15% of HCAs demonstrate activating mutations of the CTNNB1 gene encoding β-catenin, which is deranged in many malignant tumors, including hepatocellular carcinoma (HCC). At baseline, β-catenin is associated with a negative regulator complex in the cytoplasm that includes APC, AXIN1, and GSK3B (glycogen synthase kinase 3β) proteins. This complex phosphorylates β-catenin, leading to its cytoplasmic degradation by the proteasome machinery.
CTNNB1 mutations occur mainly in exon 3 and consist of a large in-frame deletion that excludes the amino acids normally phosphorylated by GSK3B, leading to an absence of β-catenin phosphorylation and its permanent activation.26 Unphosphorylated β-catenin is stabilized in the cytoplasm and translocates into the nucleus, promoting the transcription of a large number of genes involved in proliferation, metabolism, and hepatocyte functions.35 Uncommonly, mutations occur in exon 7,26 and they have been reported in HCC.36 In most cases, activating β-catenin mutations lead to upregulation of two main β-catenin target genes: GPR49 and GLUL. The latter encodes GS, which can be demonstrated by immunohistochemistry.
Molecular Diagnostics
As expected from the molecular changes previously described, GS is overexpressed in β-catenin–activated hepatocellular adenomas (B-HCAs) (see Fig. 44.1, C and D). The staining pattern is usually strong and diffuse (see Fig. 44.1, C), and it is associated with aberrant nuclear and cytoplasmic expression of β-catenin, which is often distributed in a random and heterogenous pattern (see Fig. 44.1, D inset). Good concordance has been demonstrated between these immunohistochemical findings (i.e., GS positivity and aberrant nuclear β-catenin staining) and the β-catenin mutations found by molecular methods.26 Overexpression of GS is usually easier to interpret than nuclear expression of β-catenin by immunohistochemistry, particularly in small biopsy samples.25 However, strong or faint GS staining can be heterogeneous (see Fig. 44.1, D), probably depending on the effects of different β-catenin mutations.37 Despite the variations of GS staining in B-HCA, the pattern is usually distinct from the characteristic maplike pattern of FNH (see Fig. 44.1, A and B).
Identification of B-HCA is important, because this group of HCAs is more frequently associated with the development of unequivocal HCC than other HCA subtypes.26,31,38–40 B-HCA is overrepresented among male patients, and specific risk factors are often found in both sexes, such as male hormone administration, glycogenosis, and familial adenomatous polyposis coli. Because B-HCA often displays cytologic abnormalities and a pseudoglandular pattern of growth, it can be difficult to distinguish it from well-differentiated HCC (see Chapter 55). In clinical practice, B-HCA is frequently diagnosed as “borderline lesion HCA/HCC.” When in doubt, assessment of β-catenin mutations in frozen tumor material is recommended.31 Activating β-catenin mutations are also detected in 10% of inflammatory hepatocellular adenomas (B/I-HCAs).
Inflammatory Hepatocellular Adenoma
Molecular Changes
Inflammatory hepatocellular adenomas (I-HCAs) account for more than 50% of HCAs.31,41–43 This group of HCAs is characterized by overexpression of molecules of the acute-phase inflammatory response, including serum amyloid A2 (SAA2) and C-reactive protein (CRP), at the mRNA and protein levels.27 Approximately 60% of I-HCAs have gain-of-function mutations (i.e., small, in-frame, somatic deletions) in the interleukin 6 (IL6) signal transducer gene (IL6ST), causing permanent activation of the signal transducer and activator of transcription pathway (IL6/JAK/STAT) independent of the ligand.44 The IL6ST gene encodes the signaling coreceptor glycoprotein 130 (GP130), and mutant GP130 constitutively activates STAT3 signaling in the absence of IL6 binding.44
In I-HCA subsets lacking GP130 mutations, derangements of two other molecular pathways have been identified. Twelve percent of I-HCAs harbor somatic mutations of the signal transducer and activator of transcription 3 gene (STAT3).45 Most mutations result in amino acid substitutions in the SRC homology 2 (SH2) domain that directs STAT3 dimerization. In contrast to wild-type STAT3, I-HCA STAT3 mutants constitutively activate the IL6 signaling pathway independent of ligand.
An additional 5% of I-HCAs have somatic activating mutations of the G protein α-subunit gene (GNAS),46 which are exclusive of the GP130 and STAT3 activating mutations in the IL6ST and STAT3 genes. Mutated GNAS is found in various tumors and is known to cause the McCune-Albright syndrome. Whereas IL6ST mutations directly activate the IL6/JAK/STAT pathway, GNAS mutations indirectly activate this pathway through various kinase pathways.
Approximately 10% of I-HCAs harbor mutations of the CTNNB1 (β-catenin) gene, signifying a risk of malignant transformation.31 On the other hand, some HCCs demonstrate GP130-activating mutations of the IL6ST gene. These tumors arise in normal liver and accumulate β-catenin–activating mutations. It is reasonable to hypothesize that these HCCs result from malignant transformation of HCAs. These data underline the role of inflammation in hepatic tumorigenesis and suggest that activation of the IL6/JAK/STAT pathway alone promotes benign tumorigenesis, whereas participation of β-catenin activation may lead to malignant transformation.
Molecular Diagnostics
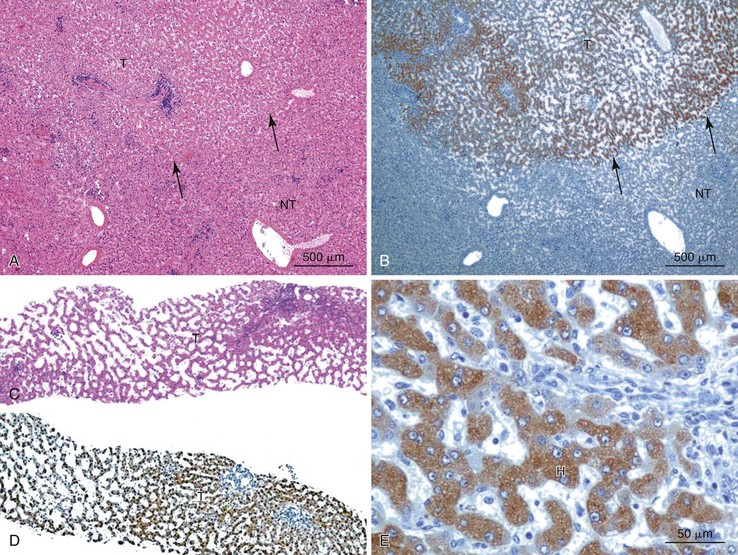
Positive immunohistochemical staining for inflammation-associated proteins (i.e., SAA and CRP) in tumor cells with a sharp demarcation from the surrounding nontumorous liver is characteristic of I-HCAs (Fig. 44.3).27,43 SAA and CRP staining is restricted to hepatocytes, without any staining of the sinusoidal lining and inflammatory cells (see Fig. 44.3, E). I-HCAs with β-catenin activation also express GS and aberrant β-catenin, as described earlier for B-HCAs.
Unclassified Hepatocellular Adenoma
Unclassified hepatocellular adenomas (U-HCAs) represent less than 10% of HCA cases.26,27 U-HCAs do not have HNF1A or CTNNB1 gene mutations, and they do not express inflammatory proteins encoded by mutated IL6ST, STAT3, or GNAS genes. This group of tumors lacks the immunophenotypic features described for the other HCA groups.
Use of Molecular Diagnostics
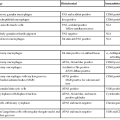
Molecular studies of FNH and HCA have significantly clarified the pathogenesis of these tumors and have provided immunohistochemical markers (i.e., FABP1, GS, β-catenin, SAA, and CRP) for routine diagnostic use. The markers are used to distinguish FNH from HCA and to classify HCAs (Table 44.1). An important contribution of these scientific advances was the reclassification of telangiectatic FNH as I-HCA.11,47
Table 44.1
Genotypic and Immunophenotypic Characteristics of Benign Hepatocellular Tumors, in Comparison with Normal Liver
| Diagnosis | Altered Gene | Usual Characteristics of Immunohistochemical Markers | |||
| GS | β-Catenin | SAA/CRP | FABP1 | ||
| Normal liver | Positive in a few rows of hepatocytes around centrilobular veins | − | − | + | |
| FNH | CTNNB1 activation without mutations | +, maplike pattern | − | − | + |
| B-HCA | CTNNB1 activation with mutations | +, strong and diffuse or heterogenous | +, nuclei ± cytoplasm (usually focal) | − | + |
| H-HCA | HNF1A biallelic inactivating mutations (90% somatic and 10% germline) | − (or a few positive hepatocytes around veins) | − | − | − |
| I-HCA | IL6ST (gp130) activating mutations (60%) STAT3 activating mutations (12%) GNAS activating mutations (5%) |
− (or a few positive hepatocytes around veins) | − | + | + |
| B/I-HCA (10% of I-HCAs) | Same as B-HCA and I-HCA | + | + | + | + |
| U-HCA | Unknown | − | − | − | + |
B-HCA, β-Catenin–activated hepatocellular adenoma; B/I-HCA, β-catenin–activated inflammatory HCA; CRP, C-reactive protein; FNH, focal nodular hyperplasia; GS, glutamine synthetase; HCA, hepatocellular adenoma; H-HCA, hepatocyte nuclear factor 1α–inactivated hepatocellular adenoma; I-HCA, inflammatory hepatocellular adenoma; FABP1, liver-type fatty acid binding protein; SAA, serum amyloid A; U-HCA, unclassified hepatocellular adenoma; + or −, presence or absence of characteristic features that help to ascertain the diagnosis.
Standard microscopic features (see Chapter 55) often allow accurate differentiation, but because of the significant clinical and prognostic differences among the HCA groups, immunophenotyping is considered mandatory to ascertain the HCA subtype in each case.43 Immunophenotyping may become an important tool for HCA management in the near future.31,48 When feasible, tumor tissue should be frozen for molecular characterization of difficult cases, particularly to detect B-HCA, which has a higher risk of HCC transformation.
Hepatocellular Carcinoma and Its Precursors
HCC usually arises in a background of chronic liver disease. In autopsy series, 80% of HCCs are found in livers with cirrhosis;49 most of the remaining cases arise in chronically diseased rather than normal livers. On a worldwide scale, infection with the hepatitis B virus (HBV) and the hepatitis C virus (HCV) are the most common predisposing factors, with at least 350 million and 170 million infected people, respectively. Approximately 85% of HCC cases are related to one of these viruses.50,51
The carcinogenic effects of HBV are illustrated by the fact that HCCs often develop in HBV-infected patients without cirrhosis, whereas HCCs in HCV-infected patients typically occur after cirrhosis is established. Among cirrhotic patients, the highest incidence of HCC is found for those with HCV infection (5-year cumulative incidence of 30% in Japan and 17% in the West), followed by those with hereditary hemochromatosis (5-year cumulative incidence of 21%), and those with HBV infection (5-year cumulative incidence of 15% in regions of high endemicity and 10% in the West).52 Other causes of cirrhosis, such as alcohol-induced liver disease, autoimmune hepatitis, biliary diseases, and Wilson disease, are associated with lower incidences of HCC. HCC may occasionally arise in normal liver, and some of these cases may be derived from HCA. The risk factors for HCC are listed in Box 44.1.
Molecular Changes in Hepatocarcinogenesis
Many factors favor initiation of hepatocarcinogenesis in chronic liver diseases53–57:
• Increased hepatic cell proliferation due to hepatocyte loss
• Deregulation of genes due to HBV DNA insertion into the host genome
HBV DNA insertion causes genomic instability and deregulates genes involved in cell signaling and replication, such as human telomerase reverse transcriptase (TERT), platelet-derived growth factor receptor (PDGFR), and calcium signaling–related genes.58,59 The HBV X protein acts as an oncoprotein, transactivating various genes involved in signal transduction pathways and inhibiting expression of the tumor suppressor gene TP53. However, a specific TP53 mutation in codon 249 is characteristic of aflatoxin B1 exposure.60
Due to vascular changes causing parenchymal loss in cirrhotic livers, the rate of hepatocyte loss and replication is often significantly enhanced compared with the precirrhotic stages of chronic liver diseases. Simultaneously, hepatocytes become senescent, and hepatic progenitor cells are activated to repopulate the dwindling parenchymal regions.61,62 In the course of chronic liver diseases, clonal cell populations of increasing size are established,63 some of which may have accumulating genetic and epigenetic changes favoring neoplastic transformation (Fig. 44.4). Clonal cell populations with significant molecular alterations may be identified on microscopic examination as precancerous lesions. The cell of origin of these lesions may be the hepatocyte or the hepatic progenitor cell.64 Molecular analysis of precancerous lesions and HCCs reveals a sharp increase in structural DNA changes compared with the surrounding hepatic parenchyma.54
The diverse molecular changes in HCC cells include chromosomal amplifications and deletions, point mutations, and epigenetic alterations. The most common cytogenetic abnormalities include gains of 8q and 1q and losses of 16q.65,66 The cancer-related genes have been identified and validated for some loci; examples include MYC (8q), hepatoma-derived growth factor (HDGF, 1q), RB1 (13q), and TP53 (17q).55,57 The large regional gains and losses suggest that additional cancer-related genes may reside in these hotspots, but for most amplified and deleted foci, the genes involved are uncertain.55
Molecular changes are crucial in deregulating important signal transduction pathways. Commonly disrupted pathways include the vascular endothelial growth factor (VEGF) and PDGF signaling cascades, the WNT/β-catenin pathway, the epidermal growth factor (EGFR)/RAS/mitogen-activated protein kinase (MAPK) pathway, the hepatocyte growth factor (HGF) and insulin-like growth factor (IGF) signaling cascades, and the phosphatidylinositol-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway.57,67–69 Most HCCs have activated telomerase, which provides the tumor cells with limitless replicative potential.70
Global genomic analysis has provided evidence that certain molecular changes are associated with clinicopathologic features and prognosis, providing a basis for molecular classification of HCC. Lee and colleagues71 identified a subtype of HCC with a poor prognosis that expressed a fetal hepatoblast gene signature, suggesting a progenitor cell origin. Boyault and associates suggested that HCC could be classified in six groups (G1 to G6).72 The primary clinical determinant of group membership was HBV infection, and other major determinants included chromosomal instability (in G1, G2, and G3 tumors but not in G4, G5, and G6 tumors), CTNNB1 and TP53 mutations, and parental imprinting.
Hoshida and coworkers73 performed a meta-analysis of gene expression data from studies around the world and suggested that HCC could be grouped in three subclasses (S1, S2, and S3). S1 tumors were characterized by aberrant activation of the WNT/β-catenin pathway due to TGF-β stimulation; S2 tumors were characterized by MYC and AKT activation; and S3 tumors had gene expression patterns that suggested differentiated hepatocyte function.
These studies are providing the foundation for targeted therapies for a neoplasm that has traditionally had a poor prognosis due to late diagnosis and a lack of response to systemic therapy. One targeted therapy is already in use. Sorafenib, a multikinase inhibitor, can increase the survival of patients with HCC.74 Nevertheless, early detection still provides the best opportunity for successful HCC treatment. Advances in hepatic imaging methods can provide efficient screening of populations at risk. Surveillance is recommended for cirrhotic patients and those with chronic HBV infection.75
The molecular pathogenesis of fibrolamellar carcinoma is obscure. This tumor usually occurs in children and young adults without underlying liver disease. The incidence of chromosomal gains and losses is lower in fibrolamellar carcinoma than in classic HCC.76 Gains in 1q and 8q and losses in 18q are the most frequent genomic alterations. Molecular changes of the major pathways involved in classic HCC have not been detected in fibrolamellar carcinoma.77 Low levels of methylation of tumor suppression gene promoters have been found in this tumor.78 Immunohistochemical studies have found that fibrolamellar carcinomas overexpress TGF-β and anterior gradient 2 protein (AGR2).79,80
Precancerous Lesions in Hepatocarcinogenesis
In the past 25 years, many studies have investigated the morphologic features of candidate HCC precursors in series of resected and explanted livers. The biologic nature of these lesions was then investigated by epidemiologic, molecular, radiologic, and clinical follow-up studies. The terminology for hepatocellular precancerous lesions has been established through international consensus meetings.81–83
Two types of lesions are detected in chronically diseased, usually cirrhotic livers, and they are often multiple (Fig. 44.5). Dysplastic foci consist of groups of hepatocytes with dysplasia. They are less than 1 mm in diameter and are incidental findings on microscopic examination of hepatic specimens. Dysplastic nodules are a few millimeters to a few centimeters in diameter and can be detected on gross examination and sometimes on hepatic imaging. They must be differentiated from HCC, and in all cases, the diagnosis relies on microscopic examination. In patients without chronic liver disease, HCA (mainly B-HCA) is recognized as a potential HCC precursor (see Hepatocellular Adenoma).27
Dysplastic Foci
Small cell change (SCC) of hepatocytes, originally described as small cell dysplasia, is the most common cytologic change found in dysplastic foci.84 SCC is characterized by a decreased cell volume, an increased nucleus-to-cytoplasm ratio, mild nuclear pleomorphism and hyperchromasia, and cytoplasmic basophilia (Fig. 44.6, A). Hepatocytes with SCC have increased expression of proliferation markers compared with adjacent hepatocytes. They have molecular alterations that indicate a precancerous nature, including chromosomal gains and losses similar to those of adjacent HCCs, telomere shortening, and CDKN1A-regulated cell cycle checkpoint inactivation.85–87 SCC has great cytologic similarity to early HCC.88
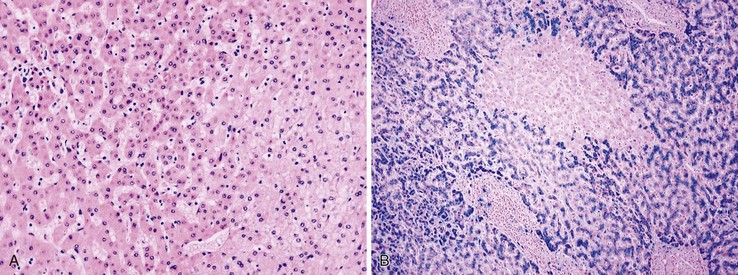
Iron-free foci have been associated with the development of HCC on follow-up of patients with hereditary hemochromatosis (see Fig. 44.6, B).89 In addition to resistance to iron accumulation, iron-free foci may demonstrate SCC or large cell change (LCC) of hepatocytes.
Large Cell Change in Hepatocytes
LCC, originally described as liver cell dysplasia,90 is characterized by nuclear and cytoplasmic enlargement (with a preserved nucleus-to-cytoplasm ratio), nuclear pleomorphism and hyperchromasia, and multinucleation (Fig. 44.7). LCC is commonly seen in livers with chronic HBV infection or cirrhosis of various causes. In cirrhosis due to chronic HBV or HCV infection, LCC is an independent risk factor for HCC.91,92
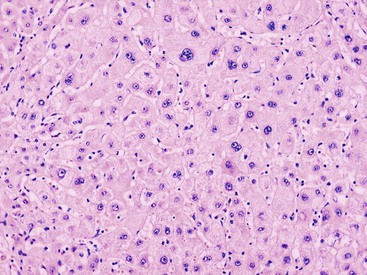
The nature of this cytologic change has been disputed, and it is possible that LCC is biologically heterogeneous. In most cases of cirrhosis, LCC appears to be a degenerative change due to senescence or cholestasis.93,94 LCC usually occurs diffusely rather than focally, which does not favor a precancerous characterization.95 However, a subset of LCC in cases of chronic hepatitis B may be precancerous. These lesions are characterized by shortened telomeres, CDKN2A- and CDKN1A-regulated cell cycle checkpoint inactivation, increased DNA damage, and a higher proliferation index compared with the adjacent cirrhotic parenchyma.96
Dysplastic Nodules
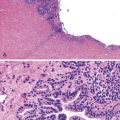
Dysplastic nodular lesions are characterized by cytologic or structural atypia that suggest a precancerous nature. Dysplastic nodules were originally classified as low or high grade on the basis of the degree of atypia.81 In practice, making the histologic distinction between low-grade dysplastic nodules and large regenerative nodules is difficult or impossible,82 but molecular markers may provide a way in the future. A diagnosis of high-grade dysplastic nodule has been assigned to lesions with cytologic or structural atypia that does not reach the degree seen in HCC (Fig. 44.8, A and B). The histologic features of dysplastic nodules are described in Chapter 55. The blood supply of these lesions is commonly derived from portal tracts and small, nontriadal (unpaired) arteries (see Fig. 44.8, B).
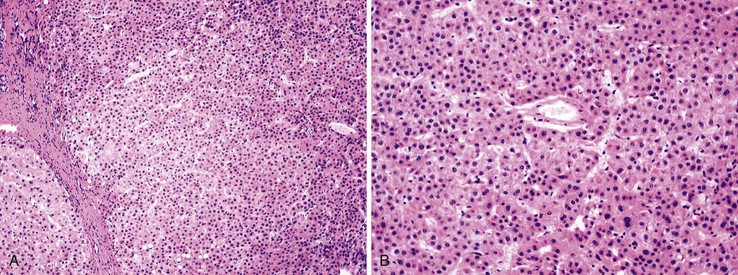
Follow-up studies of cirrhotic patients have shown that dysplastic nodules represent a risk factor for HCC.97–99 Subnodules of HCC arising in dysplastic nodules are occasionally found on histologic examination (Fig. 44.9, A).100 The precancerous nature of the dysplastic nodules has been confirmed by several molecular studies. These lesions and adjacent HCCs had similar chromosomal gains and losses and a loss of heterozygosity in microsatellite foci.101–103 Telomere shortening, telomerase activation, and CDKN1A-regulated cell cycle checkpoint inactivation have been found in dysplastic nodules.104–107 Dysplastic nodules can be distinguished from HCC on the basis of gene expression profiles.108
Prognostic and Therapeutic Implications
Detection of dysplastic foci and dysplastic nodules in liver biopsy and resection specimens signifies an increased risk of HCC, and all findings should be mentioned in pathology reports.64 The same applies for LCC.
Although there is no consensus regarding the most appropriate treatment of dysplastic nodules, it is reasonable to suggest that high-grade dysplastic nodules should be treated by ablation (similar to early HCC) because of the high risk of malignant progression.64
Early Hepatocellular Carcinoma
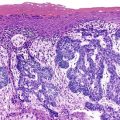
International consensus regarding the defining features of early HCC was reported in 2009.82 Early HCC is a small, low-grade, early-stage tumor.99,109 However, not all small HCCs (<2 cm in diameter81) are early HCCs; some small HCCs have histologic features similar to those of larger, classic HCCs. The classic lesions may be associated with vascular invasion (27% of cases) and intrahepatic metastases (10% of cases), whereas early HCC lacks the ability to invade vascular structures or metastasize.109 However, foci of classic HCC may arise in early HCC, and in time, the classic HCC replaces the early HCC.
Most early HCCs consist of small hepatocyte-like neoplastic cells, arranged in irregular, thin trabeculae and pseudoglandular structures (Fig. 44.10, A and B).109–111 Cellular crowding is a characteristic finding on low-power examination of these lesions. Poor circumscription is another characteristic feature. The neoplastic cells at the tumor margin merge imperceptibly with the adjacent hepatocytes (see Fig. 44.10, C), and on gross examination, the lesion usually appears as a nodule with indistinct margins. Diffuse fatty change is common and has been attributed to insufficient vascular supply.112
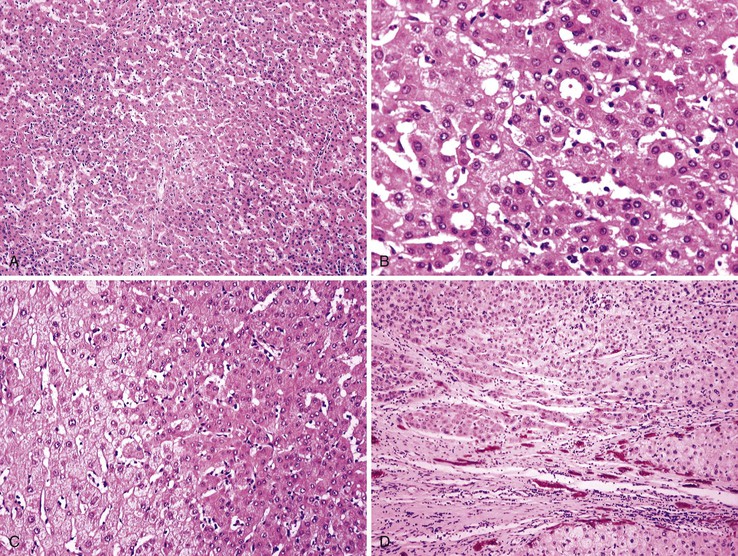
Similar to a high-grade dysplastic nodule, early HCC may receive its blood supply from the portal tracts and small nontriadal (unpaired) arteries. On the arterial phase of contrast-enhanced imaging studies, early HCCs usually appear isovascular or hypovascular compared with the adjacent hepatic parenchyma. Classic HCCs typically appear hypervascular because of well-developed nontriadal arteries.82 Lesions consisting of classic HCC arising in early HCC (Fig. 44.11) may provide a characteristic nodule-in-nodule sign on contrast-enhanced imaging studies.
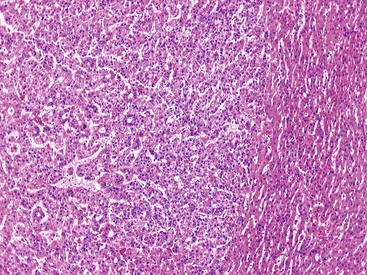
Stromal invasion into the portal tract or septal stroma has emerged as the most important feature distinguishing early HCC from high-grade dysplastic nodules.82 A ductular reaction is found around noninvasive hepatocellular nodules but is characteristically lacking in areas of stromal invasion by HCC. Immunohistochemical stains for cytokeratin 7 (CK7) or CK19 can be used to identify a ductular reaction (see Fig. 44.10, D).113 Other immunohistochemical stains that may be useful in this differential diagnosis are discussed in the following section.
Molecular Diagnostics
The many structural DNA changes identified in HCCs and precancerous lesions have elucidated the molecular evolution of hepatocarcinogenesis, but they are not used for routine diagnostic purposes. Some chromosomal abnormalities, such as gains of 1q and 8q, which can be demonstrated by fluorescent in situ hybridization or comparative genomic hybridization, may be useful in distinguishing well-differentiated (adenoma-like) HCC from HCA.114–116 The large-scale studies of gene expression performed in the past decade have provided new immunohistochemical markers for the pathologist’s armamentarium.
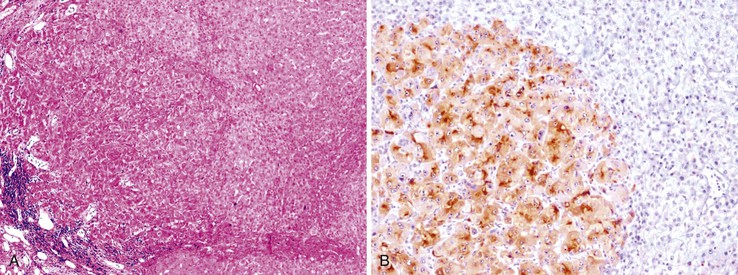
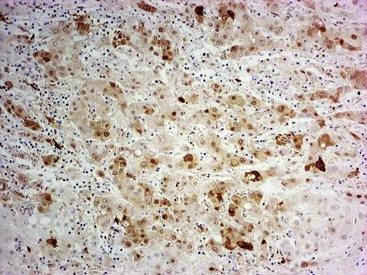
Heat shock protein 70 (HSP70) is significantly upregulated in early and classic HCCs compared with cirrhotic liver and precancerous lesions.117 Using immunohistochemical methods, approximately 80% of early HCCs are positive for HSP70, compared with only 5% of high-grade dysplastic nodules, making this protein a useful marker in the differential diagnosis. Staining is nuclear and cytoplasmic (Fig. 44.12). Lesions are considered positive when 10% or more of the cells express HSP70.83 Normal hepatocytes are negative for HSP70, and biliary epithelial cells provide a useful internal positive control.
The oncofetal antigen glypican 3 (GPC3) is an established serum and tissue marker of HCC. Immunohistochemical staining for GPC3 is positive in 70% to 90% of HCCs overall and more often positive in poorly differentiated than well-differentiated tumors.118–120 Dysplastic nodules occasionally may be positive for GPC3. Nonseminomatous germ cell tumors and malignant melanoma also express GPC3.116 Staining is usually cytoplasmic, but it may be membranous or canalicular (see Fig. 44.9, B).82 GPC3 is not expressed in HCAs and is therefore a useful marker for distinguishing between well-differentiated HCCs and HCAs.120 However, negative immunohistochemical results do not rule out HCC.
The GS protein is often detected in HCCs. The characteristic GS immunostaining patterns of normal liver, FNH, and HCA were described earlier (see Benign Hepatocellular Tumors). GS immunopositivity is usually strong and diffuse but is sometimes heterogenous in B-HCAs and β-catenin–activated HCCs (Fig. 44.13).27,121 A small percentage of high-grade dysplastic nodules may be focally positive.
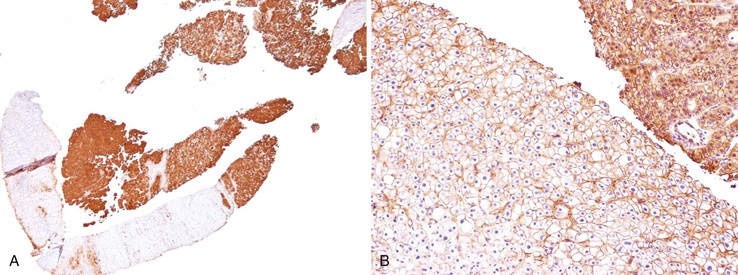
Assessment of the combination of HSP70, GPC3, and GS in distinguishing between high-grade dysplastic nodules and early or well-differentiated HCCs showed that positivity for any two of the three immunohistochemical markers was a feature of HCC (100% specificity), with modest sensitivity.122,123 Although the sensitivity was lower in biopsy (57%) than resection specimens (72%), this three-stain panel can be useful in this difficult differential diagnosis when stromal invasion is not detected.83 Additional diagnostic markers (e.g., clathrin heavy chain124) are being developed and tested as molecular data find their way to diagnostic pathology.
Ongoing research relevant to targeted therapies for HCC is likely to provide prognostic and predictive markers in the near future. Currently, CK19 expression suggesting the progenitor cell origin and detected by immunohistochemistry in a minority of HCCs is thought to confer an adverse prognosis. CK19-positive HCCs have a higher incidence of recurrence and lymph node metastases than CK19-negative ones.125,126