Molecular Diagnostics of Tubal Gut Neoplasms
Mark Redston
John Iafrate
Introduction
Molecular diagnostic assays comprise three broad categories. Molecular microbiology is used for infectious disease diagnosis and classification. Genetic testing is applied to germline DNA for detecting heritable diseases, pathogenic mutations, and disease-associated single nucleotide polymorphisms. Molecular oncology methods are used for testing somatic tumor cell samples or their derived cellular or molecular constituents. This chapter focuses on the methods and applications of molecular oncology to the gastrointestinal (GI) tract and highlights the genetic and infectious disease testing used in the clinical practice of diagnostic pathology (Table 23.1).
Table 23.1
Validated Molecular Genetic Tests in GI Pathology
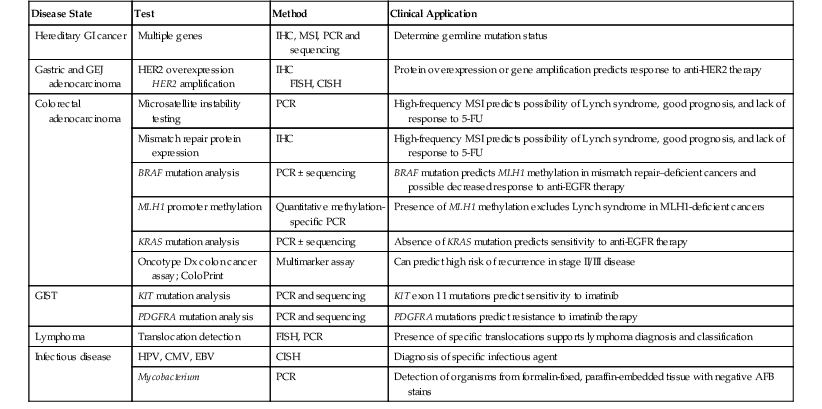
| Disease State | Test | Method | Clinical Application |
| Hereditary GI cancer | Multiple genes | IHC, MSI, PCR and sequencing | Determine germline mutation status |
| Gastric and GEJ adenocarcinoma | HER2 overexpression HER2 amplification |
IHC FISH, CISH |
Protein overexpression or gene amplification predicts response to anti-HER2 therapy |
| Colorectal adenocarcinoma | Microsatellite instability testing | PCR | High-frequency MSI predicts possibility of Lynch syndrome, good prognosis, and lack of response to 5-FU |
| Mismatch repair protein expression | IHC | High-frequency MSI predicts possibility of Lynch syndrome, good prognosis, and lack of response to 5-FU | |
| BRAF mutation analysis | PCR ± sequencing | BRAF mutation predicts MLH1 methylation in mismatch repair–deficient cancers and possible decreased response to anti-EGFR therapy | |
| MLH1 promoter methylation | Quantitative methylation-specific PCR | Presence of MLH1 methylation excludes Lynch syndrome in MLH1-deficient cancers | |
| KRAS mutation analysis | PCR ± sequencing | Absence of KRAS mutation predicts sensitivity to anti-EGFR therapy | |
| Oncotype Dx colon cancer assay; ColoPrint | Multimarker assay | Can predict high risk of recurrence in stage II/III disease | |
| GIST | KIT mutation analysis | PCR and sequencing | KIT exon 11 mutations predict sensitivity to imatinib |
| PDGFRA mutation analysis | PCR and sequencing | PDGFRA mutations predict resistance to imatinib therapy | |
| Lymphoma | Translocation detection | FISH, PCR | Presence of specific translocations supports lymphoma diagnosis and classification |
| Infectious disease | HPV, CMV, EBV | CISH | Diagnosis of specific infectious agent |
| Mycobacterium | PCR | Detection of organisms from formalin-fixed, paraffin-embedded tissue with negative AFB stains |
AFB, Acid-fast bacillus; CISH, chromogenic in situ hybridization; CMV, cytomegalovirus; EBV, Epstein-Barr virus; EGFR, epidermal growth factor receptor; FISH, fluorescence in situ hybridization; 5-FU, 5-flurouracil; GEJ, gastroesophageal junction; GI, gastrointestinal; GIST, gastrointestinal stromal tumor; HER2, human epidermal growth factor receptor 2; HPV, human papillomavirus; IHC, immunohistochemistry; KIT, CD117; MSI, microsatellite instability; PCR, polymerase chain reaction; PDGFRA, platelet-derived growth factor receptor-α.
Methodologies
Sample Collection and Processing
Most routine genetic testing is performed on high-molecular-weight genomic DNA and RNA extracted from white blood cells in whole blood samples. Improved methods use more limited samples, such as blood spots and buccal smears. Because a thorough discussion of all methods is beyond the scope of this chapter, we focus on testing of surgical pathology samples and other samples that may harbor neoplastic cells, DNA, or microsomes that may be used for molecular screening and diagnosis.
Tissue
Historically, extraction of high-quality DNA and RNA has required fresh or frozen tissue samples. If processed very quickly after surgical removal, these samples provide excellent nucleic acids for subsequent investigations. However, degradation of nucleic acids may occur. Degradation depends on several factors: tissue necrosis, duration of acute surgical ischemia, the time from resection to tissue harvesting, DNase and RNase activity during extraction, and improper storage. Attempts to use formalin-fixed, paraffin-embedded tissue samples create unique challenges and usually are less successful because of DNA fragmentation, DNA cross-linking, and contaminants. However, these samples are much more widely available, and improvements in nucleic acid recovery and subsequent testing have greatly increased the utility of these samples.
Microdissection
Tumor samples, particularly from carcinomas, are a complex mixture of neoplastic cells, stromal cells, and inflammatory cells. Some carcinomas, especially those of the pancreas and biliary tree, are characterized by scattered, small neoplastic glands embedded in an abundant desmoplastic stroma. Other cancers may be infiltrated by a dense lymphocytic response. As a result, unenriched tumor samples may vary in neoplastic cellularity from a high of approximately 80% to a low of only 10% to 20% tumor cells. The success of some molecular diagnostic assays depends on a neoplastic cellularity of at least 50%, and microdissection may be required to enrich the sample for neoplastic cells. Manual microdissection involves trimming frozen tissue blocks under the direct guidance of frozen section histology or scraping specified regions of neoplastic cells from an unstained section of paraffin-embedded tissue that is aligned with the adjacent hematoxylin and eosin (H&E) staining.1 These approaches can produce marked improvements in neoplastic cellularity.
Laser microdissection methods may be used to dramatically enrich neoplastic populations, although the yield of tissue is significantly reduced in these labor-intensive situations.1 Assay requirements must be tailored to the tissue parameters to determine which enrichment methods are needed. In liquid-phase enrichment, cells are digested to release them in a suspension and then purified by bead-tagged antibodies that target cell surface antigens. These approaches are common in research applications but are used much less frequently in clinical practice.
In addition to tissue samples obtained from sections cut from paraffin blocks, cytologic preparations may be used in molecular diagnostic assays. The number of cells and quantity of DNA available for testing are usually limited with these methods. The use of cytologic specimens in molecular assays needs to be closely supervised by a pathologist because the process may consume the permanent diagnostic record.
Circulating DNA
Small amounts of detectable, cell-free DNA circulate in the plasma and serum of healthy individuals and in persons with a variety of disease states. In patients with cancer, this includes DNA and RNA from tumor cells. Detection of a variety of tumor mutations, microsatellite alterations, methylation abnormalities, and chromosomal alterations has laid the foundation for development of assays for use in cancer screening, diagnosis, and therapeutic monitoring.2 In a similar manner, DNA from other biofluids (e.g., effusions, fecal samples, biliary aspirations, luminal washings) may be used in molecular diagnostic assays. These findings and technologies are an extremely active area of research and hold promise for the development of a variety of novel assays, but there currently are no validated tests in routine use for GI diseases that use these approaches.
Nucleic Acid Extraction and Purification
Traditional extraction of good-quality high-molecular-weight DNA requires a three-step extraction procedure. First, cell and tissue lysis use detergents, proteinase K, and RNase. Second, proteins are removed, avoiding RNA and chemical contamination. Third, DNA is purified. Conventional manual extraction techniques use phenol-chloroform purification and precipitation by using ethanol or isopropanol. Newer, kit-based approaches use a variety of bead-based elution technologies to purify DNA.3 Some methods yield DNA of lesser purity and lower molecular weight, although this result is acceptable for many applications. The methods used must appropriately balance ease of use, expense, quality, and downstream assay requirements.
RNA is extracted by two predominant methods: phenol-based extraction (e.g., TRIzol reagent) and silica matrix or glass fiber filter-based binding. TRIzol methods retain small RNAs, including microRNA (miRNA) and small interfering RNA (siRNA). TRIzol reagent also includes guanidine isothiocyanate to maintain the integrity of RNA while disrupting cells and dissolving cell components.
After extraction, nucleic acids must be accurately quantified to assess the success of the extraction and to determine the correct quantity of template for subsequent applications. Quantification is typically performed by using an ultraviolet spectrophotometer with a known control, usually calf thymus DNA.
Detection of Sequence Variants
Detection of single nucleotide changes and small insertions and deletions in an unknown nucleic acid sample is the most common assay performed in molecular diagnostics. These assays are performed to identify hereditary germline disease-associated mutations, single nucleotide sequence variants (i.e., polymorphisms), and somatic aberrations in tumor cells. Although sequencing is the gold standard method, it has not always been the most practical approach because of the size of the target gene that needs to be analyzed, the limited DNA sample that may be available, or the rarity of the variant sequence in the nucleic acid population to be analyzed. Many different approaches to screening for mutations have evolved in the past 20 years in response to improved technologies.
For simplicity, sequence variant detection methods may be divided into polymerase chain reaction (PCR)–based technologies and sequencing. Methodologic approaches may be tailored to the expected result. For instance, the approaches to detect a common known sequence variant at a set nucleotide (e.g., BRAF V600E mutation in colorectal cancer) are very different from mutation screens applied to an entire exon, gene, or panel of genes. PCR-based approaches use a variety of different techniques. Common approaches include PCR primers that are designed for specific recurrent mutations and real-time PCR melting curve analysis. As sequencing technologies have improved and reactions have become simpler and cheaper to perform and analyze, sequencing approaches have become the first choice for sequence variant detection.
Sanger Sequencing
Now known as first-generation sequencing, automated Sanger sequencing was introduced in 1977. It uses electrophoretic separation of randomly terminated linear sequence extensions and remains the most widely available technology today.4 It is accurate, has well-defined chemistry, and is best suited for reading 500-base pair (BP) to 1-kilobase (kb) DNA fragments in a single reaction. Modern platforms use automation, fluorescent chemistry, and capillary electrophoresis. They have served as the primary machine for single-gene diagnostics in molecular diagnostics laboratories and as the workhorse for the first generation of the genome sequencing project.
Sanger sequencing does have technical limitations, particularly throughput (i.e., number of bases per second that can be read). Electrophoretic separation is the predominant rate-limiting step, severely limiting the speed and cost of assays. For example, it has been estimated that one automated sequencer would require several decades and tens of millions of dollars to sequence a single genome.4
Next-Generation Sequencing
Also known as second-generation sequencing and massively parallel sequencing, next-generation sequencing (NGS) technologies are capable of sequencing large numbers of different DNA sequences in parallel (i.e., in a single reaction).4 NGS technologies were developed in part from focused investment by the National Human Genome Research Institute (NHGRI) in an effort to markedly reduce the expense of large-scale sequencing and thereby rapidly advance the realization of personalized medicine.
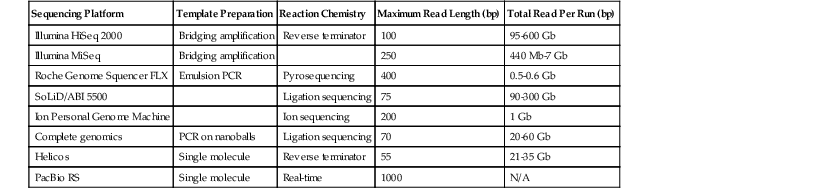
NGS assays the addition of nucleotides to immobilized and spatially arrayed DNA templates. Although the various platforms use four basic steps (i.e., sample collection, template generation, sequencing reaction, and detection), they use substantially different methods of template generation and sequence interrogation (Table 23.2). Because NGS represents the most important technologic development emerging in molecular diagnostics, it is considered in more detail in the following sections.
Table 23.2
Next-Generation Sequencing Platforms
| Sequencing Platform | Template Preparation | Reaction Chemistry | Maximum Read Length (bp) | Total Read Per Run (bp) |
| Illumina HiSeq 2000 | Bridging amplification | Reverse terminator | 100 | 95-600 Gb |
| Illumina MiSeq | Bridging amplification | 250 | 440 Mb-7 Gb | |
| Roche Genome Squencer FLX | Emulsion PCR | Pyrosequencing | 400 | 0.5-0.6 Gb |
| SoLiD/ABI 5500 | Ligation sequencing | 75 | 90-300 Gb | |
| Ion Personal Genome Machine | Ion sequencing | 200 | 1 Gb | |
| Complete genomics | PCR on nanoballs | Ligation sequencing | 70 | 20-60 Gb |
| Helicos | Single molecule | Reverse terminator | 55 | 21-35 Gb |
| PacBio RS | Single molecule | Real-time | 1000 | N/A |
bp, Base pairs; Gb, giga base pairs; Mb, mega base pairs; N/A, not applicable; PCR, polymerase chain reaction.
From Rizzo JM, Buck MJ. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev Res. 2012;5:887-900.
Template Generation
The general starting point for all NGS assays is double-stranded DNA (dsDNA). It is obtained from a variety of sources, including genomic DNA and reverse-transcribed RNA (i.e., complementary DNA [cDNA]). All starting dsDNA must be converted into a sequencing library, wherein fragmentation, size selection, and adapter ligation are used to generate an unbiased representation of the DNA population to be sequenced. Template generation spatially separates and immobilizes DNA fragments by attaching them to solid surfaces or beads. Limitations in the detection sensitivity require that sequencing reactions be amplified before sequencing, which may introduce bias (e.g. loss of representation of rare sequences) and errors (e.g., particularly problematic in clinical applications). The latest technologic platforms can sequence from single-molecule templates, which represents such a significant improvement that some authorities refer to them as third-generation platforms.4
Sequencing Reaction
NGS platforms use a series of repeating chemical reactions that apply DNA polymerase or ligase to add and detect nucleotides on a repetitive nucleotide-by-nucleotide basis. This process, referred to as sequence by synthesis, represents a dramatic improvement to Sanger sequencing, which requires discrete separation and detection of fragments that differ in length by 1 bp. Thee simultaneous analysis of massive numbers of reactions by NGS is largely responsible for the more than 100,000-fold decrease in per base sequence cost during the past 5 years.
Data Analysis
Compared with Sanger sequencing (which typically reads 500 bp to 1 kb in a single reaction), most NGS platforms offer shorter average read lengths (30 to 400 bp). Because NGS reactions generate millions of these short reads, data analysis is a much more difficult task, making heavy demands on data acquisition, storage, tracking, quality control, analysis, and interpretation.4 NGS data generation can vastly outpace analytic and interpretation resources.
Typically, the first phase of analysis is base calling, which is usually performed by the proprietary software associated with the sequencing platform. Base calling is followed by sequence alignment and assembly, an area of extremely active computational research.5 The last phase of analysis requires interpretation of the final sequence data. Third-generation platforms may offer longer reads, which could vastly reduce the challenges of data analysis, but these underdeveloped platforms are not widely available.
Sequencing Coverage
All NGS platforms have inherent errors in base calling, and the rate of error varies with each platform and chemistry. To counter these qualitative errors, each base pair is sequenced multiple times in separate, parallel sequencing reactions. The degree to which each nucleotide is quantitatively resequenced is referred to as the coverage or coverage depth of sequencing.
Because there may be biases in coverage depth during any NGS experiment, evolving standards suggest a need for 30 to 100 times coverage to ensure 100% accuracy in detection of single-nucleotide sequence variants. Deep sequencing produces very high coverage, meaning that each nucleotide may be sequenced hundreds to thousands of times, enabling the quantitative detection of rare variants in a DNA population. This method has clinical application in the detection of rare mutations. The occurrence of errors and the need for coverage contribute to the plateau in the cost reduction of whole-genome sequencing. This plateau may be overcome by the increased fidelity associated with third-generation technologies that use single-copy DNA.
Clinical Applications
NGS platforms offer the potential for large-scale genotyping and tumor profiling to characterize human disease at an individual level. Despite the explosion of research studies, validated clinical applications remain limited and are targeted at the small number of disease-associated genes. Targeted sequencing with NGS technology requires enriching DNA regions of interest before sequencing. Enrichment strategies include hybrid capture, microdroplet PCR, and array capture. Abnormalities identified in NGS screens are typically confirmed with traditional Sanger sequencing.
Targeted Sequencing
NGS is used to target panels of genes that have relevant mutations in the diagnosis or management of disease. NGS becomes cost-effective compared with Sanger sequencing diagnostics when multiple genes need to be sequenced. Examples include panels of genes for the diagnosis of genetic diseases and for the diagnosis of oncogenic driver mutations in cancer.
Multigene oncogenic assays have been established for lung cancer, for metastatic disease that has failed other therapeutic options, and for a variety of research studies, and they have offered significant advantages to traditional molecular diagnostic approaches.6,7 A panel developed for screening known genes associated with colorectal polyposis and Lynch syndrome has been a powerful and cost-effective approach that eliminates the traditional stepwise approach to genetic characterization.8,9 GI tract, liver, and pancreaticobiliary malignancies do not have other sufficiently validated gene targets to warrant additional NGS gene panels, and although current applications are mostly limited to the research setting, this situation is expected to change dramatically in the next 5 years.
Whole-Exome Sequencing
Exome sequencing refers to large-scale sequencing of genomic coding regions, which comprise only approximately 2% of the genome. Exome sequencing may be directed at the whole exome or may target the approximate 3000 genes or smaller subsets known to be involved in human disease. Although exome sequencing has not been validated for routine clinical use, it has revolutionized research into the genetic basis of human disease and the comprehensive identification of novel gene mutations in human tumors.
Whole-Genome Sequencing
Although whole-genome sequencing (WGS) does not have routine validated clinical applications, it is technologically feasible on an individual basis and has been used with great success in selected cases and research studies. WGS has been used in the analysis of tumors and has resulted in the rapid identification of novel translocation fusion proteins not identified by other methods. For example, WGS has been used in a clinical trial setting for an individual patient with metastatic colorectal cancer in whom a novel experimental therapy was used based on the mutation profile of the tumor.10
Rare Variant Detection
In most studies and applications, NGS has been used to broadly sequence the entire genome or large portions of the genome to detect variants that are present at high frequency in the sample DNA. NGS also may be used to repeatedly sequence large numbers of the same template and identify rare variants. These deep sequencing studies use very dense coverage of a small portion of the genome. Examples of potential applications include identification of circulating donor DNA in patients with very early transplant rejection and identification of fetal Down syndrome in circulating DNA of the pregnant mother of the fetus.11 The technique has many potential applications in screening and early detection of cancer.
Rare Mutation Detection
Technologies designed to identify rare mutation loads in clinical biospecimens can be applied to screening for malignancy (e.g., detection of KRAS mutations indicating colorectal adenomas and carcinomas in fecal samples), monitoring of the cancer burden and minimal residual disease (e.g., reverse transcriptase PCR detection of leukemia-specific translocations in bone marrow and blood samples), and detection of secondary oncogenic mutations leading to resistance to targeted therapy (e.g., Beads, Emulsion, Amplification, and Magnetics [BEAMing] technology to identify resistance mutations in KIT). The potential superiority of these approaches in blood and plasma samples compared with tissue biopsies has generated a concept referred to as liquid biopsy.
BEAMing Technology
BEAMing is a proprietary technology (Inostics, Baltimore, Md.) that allows the quantitative detection of oncogenic mutations.12 The technology allows performance of single-molecule PCR on magnetic beads in an emulsion and can detect mutant DNA at low levels compared with wild-type DNA (ratios as low as 1 : 10,000). BEAMing has been used to detect tumor mutations in the circulating DNA and fecal DNA of colorectal cancer patients and to monitor the evolution of resistant KIT mutations in gastrointestinal stromal tumors (GIST) patients undergoing targeted therapies.13,14
Detection of Minimal Residual Disease
The term “minimal residual disease” refers to persistence of neoplastic cells below the threshold of conventional morphologic detection. The rapid development of molecular technologies in the past decade has revolutionized the ability to monitor minimal residual disease. These molecular technologies have been particularly important to the management of acute myeloid leukemias and chronic myelogenous leukemia.15 Although the potential exists to apply these technologies to the management of many cancers, there have been no validated practical applications to date in management of GI cancers.
Detection of Translocations and Genomic Copy-Number Alterations
Chromosomal structural variations are a hallmark of cancer and include abnormal chromosomal number (i.e., aneuploidy), translocations, focal amplifications, and subchromosomal deletions.16 Although deletions and abnormalities in chromosome number are among the most common changes, particularly in solid tumors, translocations and focal amplifications have garnered the most interest because of their association with oncogenic activations that may be susceptible to targeted inhibition. Translocations and other rearrangements also are important in the routine characterization and diagnosis of hematologic malignancies.
Methods used in the characterization of chromosomal structural variations include traditional cytogenetics, fluorescence in situ hybridization (FISH), comparative genomic hybridization (CGH) array-based methods (e.g., CGH array), and PCR. The remainder of this discussion focuses on the methods most commonly applied to tissue samples in clinical molecular diagnostics: FISH and PCR. CGH arrays are briefly discussed (see Microarray-Based Platforms).
Fluorescence and Chromogenic In Situ Hybridization
FISH and chromogenic in situ hybridization (CISH) are widely used methods that can be applied to the detection of DNA and RNA. Metaphase FISH is a cytogenetic technique that requires culture of live cells and allows detection of much smaller abnormalities than are detected by conventional karyotyping. Interphase FISH does not require culture of live cells, and it is therefore a practical method that is commonly used to study routinely fixed tumors and hematologic malignancies.
For interphase FISH analyses, DNA probes in the 60- to 200-kb range are covalently attached to a fluorescent molecule, hybridized to a complementary target sequence in cellular DNA, and visualized under a fluorescent microscope as a point of fluorescent light in the nucleus of the cell. Probes are designed with specificity to any region of interest within the genome and are widely available for centromeric regions (i.e., CEN probes) and telomeres. Multiple probes may be hybridized simultaneously and analyzed separately by using different colors of fluorescent dyes.
Known, recurrent oncogenic translocations are detected by FISH by using dual-fusion probes or break-apart probes. Dual-fusion probes contain two probes, each labeled with a different fluorescent dye and designed to bind the regions spanning the breakpoint of each translocation partner. In the absence of a translocation, two distinct nuclear signals are observed for each colored probe. For a translocation, there is a single, distinct signal for each color from the nontranslocated chromosomes, and the two signals then combine both colors for the translocation and its reciprocal. Dual-color, break-apart probes are particularly useful in detecting translocations in which one chromosome can recombine with multiple partners. This approach consists of two probes that bind to the intact chromosome flanking the breakpoint. In the setting of a translocation, the two probes break apart from each other and yield two distinct signals rather than a single hybrid signal.
Copy number aberrations are well suited to detection by FISH. Centromeric probes give an accurate count of chromosome number and are useful in the detection of aneuploidy. When centromeric probes are used in combination with probes targeting hotspot chromosomal regions, they also allow a readout of regions of genomic deletion or amplification. Diploid tumor cells have two distinct centromeric probe signals and only one chromosomal arm signal in the setting of a deletion but more than two in the case of amplification. Multiple cells are typically scored, and a signal ratio is calculated and compared with validated positive and negative control ratios.
Polymerase Chain Reaction Detection of Translocations
Known, recurrent translocations are readily detectable by PCR. Different methods use the same general principles; several primer pairs and sets of primer pairs are designed to detect all possible known breakpoints for a given translocation. Multiple primers are needed because of the limitation in the size of PCR products and the large genomic variability in known breakpoints for some translocations. Assays usually are performed by real-time PCR with the use of standard curves for known controls, and results are reported as the percentage of translocated cells in the specimen tested.
Molecular Detection of Copy Number Aberrations
Copy number abnormalities may be detected by semiquantitative or quantitative PCR. For instance, PCR amplification of a polymorphic microsatellite reveals two bands in normal tissue. In the presence of an intragenic deletion in a tumor sample that spans the microsatellite, there is a reduction in amplification of the deleted allele. Referred to as loss of heterozygosity (LOH), this standard research approach characterizes genomic deletions in tumor samples and has been used in some clinical trials (e.g., evaluation of 18q LOH as a marker of adverse outcome in colorectal cancer). These types of assays are not being routinely used in validated tests in GI pathology.
Microarray-Based Platforms
Array-based hybridization is a technology that uses a large number of targets that are densely arrayed on a small, readable substrate, allowing multiple, simultaneous hybridizations. Array targets are immobilized on glass slides or other materials and may consist of DNA, cDNA, PCR products, oligonucleotides, RNA, or proteins.
Array Technologies
Array-based hybridizations were originally developed on nitrocellulose and nylon membranes and were moved to treated glass slides in 1987. The development of technology to deposit small spots of target on glass substrates led to a rapid increase in the miniaturization of spots and the array density of spots. The first automated arrayer was described in 1995, and it used a pen-type device to dot target material onto substrate.17 These technologies have rapidly evolved and are now capable of arraying as many as 100,000 spots on a substrate the equivalent size of a standard microscope slide.
Oligonucleotide arrays use technologies that directly synthesize DNA on glass or silicon substrates.18 These proprietary arrays (e.g., Affymetrix) use the efficient synthesis of short oligomers (10 to 25 bp), and samples can be arrayed at very high density. The selection of oligonucleotides to be arrayed varies with the intended application of the array.
Samples to be arrayed are typically prepared by fluorescent labeling, and several methods may be used. Some array technologies use single-color fluorescent labeling, whereas others (typically expression arrays) depend on dual-color competitive hybridizations in which a control sample is quantitatively compared with a test sample.
Reading microarrays requires a fluorescent reader and analysis software. Array results are corrected for background noise and normalized with standards. Results from duplicate or triplicate sample data are averaged by the analytic software.
Although very powerful in research settings, applications of array technology in the clinical molecular diagnostic laboratory remain limited, in large part because of a lack of established standards and controls and a high degree of noise and variability. The commercial development of many of these platforms has addressed many of these challenges, and array-based assays have recently seen increased use in clinical laboratories, particularly in the area of cytogenetics.
Expression Arrays
Expression arrays are designed to determine the relative expression level of a vast number of genes in a single sample. Typical experiments use labeled mRNA in a competitive, two-color fluorescent hybridization with a known control sample.19 These tests simultaneously measure the transcript level of thousands of genes compared with a control or normal specimen. This transcriptional profiling (i.e., transcriptome analysis) is commonly applied to human neoplasms.
Comparative Genomic Hybridization
CGH arrays are designed to test DNA. Specific genomic DNA sequences are spotted onto an array corresponding to loci known to be amplified or deleted in human tumors or possibly encompassing a much more comprehensive representation of the human genome.20 Genomic DNA from the test sample is purified, fragmented, fluorescently labeled, and hybridized, typically in a competitive two-color hybridization with a known normal or control sample. To facilitate application to limited tissue samples, several methods have been developed to globally amplify test DNA before CGH analysis. Competitive hybridization allows a readout of the relative genomic copy number across all assayed genomic loci.
Multimarker Assays
The preceding discussions on sequencing and array technologies provide insight into the depth of genomic and gene expression data that can be harvested from analyses of a pathology specimen. For example, the Cancer Genome Anatomy Project recently concluded many years of work detailing the genetic and epigenetic aberrations of a panel of colorectal cancer tumors assayed by many technologic platforms.21 In combination with the efforts of the pharmaceutical industry to develop targeted therapies, research in the development of multimarker assays is likely to yield significant progress in the coming years.
Molecular Diagnostics of Hereditary Gastrointestinal Cancer
Molecular diagnostics were applied to hereditary GI cancer syndromes, with germline testing for familial adenomatous polyposis and Lynch syndrome beginning in the early and mid-1990s, respectively.22 In the past 20 years, hereditary cancer testing has expanded to include all of the polyposis syndromes and familial gastric cancer (Table 23.3).
Table 23.3
Molecular Genetic Diagnosis of Polyposis and Other Hereditary GI Cancer Syndromes
| Entity | Gene | Proportion of Cases Attributable to Gene |
| Polyposis Syndromes | ||
| Familial adenomatous polyposis (FAP) | APC | >95% |
| Attenuated adenomatous polyposis (AFAP) | APC | 20-30%* |
| MUTYH (MYH)† | 20-50% | |
| Peutz-Jeghers syndrome (PJS) | STK11 (LKB1) | Familial 100%; simplex 90% |
| Juvenile polyposis (JPS) | SMAD4 | 20% |
| BMPR1A | 20% | |
| Cowden syndrome | PTEN‡ | 85% |
| Bannayan-Riley-Ruvalcalba syndrome (BRRS) | PTEN‡ | 65% |
| Proteus or proteus-like syndrome | PTEN‡ | 20-50% |
| Nonpolyposis Hereditary Cancer | ||
| Lynch syndrome | MLH1 | 50% |
| MSH2 | 40% | |
| MSH6 | 7-10% | |
| PMS2 | 5% | |
| EPCAM (TACSTD1)§ | 1-3% | |
| Hereditary diffuse gastric cancer | CDH1 | 30-50% |
| Genetic Basis Not Defined | ||
| Serrated polyposis syndrome | Unknown | |
| Familial colorectal cancer type X | Unknown | |
| Familial esophageal cancer | Unknown | |
* The proportion of cases with APC mutations increases with increasing numbers of adenomas.
† AFAP caused by MUTYH mutations is also known as MUTYH-associated polyposis (MAP). Some MUTYH mutation carriers have more than 100 polyps at presentation, and these cases are differentiated from FAP by the absence of an autosomal dominant family history and patient age older than 35 years.
‡ The term PTEN hamartoma tumor syndrome (PHTS) is used to describe cases with PTEN mutations.
§ Hereditary EPCAM mutations cause Lynch syndrome by inducing MSH2 methylation and gene silencing in colonic epithelial cells.
With the exception of Lynch syndrome (discussed later), hereditary GI cancer molecular testing does not involve anatomic pathology specimens. Germline testing for hereditary cancer mutations is performed on genomic DNA extracted from white cells obtained from peripheral blood. Mutational testing is typically performed by PCR and Sanger sequencing or pyrosequencing. With the advent of other NGS technologies, much of this single-gene mutational testing is being combined into panels of genes, allowing significant reductions in cost and turnaround time.8,9 In addition to the sequence variants identified by sequencing, some inherited defects in the hereditary cancer genes consist of much larger genomic deletions or duplications that are not routinely detected by sequencing. They are particularly common in familial adenomatous polyposis, accounting for approximately 10% of cases or families.23 If sequencing fails to identify a pathogenic variant, comprehensive adenomatous polyposis coli gene (APC) testing programs include assays to detect larger aberrations, including Southern blot, quantitative PCR, cytogenetics, and CGH arrays.
The surgical pathologist should be aware that testing is available for most of the hereditary GI cancer syndromes. Because the pathologist may be the first clinician to recognize that a patient is affected by one of these syndromes, he or she can play a key role in suggesting referral to a genetics counselor for assessment and testing.
Lynch Syndrome
Individuals with Lynch syndrome have a hereditary predisposition for colorectal cancer caused by germline mutations in one of the DNA mismatch repair genes (MLH1, MSH2, MSH6, or PMS2) or the EPCAM gene, which induces methylation silencing of MSH2. Lynch syndrome causes approximately 3% to 4% of all colorectal cancers.24 Originally described as Lynch syndrome I (site-specific colorectal cancer), Lynch syndrome II (colorectal and extracolonic cancer), and Muir-Torre syndrome (skin tumors and visceral malignancies), it is now understood that all three syndromes share the same basic underlying molecular pathogenesis. The genetic basis for differences in the tumor spectrum between individuals and families most likely reflects genotype-phenotype relationships of different mutations or the effect of other modifier genes or environmental influences that are not fully understood.
Many Lynch syndrome patients have family histories with strong autosomal dominant patterns and therefore meet Amsterdam I or II family history criteria for hereditary nonpolyposis colorectal cancer (HNPCC). The HNPCC criteria describe a family history of colorectal cancer predisposition, and not all HNPCC patients therefore have Lynch syndrome. As many as 50% of HNPCCs (e.g., familial colorectal cancer type X) may be caused by unknown genes.24 The clinical, pathologic, and pathogenetic aspects of Lynch syndrome are discussed in Chapter 27.
Universal Molecular Screening for Lynch Syndrome
Despite the identification of germline mismatch repair gene mutations as the cause of Lynch syndrome 20 years ago, identification and genetic testing of possible Lynch syndrome patients has been slow to catch on in clinical practice.24 Although some of this disparity is related to a failure of the clinical community to rigorously screen all newly diagnosed colorectal cancer patients for a family history of cancer, many Lynch syndrome patients do not meet the traditional family history criteria. Approximately 40% of Lynch syndrome patients do not meet Amsterdam I or II criteria, including approximately 25% who do not meet the much less stringent Bethesda criteria.25
Because effective screening tests are readily available and the morbidity and mortality associated with Lynch syndrome are greatly reduced with intensive colonoscopic screening, several authorities, including the American Molecular Pathology Association, recommend universal screening of newly diagnosed colorectal cancer by using microsatellite instability testing or immunohistochemistry.26,27 Both tests have approximately 90% to 95% sensitivity for detection of a mismatch repair defect; in the setting of a strong family history, a second test is recommended if the initial screening test result is negative. Some universal screening programs use upper age cut-off values of 65 to 75 years. Figure 27-29 in Chapter 27 shows a sample screening algorithm that uses immunohistochemistry as an initial screening test. Immunohistochemistry offers several advantages as an initial screening test. It is rapid and inexpensive, does not require tissue microdissection or DNA extraction, can be readily preformed on a biopsy sample, and can be used to guide subsequent genetic testing in the case of an abnormality. Universal mismatch repair analysis is also useful for predicting patient outcomes and tailoring disease management.
Microsatellite Instability Testing
Microsatellite instability (MSI) testing is an accepted method of screening for Lynch syndrome, and MSI is the original molecular genetic signature by which the involvement of DNA mismatch repair defects in human cancer were first identified.24 MSI testing is a PCR test that amplifies a panel of DNA microsatellites in tumor DNA and compares the results with normal DNA from the same patient. Because DNA mismatch repair deficiency is associated with a dramatic decrease in the fidelity of DNA repair, particularly at short repetitive DNA sequences, tumors with DNA mismatch repair deficiency have widespread alterations in the sizes of microsatellites, which are readily identified in an MSI test.
MSI testing is routinely performed on paraffin-embedded tissue and requires only crude tissue microdissection. After PCR amplification, amplified products are typically analyzed on an automated sequencing platform by capillary gel electrophoresis to resolve 1- and 2-bp shifts in microsatellite sizes (see Fig. 27-26 in Chapter 27). The use of relatively monomorphic mononucleotide repeats allows MSI testing to be performed on tumor DNA samples without accompanying normal DNA. A positive MSI test result has extremely high specificity for an underlying DNA mismatch repair deficiency.
Mismatch Repair Immunohistochemistry
The DNA mismatch repair protein complex is involved in postreplicative DNA repair and is therefore expressed in all proliferating cells, including normal crypt base epithelium and most colorectal cancers. Patients with Lynch syndrome inherit a mutant copy of one of the DNA mismatch repair genes, typically resulting in protein truncation or major impairment in protein function. Because the second allele is normally expressed in somatic tissues, there is no appreciable phenotype in normal cells. In tumorigenesis, the second wild-type allele is inactivated by point mutation, deletion, or methylation silencing, resulting in total loss of the normal mismatch repair function. Most inactivating mismatch repair gene defects result in total loss of mismatch repair protein expression, which is readily identifiable by immunohistochemisty.24 The interpretation of mismatch repair protein immunohistochemistry is discussed further in Chapter 27.
Characterization of MLH1-Deficient Colorectal Cancers
Most MLH1-deficient colorectal cancers are caused by promoter hypermethylation of MLH1, which occurs in approximately 12% to 15% of colorectal cancers. The pathogenesis of methylation abnormalities in colorectal cancer is discussed in Chapter 27. MLH1 methylation silencing presents a distinct problem in the interpretation of MLH1 loss in universal screening programs; only approximately 15% of these cases are caused by Lynch syndrome.24 Fortunately, two other extremely helpful tests can be used to determine which MLH1-deficient cases are likely to be caused by Lynch syndrome, and therefore need referral for genetic counseling and evaluation.
BRAF Testing
Although BRAF mutations occur in only 5% to 10% of colorectal cancers, they are associated with MLH1 promoter methylation in approximately 50% of these cases. BRAF mutations, MLH1 methylation, and the serrated pathway are discussed in Chapter 27.
BRAF mutations are almost never identified in Lynch syndrome-associated colorectal cancers. Identification of a BRAF mutation in an MLH1-deficient colorectal cancer excludes the need for further evaluation for Lynch syndrome.28 In colorectal cancer, BRAF mutations usually are limited to the V600E variant, which is readily evaluated in paraffin-embedded tissue by sequencing, mutation-specific PCR, or real-time PCR.
MLH1 Methylation Testing
Testing for MLH1 promoter methylation is a more direct approach to definitively determine that MLH1 loss is not caused by Lynch syndrome. Because MLH1 methylation testing is a more complicated analysis than BRAF V600E testing, it is typically performed only in cases that have been determined to have the wild-type BRAF sequence.27
MLH1 methylation analysis is usually performed by methylation-specific or real-time PCR or sequencing; both are performed after bisulfite treatment of template DNA. Methylated DNA is characterized by the conversion of cytosine to 5-methylcytosine, which results in suppression of gene transcription when it occurs in the regulatory promoter region. During bisulfite treatment, unmethylated cytosine residues are deaminated, which converts them to uracil, whereas 5-methyl cytosines are unchanged. Possible methylation of key DNA sequences may then be evaluated by methylation-specific PCR, which uses sets of primers that are complementary to T residues for unmethylated cytosines or C residues for methylated cytosines. In a similar manner, sequencing across a potentially methylated bisulfite-treated DNA sequence yields a T for an unmethylated cytosine and a C for a methylated cytosine.
Germline Testing
Putative Lynch syndrome patients should be sent for genetic evaluation and counseling before germline testing, which is usually only undertaken only in patients whose tumors are mismatch repair deficient or have MSI. Rarely, tumors from Lynch syndrome patients have no detectable abnormalities by MSI testing and mismatch repair immunohistochemistry and some with normal screening results and a strong family history may still be referred for germline testing.
Genetic testing is performed by PCR and sequencing by using genomic DNA obtained from white cells from peripheral blood. Most mutations occur in MLH1 and MSH2 (see Table 23.3).24 In addition to germline sequence variants and genomic deletions, rare families have heritable methylation silencing of MLH1 and MSH2 expression.28,29
Hereditary Diffuse Gastric Cancer
An estimated 10% to 15% of gastric cancers are familial. Whereas several hereditary cancer syndromes are associated with an increase in gastric cancer, including Lynch syndrome and familial adenomatous polyposis, hereditary diffuse gastric cancer (HDGC) is a form of site-specific familial gastric cancer. Approximately 30% to 50% of HDGC families have inherited germline mutations in CDH1, which encodes E-cadherin, a transmembrane calcium-dependent adhesion molecule involved in cell adhesion.30 The average age at diagnosis of gastric cancer in CDH1 mutation carriers is 37 years.
Genetic testing is recommended for young gastric cancer patients with a positive family history. Because of the difficulty in identifying early gastric cancer in mutation carriers, prophylactic gastrectomy has been recommended and performed in many affected individuals. Mutation analysis is performed by PCR and sequencing by using genomic DNA extracted from white cells from peripheral blood.
Molecular Diagnostics in Targeted Therapy
Principles of Targeted Therapy
Cytoreductive chemotherapy uses toxic compounds that affect all proliferating cells and attempts to find drug combinations and dosage limits that allow selective killing of neoplastic cells instead of non-neoplastic cells. Although the past 50 years have seen major strides in cancer management, these approaches do not take advantage of the exceptional biologic understanding that now exists for most human cancers. Targeted therapies attempt to exploit the inhibition of selected target genes that are thought to be essential for tumorigenesis. Targeted therapies are individualized to the genetic aberrations in a specific tumor and have transformed trial-driven oncology into one of the leading frontiers of personalized therapy.31 Because targeted therapies are based on specific gene aberrations in a given tumor, these approaches have transformed diagnostic pathology and prompted development of a wide range of companion tests that support the application of tailored therapeutic options.32
Driver Mutations and Oncogene Addiction
Targeted therapies are successful in large part because of the genetic mechanisms that underlie the development and progression of human tumors. Human neoplasia is a genetic disease driven by the acquisition of clonal genetic alterations that confer a growth advantage. Some of these mutations are major drivers of the transformation process.33 These oncogenic drivers have diverse effects in cells and may drive some inhibitory pathways as well as stimulatory pathways. The long-term proliferative effect of driver mutations may be associated with a decrease in activity in other normal cellular growth-stimulatory pathways. The cellular tolerance of subsequent downstream mutations may depend entirely on the ongoing effects of these driver mutations.
The net result of these processes is that cancer cells may become dependent on driver mutations, and shutting off the drivers could result in a sudden increase in inhibitory signals, an absence of compensatory stimulatory signals, and unopposed activities of other genes that become detrimental to the cell.34 Shutting drivers off may result in much more striking antiproliferative and apoptotic effects than otherwise predicted. This concept known as oncogene addiction was developed more than 15 years ago and has been demonstrated in cell culture and animal models of human neoplasms.34,35
The dramatic effects of several early targeted therapies, including the use of the tyrosine kinase inhibitor imatinib in chronic myeloid leukemia (CML) with BCR-ABL activation and GIST with activating KIT mutations and the use of the epidermal growth factor receptor (EGFR) inhibitor gefitinib in lung cancers with EGFR mutations, have further supported the exploitable nature of this oncogene addiction.36,37 This therapeutic paradigm of targeting and shutting down the key oncogenic driver mutations in human cancer forms the cornerstone of modern developmental cancer therapeutics.
Tyrosine Kinase Inhibitors
The nature of signaling cascades has made protein kinases very attractive targets for the development of therapeutic compounds, particularly tyrosine kinase inhibitors. These small-molecule inhibitors are designed to compete with ATP or substrate binding or to bind in an allosteric fashion and alter activity because of a conformational change of the protein.
The first tyrosine kinase inhibitor was found in 1988 in a search to identify small molecules that specifically inhibit the activity of EGFR but not other receptor tyrosine kinase inhibitors.38 This early work paved the way for the development of imatinib, an inhibitor of ABL, the oncogenic target of BCR-ABL translocations in CML.39 The therapeutic efficacy of imatinib in CML and, later, in GIST has been one of the major driving factors in the development of new tyrosine kinase inhibitors throughout the pharmaceutical industry. A steady flow of new inhibitors is moving into early clinical trials, with many more in the development pipeline.
Monoclonal Antibody Therapies
The search to produce therapeutic antibodies has been ongoing for more than 100 years. Early therapeutic efforts focused on nonhuman monoclonal antibodies, but they are highly immunogenic and therefore associated with a very short half-life. The first successful therapeutic antibody was the chimeric mouse/human anti-CD20 drug rituximab (Rituxan).40 Chimeric antibodies are approximately 65% human, resulting in decreased immunogenicity and a much longer serum half-life. Cetuximab (anti-EGFR) is also a chimeric antibody.
Second-generation therapeutic monoclonals, referred to as humanized monoclonals, are 95% human, resulting in an even greater reduction in immunogenicity. Trastuzumab (anti-HER2) and bevacizumab (anti-VEGF) are examples of humanized antibodies.
Later methods allowed the development of fully human monoclonal antibodies, including panitumumab (anti-EGFR) and ipilimumab (anti-CTLA4).41 The fully human monoclonals have an extended half-life and improved pharmacokinetics, resulting in increased dosage intervals for these expensive therapies. Monoclonal antibody therapies are particularly useful for targeting cell surface receptors, and they have been among the most successful targeted therapies of the past 10 years.
Tumor Heterogeneity and Acquired Resistance to Targeted Therapy
Despite good initial tumor responses, it has been a general theme for most targeted therapies that acquired resistance develops, often after relatively short intervals of response. The resistant tumor clones typically are characterized by two types of genetic alteration not found in the original tumor analysis. First, mutations in the target gene block the drug binding site or confer new activity that is no longer responsive to the targeted therapeutic agent.42 Second, alterations bypass the targeted signaling blockade by activation of downstream or parallel pathways.43
Although the term acquired resistance implies that the tumor has evolved during the course of treatment, mathematical models suggest that the resistant clones may exist at the outset of therapy as very small subpopulations within the original tumor.44 The course of therapy eliminates the bulk of sensitive cells, but the resistant subclones are unaffected and continue to grow in the same location, resulting in the emergence of recurrent tumors. The significant degree of heterogeneity in most human tumors has been the subject of several investigations.45
These concepts about tumor heterogeneity are shifting the way we think about the diagnosis and management of cancer. Rather than administering targeted therapeutic agents consecutively, it may be advantageous to use two or even three targeted agents simultaneously.44 It may become important for the molecular diagnostics laboratory to characterize the dominant genetic aberrations and the subclones that may harbor mutations important for the development of resistant tumors.45 The need for more comprehensive genomic profiling may drive the development of assays better able to survey the entire tumor, such as those based on circulating tumors cells or DNA.
HER2 Testing in Gastric and Gastroesophageal Cancer
The human epidermal growth factor receptor 2 gene (HER2, also known as HER2/NEU or ERBB2) is a member of the EGFR receptor tyrosine kinase family that activates a downstream signaling cascade, including RAS, RAF, MAPK, PI3K, and mTOR. The HER2 gene is amplified and the protein overexpressed in several human cancers, most notably breast cancer, and promotes tumorigenesis through important biologic effects on cell growth, differentiation, survival, and migration.46 HER2 activation is a driver mutation in breast cancer, which led to the development of a highly efficacious targeted monoclonal antibody therapy.47 In breast cancers with HER2 amplification, trastuzumab (Herceptin) binds to the extracellular domain of HER2, thereby blocking HER2 activation and silencing downstream signaling.
HER2 overexpression and amplification occurs in approximately 20% of gastric cancers and has been investigated as a possible adverse prognostic marker.48,49 In 2010, trastuzumab was shown to offer a significant benefit to metastatic gastric and gastroesophageal cancer patients whose tumors had HER2 overexpression or amplification.50 Assessment of HER2 status is a routine component of management of advanced gastric and gastroesophageal cancers, with many experts recommending initial testing of all newly diagnosed patients as an important component of therapeutic planning.51 HER2 status may be analyzed by immunohistochemistry and FISH, and many laboratories offer a two-step screening similar to that used in the characterization of breast cancers (Fig. 23.1).
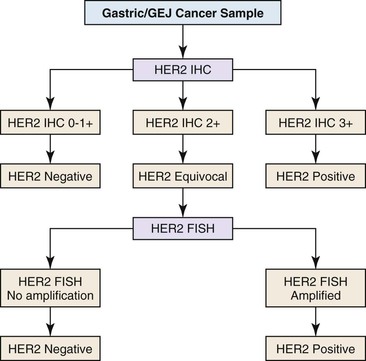
Immunohistochemical Analysis of HER2 Expression
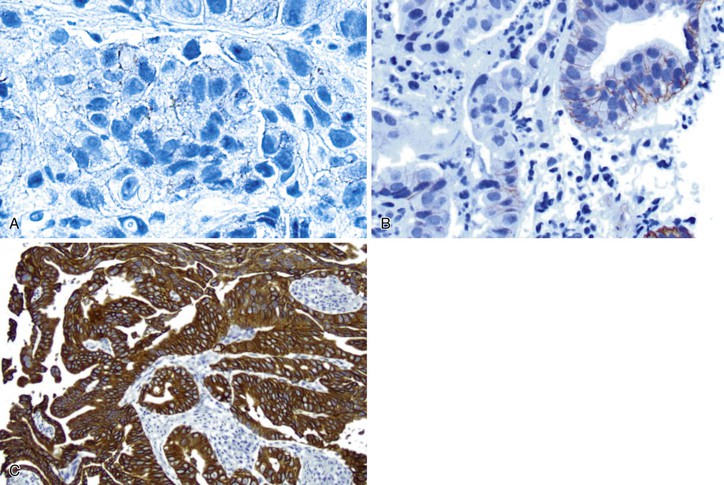
Immunohistochemistry is the first test offered by most laboratories in the characterization of cancers for HER2, and the College of American Pathologists (CAP) guidelines state that algorithms should be accurate, reproducible, and performed in a CAP-accredited laboratory, thereby requiring rigorous validation and ongoing verification.52 Guidelines for scoring HER2 expression in gastric cancer are summarized in Table 23.4.50,53 Briefly, the degree of lateral or basolateral membranous positivity is scored on a scale from 0 to 3+ (Fig. 23.2).
Table 23.4
Interpretation of HER2 Immunohistochemistry in Gastric Cancer
| IHC Score | Staining Pattern | Biopsies | Resections | HER2 Overexpression Result |
| 0 | No reactivity | All tumor cells | All tumor cells | Negative |
| 0 | Membranous reactivity | No tumor cell clusters (≥5 cells)* | <10% of tumor cells | Negative |
| 1+ | Faint or barely perceptible incomplete membranous reactivity (visualized with 40× objective) | Tumor cell clusters (≥5 cells)* | ≥10% of tumor cells | Negative |
| 2+ | Weak to moderate complete, basolateral, or lateral membranous reactivity | Tumor cell clusters (≥5 cells)* | ≥10% of tumor cells | Equivocal |
| 3+ | Strong complete, basolateral, or lateral reactivity | Tumor cell clusters (≥5 cells)* | ≥10% of tumor cells | Positive |
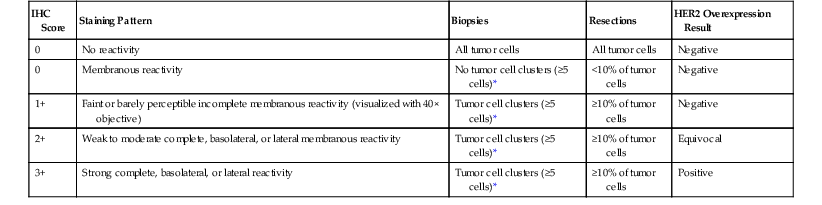
* Irrespective of the percentage of positively staining tumor cells.
HER2, Human epidermal growth factor receptor 2; IHC, immunohistochemistry.
Modified from Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2 positive gastric or gastroesophageal junction cancer (ToGA): a phase 3, open-label, randomized controlled trial. Lancet. 2010;376:687-697.
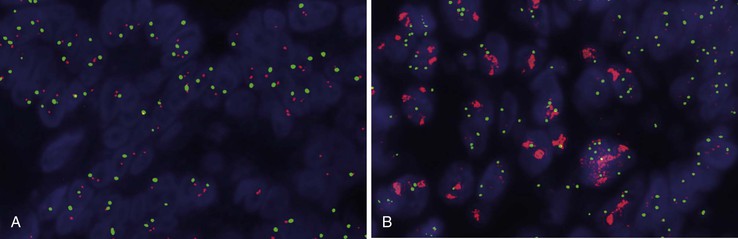
Fluorescence In Situ Hybridization Analysis of HER2 Copy Number
FISH is the gold standard for assessing the copy number of the HER2 gene. Dual-color, Food and Drug Administration (FDA)-approved commercial probe kits consist of a chromosome 17 centromeric reference probe and a 17q12 HER2 probe. Results are enumerated by counting 20 tumor cell nuclei and the ratio of the HER2 probe signals to the centromeric probe signals, with results higher than 2.2 considered positive for amplification (Fig. 23.3). FISH results may be scored manually or by using automated image analysis systems.
HER2 Testing in Other Cancers
In addition to breast and gastric cancer, HER2 is overexpressed in a subset of other cancers, including approximately 5% of colorectal cancers and some pancreatic, ovarian, endometrial, cervical, and lung cancers.21,54 Clinical trials are ongoing for many of these cancers, and we may be entering an era in which widespread HER2 testing becomes a standard aspect of management of many advanced cancers.
Sensitivity to Anti-EGFR Therapy in Colorectal Cancer
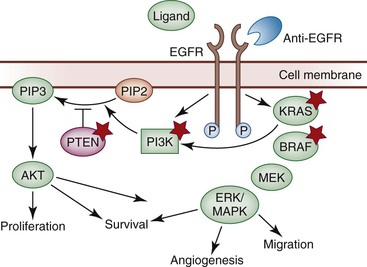
The epidermal growth factor receptor gene (EGFR; also known as HER1 or ERBB1) encodes a transmembrane glycoprotein with intrinsic protein tyrosine kinase activity. EGFR is expressed constitutively throughout the body, including many epithelial tissues, and it is a key activator of the RAS-RAF-MAPK signaling pathway (Fig. 23.4).55 EGFR is upregulated in a wide variety of cancers, is overexpressed in 60% to 80% of colorectal cancers, and has been associated with an increased risk of metastasis and poor survival.56,57 Whereas activating EGFR mutations and EGFR amplification are common in some cancers, epigenetic upregulation appears to be the predominant mechanism of activation in colorectal cancer.58 EGFR amplification occurs in only approximately 15% of colorectal cancers,59 and point mutations in EGFR have rarely been described.60
Effective inhibition of EGFR is one of the major breakthroughs of personalized medicine in the past 10 years. Small-molecule inhibitors (i.e., erlotinib and gefitinib) are highly effective in some cancers, including lung cancer, but they have not been effective in colorectal cancer, perhaps because of the absence of activating point mutations. However, anti-EGFR monoclonal antibodies (i.e., cetuximab and panitumumab) have been very effective in colorectal cancer and have come into routine use in patients with advanced disease.61,62
Monoclonal anti-EGFR antibodies bind to the extracellular domain and impedes ligand binding, which inhibits downstream EGFR signaling; prevents tumor cell growth, angiogenesis, invasion, and metastasis; and induces apoptosis.58 Although several potential predictors of resistance to anti-EGFR therapy have been suggested, only KRAS mutations are strongly supported by all lines of evidence for use in patient stratification.63
KRAS Mutation Testing
Mutations in KRAS result in oncogenic constitutive activation of the RAS-RAF-MEK-MAPK signaling pathway (see Fig. 23.4). KRAS is one of the most frequently mutated oncogenes in human cancer, and mutations occur in 40% to 50% of colorectal cancers.22 Because KRAS sits downstream from EGFR, activation of KRAS has the potential to bypass the inhibitory effect of anti-EGFR therapy. Many studies have demonstrated that cetuximab is ineffective in patients whose colorectal cancers harbor KRAS mutations (with the possible exception of KRAS G13D).64–66 KRAS mutation testing is a requirement of standardized chemotherapeutic protocols before cetuximab therapy is considered.61,62
Technical approaches to KRAS mutation testing have been guided by the relatively narrow mutation spectrum in human neoplasms, with most recurrent mutations in colorectal cancer confined to codons 12 and 13 and less frequently to codons 61 and 146.22 In addition to PCR of the genomic region encompassing these codons followed by Sanger sequencing (Fig. 23.5), the limited profile of possible mutations allows for a variety of other mutation detection methods, especially mutation-specific PCR with panels of primers that correspond to all known recurrent mutations. Pathologists should be familiar with these methods primarily because some laboratories offer more comprehensive panels of mutation detection than others. Mutation analysis is readily performed on paraffin-embedded tissue, requiring only a small amount of tumor sample, and it is therefore applicable to biopsy samples. KRAS mutations are relatively homogeneous within colorectal cancers, most likely because mutations typically occur during the development of an adenoma and are therefore present in all subsequent generations of evolving malignant and metastatic clones.
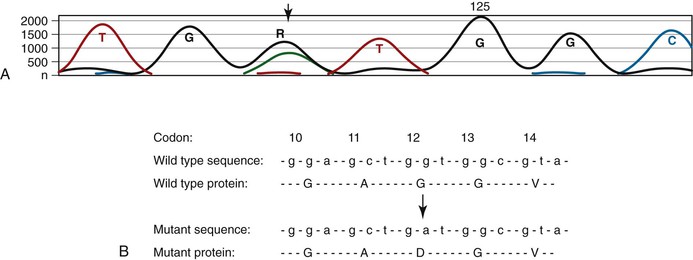
The KRAS G13D mutation is one possible exception to the lack of response to anti-EGFR therapy in colorectal cancers harboring KRAS mutations. Several studies have shown that cetuximab therapy may have some efficacy in patients whose colorectal cancers harbor G13D mutations and that these mutations should not necessarily be considered a contraindication to anti-EGFR therapy.67,68 Ongoing studies are assessing the magnitude of therapeutic benefit in this setting.
Similarly, many studies have looked at KRAS mutations only in codons 12 and 13, and there are few data regarding resistance in patients with mutations in codons 61 and 146. Although the latter two codons are not considered in some chemotherapeutic guidelines, there is evidence that they confer resistance, and it seems reasonable to assume that they will be included in future guidelines after additional validation data are available.69
Other Predictors of Resistance to Anti-EGFR Therapy
NRAS Mutation
NRAS mutations occur in approximately 3% of colorectal cancers and are mutually exclusive of KRAS mutations.22 There is some evidence to suggest that NRAS mutations are associated with resistance to cetuximab therapy, but the data are limited. NRAS mutation testing is not recommended in routine settings, and the mutation is not considered a contraindication to anti-EGFR therapy.69 With additional validation, however, it seems reasonable to expect that the finding of an NRAS mutation will be a contraindication to anti-EGFR therapy. NRAS mutations mostly occur at codon 61, rather than codons 12 and 13, and they are readily detected by PCR and sequencing or other mutation-specific PCR approaches.
BRAF Mutation
BRAF is key member of the RAS-RAF-MEK-MAPK signaling pathway (see Fig. 23.4), and it is mutated in approximately 5% to 10% of colorectal cancers.22 Because BRAF lies downstream of KRAS in the EGFR signaling pathway, mutations that constitutively activate BRAF have the potential to bypass the potential therapeutic effect of anti-EGFR therapy. Although evidence suggests that BRAF mutations are associated with decreased response to cetuximab therapy (unlike KRAS mutations), BRAF mutations are not as clearly associated with complete resistance to anti-EGFR therapy.70,71 BRAF mutations are not considered a contraindication to anti-EGFR therapy, and BRAF mutation analysis is not recommended as a routine component of standard chemotherapeutic protocols.72 Ongoing studies are assessing the magnitude of therapeutic benefit in the setting of BRAF mutations.
BRAF mutation testing also has clinical utility in the characterization of MLH1-deficient colorectal cancers (e.g., Lynch syndrome). Because almost all BRAF mutations in colorectal cancer are limited to a single recurrent mutation (i.e., V600E mutation), the mutational status is readily screened by PCR assays in addition to sequencing.
PIK3CA Mutation
Phosphatidylinositol-4,5-bisphosphate 3-kinase-α (PIK3CA), catalytic subunit of PI3K, is involved in AKT (also known as protein kinase B [PKB]) signaling downstream of EGFR (see Fig. 23.4) and may also confer resistance to anti-EGFR therapy because both are altered in a subset of colorectal cancers. PIK3CA encodes a lipid kinase that harbors activating mutations in approximately 15% to 18% of colorectal cancers.22 Some studies have found that PIK3CA mutations confer resistance to cetuximab therapy, but this result has not been confirmed in other studies.73,74 Overall, the evidence suggests that resistance may be confined to PIK3CA exon 20 mutations.75 Outside of the setting of controlled clinical trials, PIK3CA mutation status is not considered a requirement for anti-EGFR therapy. In ongoing trials, PIK3CA mutation testing is performed by PCR and sequencing.
Unrelated to anti-EGFR therapy, the PIK3CA mutation has been associated with improved survival among regular aspirin users.76 This finding suggests that PIK3CA mutation status could be used to stratify patients for eligibility for nonsteroidal antiinflammatory drug (NSAID) therapy. This provides another reason for knowing PIK3CA mutation status, but validation studies are needed before application in clinical use.
PTEN Inactivation
The phosphatase and tensin homolog (PTEN) inhibits the conversion of phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3), resulting in inhibition of EGFR signaling through AKT. PTEN alterations are relatively common in a variety of human cancers, with inactivating mutations, deletion, or other mechanisms of downregulation resulting in a loss of PTEN activity and subsequent activation of AKT signaling. Expression of PTEN is decreased in 19% to 36% of colorectal cancers, with mutation, allelic loss, and hypermethylation occurring in smaller subsets of tumors. Because PTEN lies downstream of EGFR, inactivation of PTEN has the potential to bypass the therapeutic effect of anti-EGFR therapy. Some evidence suggests that PTEN loss can confer resistance to cetuximab therapy, but the findings are not definitive, and additional studies are required before application to clinical practice.72,77
HER2 Amplification
HER2 signaling can bypass the therapeutic effect of anti-EGFR therapy by activating parallel signaling pathways. The possible role of HER2 amplification in anti-EGFR resistance was first described in the setting of acquired resistance.78 Studies have found that HER2 amplification may predict initial resistance to anti-EGFR therapy.79 Additional validation studies are required before this marker can be considered for stratification of patients in routine clinical practice. HER2 amplification status is determined by immunohistochemistry and FISH (described earlier).
Acquired Resistance to Anti-EGFR Therapy
As with many other targeted therapies used in cancer patients, despite a good initial response to anti-EGFR therapy, acquired resistance eventually develops.66,80 Although the discovery of at least two mechanisms of acquired resistance represents an exciting advance in the development of tailored colorectal cancer therapy, the current level of understanding does not warrant routine testing of tissue samples from colorectal cancer patients with emerging resistance to anti-EGFR therapy.
HER2 Amplification
Amplification of HER2, which has been observed in some colorectal cancer patients with acquired resistance to cetuximab therapy, allows positive downstream tumor cell growth signaling that bypasses the EGFR receptor complex.78 Although this raises the possibility of therapeutic benefit from the addition of an anti-HER2 agent for patients who have developed resistance to anti-EGFR therapy due to HER2 amplification, early studies with cetuximab and trastuzumab suggest that the combined toxicity of these agents outweighs the benefit.81 Ongoing studies are exploring these therapeutic options.
EGFR Mutation
Acquired EGFR mutations have been observed in some colorectal cancer patients with acquired resistance to cetuximab therapy. These mutations result in a conformational change to the extracellular domain of EGFR that prevents cetuximab binding.82 This conformational change does not appear to effect panitumumab binding, raising the possibility that these tumors may remain sensitive to panitumumab. Although preliminary evidence indicates that switching to panitumumab may have clinical benefit in this setting, additional clinical studies are required to further characterize this application.83,84
Gastrointestinal Stromal Tumor
Abundant interest was generated in 1998, when activating KIT mutations were first described in GISTs.85 This finding raised the possibility that targeting by a tyrosine kinase inhibitor could prove to be an effective management strategy and eventually led to the discovery that imatinib could produce dramatic therapeutic responses in these tumors. GISTs have subsequently become one of the premier models for personalized therapy in cancer management.
KIT and PDGFRA Mutations
Derived from interstitial cells of Cajal, GISTs share almost universal expression of the tyrosine kinase receptor KIT. Activating KIT or platelet-derived growth factor receptor-α (PDGFRA) mutations occur in 80% to 85% of tumors and 5% to 10% of tumors respectively. They result in activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositide 3-kinase (PI3K) pathways. Overexpression of insulin-like growth factor receptor (IGFR) or direct mutation of BRAF occurs in some of the tumors with wild-type KIT and PDGFRA. Testing for KIT and PDGFRA mutations is typically performed by PCR and sequencing.
Imatinib Therapy
Imatinib mesylate (Gleevec, Novartis, East Hanover, N.J.) is an oral, small-molecule receptor tyrosine kinase inhibitor that works by blocking the adenosine triphosphate binding site of the constitutively activated mutant KIT or PDGFRA.86 Binding effectively shuts down signaling. Since it was approved by the U.S. Food and Drug Administration (FDA) for use in GIST in 2002, imatinib has become first-line therapy for metastatic or unresectable GISTs. Approximately 80% of patients respond to initial imatinib therapy; however, the development of resistance is common, and progressive disease develops in more than one half within 2 years.
The type of mutation in KIT or PDGFRA is the best predictor of response to initial imatinib therapy (Table 23.5).86 Approximately 60% to 80% of KIT mutations occur in the exon 11 intracellular juxtamembrane domain, and these mutations are associated with the best response to imatinib. Approximately 5% to 20% of mutations occur in the exon 9 extracellular domain, and they are associated with resistance to imatinib. Rare remaining mutations occur in the exon 13 ATP-binding pocket or exon 17 activation loop, and they also confer resistance to imatinib. In PDGFRA, more than 90% of mutations occur in exon 18, which is structurally analogous to KIT exon 17 and also resistant to imatinib.
Table 23.5
KIT and PDGFR Mutations and Response to Targeted Therapy in Gastrointestinal Stromal Tumors
| Protein Domain | Exon | Primary Mutation (85-95% Overall) | Secondary Mutation (40-80% Overall) | First-Line Therapy | Second-Line Therapy (Imatinib Resistant) | ||||
| KIT | PDGFR | KIT | PDGFR | KIT | PDGFR | Imatinib Sensitivity* | Sunitinib Sensitivity* | Regorafenib Sensitivity* | |
| Extracellular domain | 9 | 10 | 10-20% | 0% | 0% | + | ++ | ++ | |
| Juxtamembrane domain | 11 | 12 | 60-80% | 0.7% | 0% | +++ | |||
| Tyrosine kinase domain 1 | 13 | 14 | 1-4% | 0.1% | 40-60% | + | ++ | ++ | |
| Tyrosine kinase domain 2 | 17 | 18 | 1% | 6% | 30-40% | 10-15% | − | ± | ++ |
| No mutation | No mutation | + | ++ | ||||||
| Overall response 80-90% | Overall response 50% | Response study ongoing | |||||||
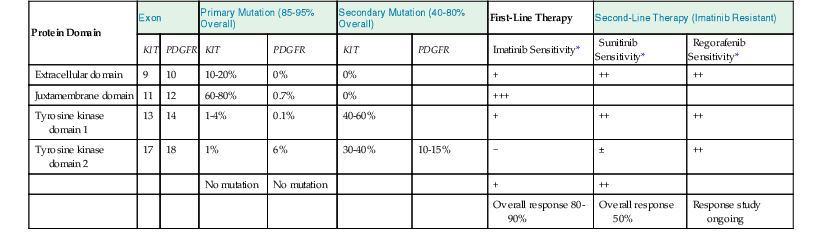
* Relative sensitivity is indicated as follows: +++, very sensitive; ++, moderately sensitive; +, minimally sensitive; ±, marginally sensitive; −, not sensitive.
Overall, approximately 10% to 15% of patients have primary resistance to imatinib therapy, defined as a lack of response or stable disease for less than 6 months. Most patients have imatinib-resistant mutations in exon 9 of KIT or exon 18 of PDGFRA or have no mutation.86 Secondary resistance develops in patients whose tumors are initially sensitive to imatinib, typically at a median of 18 to 24 months. Secondary mutations in exons 13, 14, or 17 are found in approximately 40% to 80% of those developing secondary resistance.86 Tumor heterogeneity is also seen, with different mutations identified in different lesions or within a single metastasis. Other mechanisms of secondary resistance include amplification of KIT and pharmacokinetic resistance.
Surgical resection or ablation is recommended in patients who are initially responsive but in whom limited progression occurs. By removing the imatinib-resistant clone, therapy may be continued in otherwise responsive disease. Dose escalation should be considered and may benefit as many as 30% of resistant patients.
Sunitinib Therapy
Sunitinib malate (Sutent, Pfizer Oncology, New York, N.Y.) is an oral, multitargeted receptor tyrosine kinase inhibitor that is active against KIT, PDGFRA, and other receptor tyrosine kinases. It was approved by the FDA in 2006 for use in patients with advanced GIST that is resistant to imatinib therapy and for those who did not tolerate imatinib.
Similar to imatinib, sunitinib binds the ATP-binding pocket and prevents tyrosine kinase activation. Sunitinib has a smaller molecular profile than imatinib, allowing greater activity against exon 13 and 14 KIT mutations and a broader range of exon 9 KIT mutations and wild-type KIT. Exon 17 KIT mutations and exon 18 PDGFRA mutations remain resistant. Overall, approximately 50% of patients with imatinib-resistant disease can benefit from sunitinib therapy, although the median progression-free survival is only 6 months.86 Mutational analysis of imatinib-resistant disease has been recommended by some authorities to better select patients for sunitinib therapy. Currently, sunitinib is the only agent that is FDA approved for second-line therapy in GIST.
Other Targeted Therapies
Nilotinib has shown some activity against GIST, but because it does not demonstrate any clear benefit over imatinib, further development of this compound for use in GIST is unlikely. Sorafenib and regorafenib show promising results and have greater activity than imatinib against exon 9 KIT mutations and secondary KIT mutations in exons 14 and 17.86 Regorafenib is effective in advanced GIST that has failed other standard therapies.87 Many other receptor tyrosine kinase agents are being evaluated in a variety of clinical trials.86 Targeting of downstream signaling has also been considered, including mTOR inhibitors, PI3K inhibitors, and anti-IGF1R therapies.
Resistance Monitoring during Therapy
Resistance to imatinib therapy eventually develops in most patients with GIST, even in those whose tumors are initially sensitive and have good response. Monitoring for the emergence of resistant KIT mutations plays an important role in therapeutic monitoring and decision making regarding second-line therapy. Unfortunately, the location and multiplicity of resistant disease makes biopsy sampling impractical. Emerging technologies that can detect KIT mutations in cell-free fluid samples such as serum and plasma offer significant gains in clinical utility in this setting. One such technology, BEAMing (see Rare Mutation Detection), was able to detect secondary resistant KIT mutations in the plasma of 48% of patients who had developed resistance to imatinib or sunitinib, compared with detection of secondary mutations in only 12% of tissue samples.14 This so-called liquid biopsy offers the potential for tailored monitoring of emerging resistance in any targeted therapy setting.
Molecular Diagnostics and Predictive Factors for Chemotherapy
The marginal benefit of adjuvant 5-fluorouracil chemotherapy in stage II colorectal cancer has highlighted the need for assays that better predict patients most likely to benefit from this therapy. Numerous studies have investigated the predictive power of a variety of molecular genetic aberrations (see Chapter 27), but only three tests are in clinical use.
DNA Mismatch Repair Status
DNA mismatch repair is discussed earlier in this chapter (e.g., Lynch syndrome) and in Chapter 27. Two separate biologic associations with mismatch repair deficient colorectal cancers justify the use of this marker in the routine management of colorectal cancer. First, mismatch repair–deficient cancers are associated with an improved overall survival rate. Survival is so good for stage II disease that demonstration of any additional benefit from adjuvant chemotherapy is unlikely. Second, several studies have found that mismatch repair–deficient cancers do not benefit from adjuvant 5-fluorouracil therapy and may be harmed by this therapy.
Current National Comprehensive Cancer Network guidelines state that adjuvant chemotherapy should not be offered to stage II colorectal cancer patients whose tumors are mismatch repair deficient.61,62 Mismatch repair status may be assayed by immunohistochemistry or MSI testing (discussed earlier). The use of adjuvant chemotherapy in stage III colorectal cancer patients with mismatch repair–deficient cancers remains controversial.
Proprietary Recurrence Risk Tests
Two proprietary prognostic colorectal cancer assays are available for use in the United States: Genomic Health’s Oncotype Dx Colon Cancer Assay and Agendia’s ColoPrint. Both tests are designed to identify high-risk colorectal cancer patients most likely to benefit from adjuvant chemotherapy. The Genomic Health test is available for use in stage II colorectal cancer, and the Agendia test is aimed at stage II and III colorectal cancers.
Both tests assay a number of gene markers, and both tests have demonstrated predictive power for colorectal cancer patients.88,89 The Agendia test requires fresh-frozen tissue, which limits the applicability for many patients. Both tests predict the risk of recurrence, not the potential benefit from therapy. It remains unclear whether these commercial multigene assays offer significant improvement over mismatch repair status in the evaluation of stage II and III colorectal cancer. Additional studies are ongoing.
Molecular Diagnostics of Hematologic Malignancies
A complete discussion of molecular diagnostic applications in the characterization and management of hematologic malignancies is beyond the scope of this chapter (see Chapter 31), but this section highlights some of the basic molecular genetic tests that may be used in the diagnosis and characterization of lymphoid neoplasms involving the GI tract.
Detection of Clonality
In normal lymphocytes, gene rearrangements are random and generate a population of cells with polyclonal immunoglobulin or T-cell receptor gene rearrangements. In lymphoproliferative disorders, a single rearrangement is markedly overrepresented (referred to as monotypic, monoclonal, or clonal) and is a hallmark of neoplasia.
Light-Chain Restriction
The two types of light chains in humans are κ and λ. A single B cell expresses only one class of light chain, and it remains fixed for the life of the lymphocyte. Immunohistochemistry or in situ hybridization is used to determine the mixture of κ and λ light chains expressed in a non-neoplastic population of B cells. In contrast, a monoclonal population of B cells expresses one predominant light chain, a phenomenon known as light-chain restriction. Examination of light-chain expression is useful for determining whether a given B-cell population is reactive or neoplastic.
Southern Blot
Southern blot analysis is the traditional method for identifying clonal heavy-chain gene rearrangements, and it was one of the first routinely used tests in molecular diagnostics. DNA from normal tissue and lesional tissue is digested with restriction enzymes, electrophoresed on a gel, transferred to a membrane, and hybridized with a probe to the joining region of the IGH gene. Normal tissue and polyclonal lymphoid tissue reveal the normal banding pattern generated from the known restriction sites, whereas a clonal lymphoid population reveals an abnormal, rearranged monoclonal band. The major limitation of Southern blot analysis is that it requires 20 to 30 µg of high-quality DNA, which usually is not available unless specifically collected for this analysis.
Polymerase Chain Reaction
PCR is the method of choice for detecting gene rearrangements. Different approaches vary in their ability to detect many rearrangements or all possible rearrangements. The general approach is to combine one forward PCR primer with a variety of reverse primers corresponding to different regions that may be rearranged. In a polyclonal lymphoid population, there are innumerable rearrangements, yielding a cloud or smear when the PCR products are separated by electrophoresis. The presence of a dominant monoclonal population results in a distinct PCR band.
Detection of Diagnostic Translocations and Other Cytogenetic Abnormalities
Identification of characteristic recurrent translocations is an important part of the diagnosis of many lymphomas (Table 23.6). They are typically detected by FISH or PCR and in part determined by the local expertise and the capabilities of the performing laboratory.
Table 23.6
Diagnostic Translocations in Lymphoproliferative Disorders
| Disorder | Translocation | Genetic Alteration | Detection Method |
| Marginal zone lymphoma | t(11;18) | AP12-MALT1 | Dual-color, dual-fusion FISH; PCR |
| t(14;18) | IGH-MALT1 | Dual-color, break-apart FISH | |
| t(3;14) | FOXP1-IGH | Dual-color, dual-fusion FISH; PCR | |
| Follicular lymphoma | t(14;18) | IGH-BCL2 | Dual-color, dual-fusion FISH; PCR |
| Mantle cell lymphoma | t(11;14) | IGH-CCND1 | Dual-color, dual-fusion FISH; PCR |
| Burkitt lymphoma | t(8;14) | MYC-IGH | Dual-color, dual-fusion FISH |
| t(2;8) | IGK-MYC | Dual-color, break-apart FISH | |
| t(8;22) | MYC-IGL | Dual-color, break-apart FISH | |
| Anaplastic large T-cell lymphoma | 2p33 and multiple partners | ALK upregulation | Dual-color, break-apart FISH |
FISH, Fluorescence in situ hybridization; PCR, polymerase chain reaction.
FISH approaches use dual-fusion probes or break-apart probes, whereas PCR approaches use sets of primers designed to amplify all possible known breakpoint combinations for the given translocation. In addition to the characteristic translocations, other cytogenetic abnormalities (e.g., deletion of 13q) have diagnostic and prognostic utility, and most are detected with FISH approaches.
Detection of Mutations
Mutation screening of JAK2 and MPL has become a standard component of the diagnosis and management of myeloproliferative disorders. In contrast, characteristic activating mutations in lymphomas have not been identified as components of standard diagnosis and management protocols, and there is no analogous routine mutation screening in the characterization of these neoplasms.
Infectious Disease
Molecular microbiology has seen explosive growth in the past 10 years, and molecular methods are pervasive in the identification and subtyping of infectious organisms (see Chapter 4). Applications in anatomic pathology and GI pathology are much more limited.
The most commonly performed test is CISH for viral infections (e.g., cytomegalovirus, Epstein-Barr virus, human papillomavirus). Occasionally, infection is suspected in cases with granulomatous inflammation, but no organisms are identified with special stains. In these cases, it is possible to test for mycobacterial organisms by PCR. Infectious disease testing by PCR is performed by using primers specific to microbial DNA or RNA sequences. Although these assays are straightforward conceptually, because of the challenge of a possibly very low organism burden in a tissue sample, potential contamination, and very low clinical demand, this type of testing is often performed at only a limited number of reference laboratories.