[level-membership-for-neurosurgery-category]
CHAPTER 8 Molecular Biology and Genetics of Meningiomas
INTRODUCTION
Meningiomas are slow-growing tumors and are generally diagnosed in middle aged or older individuals. They account for up to 37% of all symptomatic primary brain tumors. Small and slowly growing meningiomas may be identified only incidentally during neuroimaging for other reasons1 or at autopsy.2 The prevalence of meningioma is difficult to assess and is likely to be higher than might be concluded from the numbers diagnosed due to symptoms. In a Swedish autopsy study encompassing almost 12,000 cases, the incidence of incidental meningiomas was 1.44%.2 There are reports that the incidence has increased in the last 40 years, but this period coincides with the more widespread use of new imaging technologies, making definite conclusions difficult. Meningiomas are more common in females, with a male:female ratio of approximately 1:2 for all meningiomas at all sites, while in the spine the ratio is 1:10. Multiple tumors are frequently associated with hereditary tumor syndromes such as neurofibromatosis type 2 (NF2), as are the rare childhood meningiomas, which are more common in males. The more aggressive and malignant forms are also more common in males. These differences indicate that genetic or hormonal factors, or both, affect the frequency of the disease.
The tumors are generally fixed to the dura and can be located anywhere in the cranial cavity or spinal canal. Many histologic types of meningioma are described, with at least nine being recognized by the current World Health Organization (WHO) classification of tumors of the central nervous system (CNS) as grade I tumors (meningothelial, fibrous, transitional, psammomatous, angiomatous, microcystic, secretory, lymphoplasmacyte-rich, and metaplastic as well as intermediate and more difficult to classify types). The grade II meningiomas include chordoid, clear cell, and atypical meningiomas and the more aggressive grade III meningeal tumors include papillary, rhabdoid, and anaplastic meningiomas. Both the atypical and the anaplastic meningiomas can belong to any of the aforementioned histologic types but must also fulfill additional criteria such as increased cellularity and mitotic index, the presence of necrosis, and a malignant cytology. Meningiomas rarely metastasize and generally grow by expansion and local invasion. The pathology of meningiomas is described in detail in Chapter 5.
FAMILIAL TUMOR SYNDROMES CLINICALLY ASSOCIATED WITH MENINGIOMA
It has long been known that meningiomas are, together with bilateral acoustic schwannomas, a key feature of NF2 syndrome. However, there are additional families that show an increased susceptibility to meningiomas, but without any other evidence of the NF2 syndrome.3 More recent molecular genetic studies of some of these families has shown no linkage of the disease state to the NF2 locus,4 indicating there are one or more independent syndromes predisposing to specifically meningioma development. The type of meningioma that develops in these families is frequently meningothelial, which, as described later, is particularly interesting as sporadic meningothelial meningiomas have a low incidence of NF2 mutations compared to the other more common meningioma types.3,5 Other inherited tumor syndromes in which an increase in the incidence of meningiomas has been reported include Werner, Cowden, Gorlin, and even multiple endocrine neoplasia (MEN) type 1.3,6,7 Meningiomas have also been reported in patients suffering from both other tumor syndromes and nontumor syndromes such as Li–Fraumeni, Turner, and Hunter–MacDonald.8,9
CYTOGENETICS
Meningiomas were one of the earliest human solid tumors to be studied with regard to their cytogenetics. Studies conducted in the 1960s before the development of chromosomal banding techniques10 reported one acrocentric chromosome to be frequently lost.11,12 Use of banding techniques showed the lost chromosome to be chromosome 22.13,14 This finding was subsequently confirmed by multiple studies. As cytogenetic analysis requires short-term culture with attendant selection, it remained unknown whether the findings were representative of the majority of the meningioma tumor cells in a patient. In 1987, molecular genetic methods studying DNA from homogenized tumor tissue using polymorphic DNA markers provided evidence that loss of chromosome 22 occurred frequently in the vast majority, if not all of the cells in patients’ tumors.15–18
MOLECULAR GENETICS
Identification of the NF2 Gene as a Tumor Suppressor Gene in Meningiomas
With the establishment of molecular methods, it was rapidly determined that approximately 50% of WHO grade I meningiomas have monosomy chromosome 22, with a further 10% showing interstitial allelic losses that involve band 22q12.15,17–19 The incidence of loss of alleles on chromosome 22 was observed to vary between meningioma types.20 A recent study of 126 sporadic tumors using array-CGH reported losses to be most common in fibroblastic meningiomas (>80%) followed by transitional meningiomas (>60%), but relatively infrequent in meningothelial meningiomas (<40%).21 The figures for the other and less common variants are less well established.
In parallel with such studies, the hunt for the NF2 gene showed linkage to the same 22q12 region. In 1993 the NF2 gene was identified22,23 and with the clinically well established association of meningiomas with the NF2 syndrome, mutations of the retained NF2 allele were rapidly shown in the majority of meningiomas with loss of one copy of chromosome 22. Thus NF2 was identified as a tumor suppressor gene in meningiomas.20,24,25 In meningiomas, the loss of this tumor suppressor protein most commonly occurs by loss of one NF2 gene allele and mutation of the retained allele. Loss of the protein products of a tumor suppressor gene may occur due to inhibition of expression by promotor-methylation of a wild-type gene copy. However, in meningiomas there is little evidence to support the methylation of the NF2 promoter as an alternative mechanism.21
As there is a close relationship between the loss of alleles on chromosome 22 (encompassing one NF2 gene) and the frequency of mutation of the retained NF2 gene, the incidence of loss of both wild-type NF2 genes varies in the different subtypes of meningioma in a similar manner. Fibroblastic meningiomas have the highest incidence of NF2 mutations (>50%) while mutations in the meningothelial variant are as low as 18%.21 Loss of both wild type NF2 genes has also been shown to occur in the uncommon childhood meningiomas.26 In line with these genetic data, immunocytochemistry for a protein product of the NF2 gene—most commonly known as merlin—has been found frequently in meningothelial tumors but is commonly absent in many of the other variants.27–30
Meningiomas are not the only tumors with loss of both copies of wild-type NF2. As expected this occurs in the majority of schwannomas31,32 as well in approximately 60% of spinal ependymomas and malignant mesotheliomas.33,34 Other tumors in which mutations have been found include malignant melanomas, and thyroid carcinomas. The status of the NF2 gene in other tumor types remains poorly documented.
Thus, at least 30% of all meningiomas and the vast majority of meningothelial meningiomas do not lose alleles on 22q, nor do they have mutations or aberrant methylation of the NF2 gene. The genes involved in the oncogenesis of these meningiomas remain uncertain. As NF2 codes for a member of the 4.1 superfamily of genes other members have been investigated to some degree. Loss of heterozygosity studies have indicated losses of one copy of the DAL1 gene (also known as EPB41L3; 4.1B) at 18p11.32 and its expression in meningiomas.35,36 It is not clear how frequently this occurs, with widely differing values reported in different studies, while immunocytochemically assessed loss of expression of the protein product, has been documented in up to 76% of all meningiomas.30,35,37 However, no or very few definite mutations of retained alleles of DAL1 have been found;38 nor has any evidence for promoter methylation been reported. The EPB41 gene (1p36; coding the 4.1R protein) has also been studied and no convincing evidence for its involvement has been presented.39 No copy number abnormalities have been noted in the region of the other family members ezrin and radixin and screening for mutations in moesin has proved negative.37
Interstitial deletions of 22q not involving the NF2 locus have been interpreted to indicate another meningioma tumor suppressor gene located on this chromosomal arm. Several candidate genes have been investigated including the SMARCB1/INI1 gene (no wild-type in atypical teratoid / rhaboid tumors) and the MN1 gene, and single cases with abnormalities have been identified.40,41 A homozygous deletion at 22q12 in a sporadic meningioma resulted in the identification of a β-adaptin gene (AP1B1 or BAM22) in the deleted region. Loss of expression was found in 9/70 meningiomas, 7 of which had no detectable abnormalities of the NF2 gene. However, no further indications that this might constitute another tumor suppressor gene for meningioma on 22q have been presented.42,43
The NF2 Gene and Its Transcripts and Protein Products
The NF2 tumor suppressor gene at 22q12.2 is composed of 17 exons and covers a genomic region of 95 kb (Fig. 8-1). It encodes transcripts with at least eight well-documented alternatively spliced open reading frames. There are two dominant splice variants. The first and longest open reading frame found in transcripts is 1785 bases with the splicing out of exon 16 and encodes a protein of 595 residues, merlin (also referred to as isoform 1). The second has a slightly shorter open reading frame and splices in exon 16, altering the 3′ sequence, and encodes a protein of 590 residues (see Fig. 8-1). The other commonly expressed splice variants lack exon 2, exon 3, or both, with the remaining isoforms expressed at low levels.44 Most investigations have focused on isoform 1 and while there have been some studies of isoform 2, almost nothing is known of the function of the proteins the other splice variants code for. Transcripts of approximately 6.1 kb and 2.7 kb are expressed ubiquitously but in a ratio that is tissue specific.44 Some tissues also expressed a transcript of approximately 3.9 kb. There is also some evidence that initiation of transcription can occur at several start sites, affecting the length of the 5′ untranslated sequences while the 3′ untranslated sequences in the large transcripts consist of thousands of bases.44 The full-length splice variants encode a deduced protein with approximately 47% identity to several members of the 4.1/ERM family of proteins believed to be involved in linking cytoskeletal components to cell membrane proteins including moesin, ezrin, and radixin: thus the name merlin.23
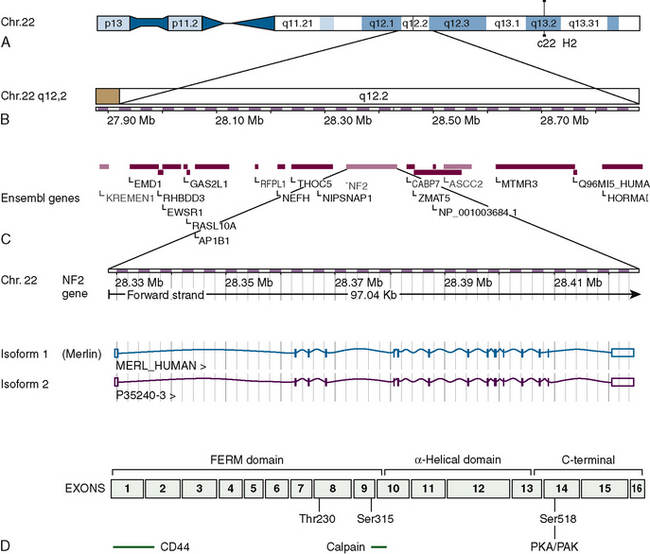
FIGURE 8-1 Overview of the location, the genetic makeup, and the two dominant splice variants of the NF2 gene as presently understood. (Data from Ensembl release 49, March 2008. http://www.ensembl.org/index.html). A and B, The location of the NF2 gene and its adjacent genes on chromosome 22. C, The two dominant splice variants commonly known as isoforms 1 and 2 showing the exons included in the their transcripts. The merlin protein (isoform 1) includes residues coded by exons 1–15 and 17. D, The merlin protein (isoform 1) outlining the domains, some of the regions believed to be involved in interactions with other proteins as well as the phosphorylation sites at threonine 230, serine 315, and serine 518 that change the conformation of the protein and are functionally important.
This family of proteins has a common structural organization. The N-terminal domain has a “globular” structure followed by an alfa-helical region and a charged C-terminal domain (Fig. 8-2). The N-terminal domains of the family members show approximately 65% homology whereas the C-terminal domains diverge, with homology decreasing to less than 30%. Merlin folds owing to the interaction of residues 8 to 121 with residues 200 to 320, forming an N-terminal globular structure containing the FERM region necessary for membrane localization (see Fig. 8-2). This region is disrupted by some NF2 missense mutations.45–47 The C-terminal residues 580 to 595 interact with residues 302 to 308 of this globular structure.45,48,49 This interaction with the N-terminal globular region is regulated by phosphorylation at serine 518. When phosphorylated merlin cannot fold, which results in the inactivation of its growth inhibitory abilities and relocalization from the cell membrane (see Fig. 8-2). This phosphorylation is believed to be carried out by p-21–activated kinases (PAKs) and cAMP-dependent protein kinase A (PKA).50–54 Dephosphorylation is believed to be carried out by the myosin phosphatase MYPT-1-PP1∂.55 Merlin has also recently been shown to be phosphorylated at two further sites (threonine 230 and serine 315) by ACT1/PKB, also resulting in its inability to take up the growth inhibitory formation of C-terminal binding to the globular N-terminal region and targeting merlin for degradation by ubiquitination.56
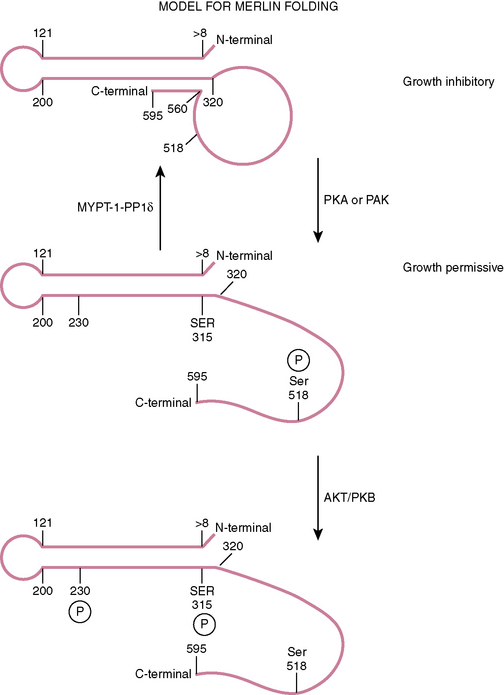
There is evidence that in its growth inhibitory conformation, merlin may inhibit the RAS-ERK pathway by blocking the ERM (ezrin, radixin, moesin)-dependent activation of RAS, which correlates with the formation of a complex comprising ERM proteins, Grb2, SOS, RAS, and filamentous actin.57 Merlin interacts with at least 34 other proteins, and how these interactions are affected or modified by the presence of similar proteins encoded by the NF2 splice variants is today difficult to envisage. For recent reviews of the complex interactions of the wild-type merlin protein and how it may control cell proliferation see refs. 58 and 59.
Much less is known about the second of the two dominant splice variants, which includes exon 16 (isoform 2) and codes for a slightly shorter protein (590 residues) with an altered C-terminal that does not appear to have the ability to interact with the N-terminal globular domain. This splice variant does not seem to have a tumor suppressor function. Its N-terminal domain appears to have a greater ability to bind to some of the merlin binding proteins such as SLC9A3R1/NHERF/EBP50.45,48,49,60 Almost nothing is known about the protein products of the other splice variants.
NF2 Mutations in Meningiomas and Their Consequences for Protein Function
Loss of one allele with mutation of the retained NF2 allele occurs in all grades of meningioma. A recent meta-analysis of NF2 mutations in published reports from both NF2 families and sporadic tumors reported 1141 NF2 mutations.61 There were 1070 small genetic changes, 42 intragenic changes involving one whole exon or larger, and 29 whole gene deletions and gross chromosomal rearrangements. Constitutional single-exon events were more likely to be nonsense or splice site changes than the somatic events. The somatic mutations favored frameshift changes affecting the coding sequence for merlin. The study included mutation data from 180 meningiomas. The somatic mutations in meningiomas were more frequent within the code for the 5′ FERM domain of the transcripts and there was a complete absence of mutations in exons 14 and 15. The somatic events differed between tumors of different pathology, that is, meningiomas and schwannomas, the latter having exon 14 and 15 mutations. The vast majority of mutations would result in the truncation of the protein encoded by isoforms 1 and 2 with fewer than 10% of all small nontruncating alterations clustered in exons 2 and 3, suggesting that this region may be especially crucial to tumor suppressor activity of merlin isoform 1. Some of the NF2 splice variants will not be affected by small mutations resulting in frameshifts in the coding sequence of exons 2 and 3 because they exclude these exons. Most mutations are either small insertions or deletions, or are nonsense mutations that affect splice sites, create stop codons, or result in frameshifts and occur mainly at the 5′ end of the gene, resulting in a truncated and presumably nonfunctional protein product.21,61 It is difficult to understand why patients with constitutive mutations of one NF2 gene mainly develop schwannomas and meningiomas when the gene is expressed ubiquitously.
Genes Involved in Progression and Atypical and Anaplastic Meningiomas
Losses on 1p are the second most common reported abnormality. The regions most commonly lost have been mapped to 1p36 (commonly lost in malignant astrocytic and many other tumors) and 1p33–34.62 Several candidate tumor suppressor genes in these regions have been screened, including P73, CDKN2C, RAD54L, and ALPL, with inconclusive results. Single cases showing mutation of the TP73 gene have been reported, and hypermethylation of the TP73 promoter has been reported in a significant number of cases retaining only one allele.63 However, most of the genes in these regions have yet to be systematically studied for mutation or hypermethylation.
The losses on 9p have been clearly associated with progression to an anaplastic meningioma. Both homozygous deletion and loss of one copy and mutation of the retained copies of CDKN2A, CDKN2B, and p14ARF at 9p21 occur. CDKN2A and p14ARF are genetically intimately entwined. Both have their own promoters and first exons, with p14ARF splicing in exons 2 and 3 of CDKN2A to form its transcript but using an alternative reading frame in these two exons to that used by CDKN2A. These three genes are also frequently involved in primary glioblastomas where homozygous deletion is a common finding.64 The proteins encoded by CDKN2A and CDKN2B both inhibit the RB1 pathway by binding to and blocking CDK4 and CDK6 forming heterodimers with cyclin D1 required for phosphorylation of RB1 at the restriction point of the cell cycle. Their loss deregulates this process, allowing inappropriate entry of cells into the S-phase of the cell cycle. If this occurs in the presence of a normal p53 pathway the cell is likely to undergo apoptosis. However, p14ARF is a negative regulator of the MDM2 protein that is involved in the degradation of p53. Loss of p14ARF deregulates the control of the MDM2 protein, permitting the inappropriate degradation of p53 protein and deregulating the p53 pathway.
Although losses on chromosome 10 are common, no targeted gene has definitely been identified. The PTEN gene at 10q23 has been studied and only very occasional mutations identified. A candidate target for the losses on 14q has been identified as the NDRG2 gene. Hypermethylation of the retained copy has been shown together with loss of expression.65 Although TP53 (17p13.1) mutations have been found to be very infrequent,66 increased copy number in the 17q21 region has been reported and the RPS6KB1 gene suggested as a potential target.67
Clonality of Solitary, Recurrent, and Multiple Meningiomas
Studies of X-chromosome inactivation and NF2 mutations have shown the same X chromosome to be inactivated or the same mutation of the retained NF2 gene in recurrent and multiple sporadic meningiomas, indicating they are monoclonal tumors, that recurrences are true recurrences of an earlier tumor, and that multiple lesions can arise through local spread.68–72 Some sporadic multiple meningiomas might be explained as situations wherein the patient is a mosaic for a constitutional NF2 mutation.73
[1] Vernooij M.W., Ikram M.A., Tanghe H.L., et al. Incidental findings on brain MRI in the general population. N Engl J Med. 2007;357:1821.
[2] Rausing A., Ybo W., Stenflo J. Intracranial meningioma: a population study of ten years. Acta Neurol Scand. 1970;46:102.
[3] Louis D.N., Ramesh V., Gusella J.F. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol. 1995;5:163.
[4] Pulst S.M., Rouleau G.A., Marineau C., et al. Familial meningioma is not allelic to neurofibromatosis 2. Neurology. 1993;43:2096.
[5] Heinrich B., Hartmann C., Stemmer-Rachamimov A.O., et al. Multiple meningiomas: Investigating the molecular basis of sporadic and familial forms. Int J Cancer. 2003;103:483.
[6] Asgharian B., Chen Y.J., Patronas N.J., et al. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1. Clin Cancer Res. 2004;10:869.
[7] Goto M., Miller R.W., Ishikawa Y., et al. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev. 1996;5:239.
[8] Schoemaker M.J., Swerdlow A.J., Higgins C.D., et al. Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. Lancet Oncol. 2008;9:239-246.
[9] Armstrong L., Graham G.E., Schimke R.N., et al. The Hunter-McDonald syndrome with expanded phenotype including risk of meningioma: an update and review. Am J Med Genet A. 2008;146:83.
[10] Caspersson T., Zech L., Johansson C., et al. Identification of human chromosomes by DNA-binding fluorescent agents. Chromosoma. 1970;30:215.
[11] Mark J. Chromosomal patterns in human meningiomas. Eur J Cancer. 1970;6:489.
[12] Zang K.D., Singer H. Chromosomal constitution of meningiomas. Nature. 1967;216:84.
[13] Zankl H., Zang K.D. Cytological and cytogenetical studies on brain tumors. 4. Identification of the missing G chromosome in human meningiomas as no. 22 by fluorescence technique. Humangenetik. 1972;14:167.
[14] Mark J., Levan G., Mitelman F. Identification by fluorescence of the G chromosome lost in human meningiomas. Hereditas. 1972;71:163.
[15] Dumanski J.P., Rouleau G.A., Nordenskjold M., et al. Molecular genetic analysis of chromosome 22 in 81 cases of meningioma. Cancer Res. 1990;50:5863.
[16] Okazaki M., Nishisho I., Tateishi H., et al. Loss of genes on the long arm of chromosome 22 in human meningiomas. Mol Biol Med. 1988;5:15.
[17] Seizinger B.R., de la Monte S., Atkins L., et al. Molecular genetic approach to human meningioma: loss of genes on chromosome 22. Proc Natl Acad Sci USA. 1987;84:5419.
[18] Dumanski J.P., Carlbom E., Collins V.P., et al. Deletion mapping of a locus on human chromosome 22 involved in the oncogenesis of meningioma. Proc Natl Acad Sci USA. 1987;84:9275.
[19] Seizinger B.R., Rouleau G., Ozelius L.J., et al. Common pathogenetic mechanism for three tumor types in bilateral acoustic neurofibromatosis. Science. 1987;236:317.
[20] Ruttledge M.H., Sarrazin J., Rangaratnam S., et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet. 1994;6:180.
[21] Hansson C.M., Buckley P.G., Grigelioniene G., et al. Comprehensive genetic and epigenetic analysis of sporadic meningioma for macro-mutations on 22q and micro-mutations within the NF2 locus. BMC Genomics. 2007;8:16.
[22] Rouleau G.A., Merel P., Lutchman M., et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515.
[23] Trofatter J.A., MacCollin M.M., Rutter J.L., et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791.
[24] Lekanne Deprez R.H., Bianchi A.B., Groen N.A., et al. Frequent NF2 gene transcript mutations in sporadic meningiomas and vestibular schwannomas. Am J Hum Genet. 1994;54:1022.
[25] Twist E.C., Ruttledge M.H., Rousseau M., et al. The neurofibromatosis type 2 gene is inactivated in schwannomas. Hum Mol Genet. 1994;3:147.
[26] Begnami M.D., Rushing E.J., Santi M., et al. Evaluation of NF2 gene deletion in pediatric meningiomas using chromogenic in situ hybridization. Int J Surg Pathol. 2007;15:110.
[27] Den Bakker M.A., van Tilborg A.A., Kros J.M., et al. Truncated NF2 proteins are not detected in meningiomas and schwannomas. Neuropathology. 2001;21:168.
[28] Hitotsumatsu T., Iwaki T., Kitamoto T., et al. Expression of neurofibromatosis 2 protein in human brain tumors: an immunohistochemical study. Acta Neuropathol (Berl). 1997;93:225.
[29] Lee J.H., Sundaram V., Stein D.J., et al. Reduced expression of schwannomin/merlin in human sporadic meningiomas. Neurosurgery. 1997;40:578.
[30] Perry A., Cai D.X., Scheithauer B.W., et al. Merlin, DAL-1, and progesterone receptor expression in clinicopathologic subsets of meningioma: a correlative immunohistochemical study of 175 cases. J Neuropathol Exp Neurol. 2000;59:872.
[31] Jacoby L.B., MacCollin M., Barone R., et al. Frequency and distribution of NF2 mutations in schwannomas. Genes Chromosomes Cancer. 1996;17:45.
[32] Ruttledge M.H., Rouleau G.A. Role of the neurofibromatosis type 2 gene in the development of tumors of the nervous system. Neurosurg Focus. 2005;19:E6.
[33] Robinson B.W., Musk A.W., Lake R.A. Malignant mesothelioma. Lancet. 2005;366:397.
[34] Ebert C., von Haken M., Meyer-Puttlitz B., et al. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol. 1999;155:627.
[35] Nunes F., Shen Y., Niida Y., et al. Inactivation patterns of NF2 and DAL-1/4.1B (EPB41L3) in sporadic meningioma. Cancer Genet Cytogenet. 2005;162:135.
[36] Gutmann D.H., Donahoe J., Perry A., et al. Loss of DAL-1, a protein 4.1-related tumor suppressor, is an important early event in the pathogenesis of meningiomas. Hum Mol Genet. 2000;9:1495.
[37] van Tilborg A.A., Morolli B., Giphart-Gassler M., et al. Lack of genetic and epigenetic changes in meningiomas without NF2 loss. J Pathol. 2006;208:564.
[38] Martinez-Glez V., Bello M.J., Franco-Hernandez C., et al. Mutational analysis of the DAL-1/4.1B tumour-suppressor gene locus in meningiomas. Int J Mol Med. 2005;16:771.
[39] Piaskowski S., Rieske P., Szybka M., et al. GADD45A and EPB41 as tumor suppressor genes in meningioma pathogenesis. Cancer Genet Cytogenet. 2005;162:63.
[40] Lekanne Deprez R.H., Riegman P.H., Groen N.A., et al. Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene. 1995;10:1521.
[41] Schmitz U., Mueller W., Weber M., et al. INI1 mutations in meningiomas at a potential hotspot in exon 9. Br J Cancer. 2001;84:199.
[42] Peyrard M., Fransson I., Xie Y.G., et al. Characterization of a new member of the human beta-adaptin gene family from chromosome 22q12, a candidate meningioma gene. Hum Mol Genet. 1994;3:1393.
[43] Ruttledge M.H., Xie Y.G., Han F.Y., et al. Physical mapping of the NF2/meningioma region on human chromosome 22q12. Genomics. 1994;19:52.
[44] Chang L.S., Akhmametyeva E.M., Wu Y., et al. Multiple transcription initiation sites, alternative splicing, and differential polyadenylation contribute to the complexity of human neurofibromatosis 2 transcripts. Genomics. 2002;79:63.
[45] Gutmann D.H., Haipek C.A., Hoang Lu K. Neurofibromatosis 2 tumor suppressor protein, merlin, forms two functionally important intramolecular associations. J Neurosci Res. 1999;58:706.
[46] Brault E., Gautreau A., Lamarine M., et al. Normal membrane localization and actin association of the NF2 tumor suppressor protein are dependent on folding of its N-terminal domain. J Cell Sci. 2001;114:1901.
[47] Gary R., Bretscher A. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol Biol Cell. 1995;6:1061.
[48] Bashour A.M., Meng J.J., Ip W., et al. The neurofibromatosis type 2 gene product, merlin, reverses the F-actin cytoskeletal defects in primary human Schwannoma cells. Mol Cell Biol. 2002;22:1150.
[49] Sherman L., Xu H.M., Geist R.T., et al. Interdomain binding mediates tumor growth suppression by the NF2 gene product. Oncogene. 1997;15:2505.
[50] Alfthan K., Heiska L., Gronholm M., et al. Cyclic AMP-dependent protein kinase phosphorylates merlin at serine 518 independently of p21-activated kinase and promotes merlin-ezrin heterodimerization. J Biol Chem. 2004;279:18559.
[51] Kissil J.L., Johnson K.C., Eckman M.S., et al. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem. 2002;277:10394.
[52] Morrison H., Sherman L.S., Legg J., et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001;15:968.
[53] Shaw R.J., Paez J.G., Curto M., et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev Cell. 2001;1:63.
[54] Xiao G.H., Beeser A., Chernoff J., et al. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002;277:883.
[55] Jin H., Sperka T., Herrlich P., et al. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature. 2006;442:576.
[56] Tang X., Jang S.W., Wang X., et al. Akt phosphorylation regulates the tumour-suppressor merlin through ubiquitination and degradation. Nat Cell Biol. 2007;9:1199.
[57] Morrison H., Sperka T., Manent J., et al. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007;67:520.
[58] Scoles D.R. The merlin interacting proteins reveal multiple targets for NF2 therapy. Biochim Biophys Acta. 2008;1785:32.
[59] Curto M., McClatchey A.I. Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. Br J Cancer. 2008;98:256.
[60] Gonzalez-Agosti C., Wiederhold T., Herndon M.E., et al. Interdomain interaction of merlin isoforms and its influence on intermolecular binding to NHE-RF. J Biol Chem. 1999;274:34438.
[61] Ahronowitz I., Xin W., Kiely R., et al. Mutational spectrum of the NF2 gene: a meta-analysis of 12 years of research and diagnostic laboratory findings. Hum Mutat. 2007;28:1.
[62] Bello M.J., de Campos J.M., Vaquero J., et al. High-resolution analysis of chromosome arm 1p alterations in meningioma. Cancer Genet Cytogenet. 2000;120:30.
[63] Lomas J., Aminoso C., Gonzalez-Gomez P., et al. Methylation status of TP73 in meningiomas. Cancer Genet Cytogenet. 2004;148:148.
[64] Collins V.P. Mechanisms of disease: genetic predictors of response to treatment in brain tumors. Nat Clin Pract Oncol. 2007;4:362.
[65] Lusis E.A., Watson M.A., Chicoine M.R., et al. Integrative genomic analysis identifies NDRG2 as a candidate tumor suppressor gene frequently inactivated in clinically aggressive meningioma. Cancer Res. 2005;65:7121.
[66] Verheijen F.M., Sprong M., Kloosterman J.M., et al. TP53 mutations in human meningiomas. Int J Biol Markers. 2002;17:42.
[67] Buschges R., Ichimura K., Weber R.G., et al. Allelic gain and amplification on the long arm of chromosome 17 in anaplastic meningiomas. Brain Pathol. 2002;12:145.
[68] Larson J.J., Tew J.M.Jr, Simon M., et al. Evidence for clonal spread in the development of multiple meningiomas. J Neurosurg. 1995;83:705.
[69] Stangl A.P., Wellenreuther R., Lenartz D., et al. Clonality of multiple meningiomas. J Neurosurg. 1997;86:853.
[70] von Deimling A., Kraus J.A., Stangl A.P., et al. Evidence for subarachnoid spread in the development of multiple meningiomas. Brain Pathol. 1995;5:11.
[71] von Deimling A., Larson J., Wellenreuther R., et al. Clonal origin of recurrent meningiomas. Brain Pathol. 1999;9:645.
[72] Zhu J.J., Maruyama T., Jacoby L.B., et al. Clonal analysis of a case of multiple meningiomas using multiple molecular genetic approaches: pathology case report. Neurosurgery. 1999;45:409.
[73] Evans D.G., Watson C., King A., et al. Multiple meningiomas: differential involvement of the NF2 gene in children and adults. J Med Genet. 2005;42:45.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 8 Molecular Biology and Genetics of Meningiomas
INTRODUCTION
Meningiomas are slow-growing tumors and are generally diagnosed in middle aged or older individuals. They account for up to 37% of all symptomatic primary brain tumors. Small and slowly growing meningiomas may be identified only incidentally during neuroimaging for other reasons1 or at autopsy.2 The prevalence of meningioma is difficult to assess and is likely to be higher than might be concluded from the numbers diagnosed due to symptoms. In a Swedish autopsy study encompassing almost 12,000 cases, the incidence of incidental meningiomas was 1.44%.2 There are reports that the incidence has increased in the last 40 years, but this period coincides with the more widespread use of new imaging technologies, making definite conclusions difficult. Meningiomas are more common in females, with a male:female ratio of approximately 1:2 for all meningiomas at all sites, while in the spine the ratio is 1:10. Multiple tumors are frequently associated with hereditary tumor syndromes such as neurofibromatosis type 2 (NF2), as are the rare childhood meningiomas, which are more common in males. The more aggressive and malignant forms are also more common in males. These differences indicate that genetic or hormonal factors, or both, affect the frequency of the disease.
The tumors are generally fixed to the dura and can be located anywhere in the cranial cavity or spinal canal. Many histologic types of meningioma are described, with at least nine being recognized by the current World Health Organization (WHO) classification of tumors of the central nervous system (CNS) as grade I tumors (meningothelial, fibrous, transitional, psammomatous, angiomatous, microcystic, secretory, lymphoplasmacyte-rich, and metaplastic as well as intermediate and more difficult to classify types). The grade II meningiomas include chordoid, clear cell, and atypical meningiomas and the more aggressive grade III meningeal tumors include papillary, rhabdoid, and anaplastic meningiomas. Both the atypical and the anaplastic meningiomas can belong to any of the aforementioned histologic types but must also fulfill additional criteria such as increased cellularity and mitotic index, the presence of necrosis, and a malignant cytology. Meningiomas rarely metastasize and generally grow by expansion and local invasion. The pathology of meningiomas is described in detail in Chapter 5.
FAMILIAL TUMOR SYNDROMES CLINICALLY ASSOCIATED WITH MENINGIOMA
It has long been known that meningiomas are, together with bilateral acoustic schwannomas, a key feature of NF2 syndrome. However, there are additional families that show an increased susceptibility to meningiomas, but without any other evidence of the NF2 syndrome.3 More recent molecular genetic studies of some of these families has shown no linkage of the disease state to the NF2 locus,4 indicating there are one or more independent syndromes predisposing to specifically meningioma development. The type of meningioma that develops in these families is frequently meningothelial, which, as described later, is particularly interesting as sporadic meningothelial meningiomas have a low incidence of NF2 mutations compared to the other more common meningioma types.3,5 Other inherited tumor syndromes in which an increase in the incidence of meningiomas has been reported include Werner, Cowden, Gorlin, and even multiple endocrine neoplasia (MEN) type 1.3,6,7 Meningiomas have also been reported in patients suffering from both other tumor syndromes and nontumor syndromes such as Li–Fraumeni, Turner, and Hunter–MacDonald.8,9
CYTOGENETICS
Meningiomas were one of the earliest human solid tumors to be studied with regard to their cytogenetics. Studies conducted in the 1960s before the development of chromosomal banding techniques10 reported one acrocentric chromosome to be frequently lost.11,12 Use of banding techniques showed the lost chromosome to be chromosome 22.13,14 This finding was subsequently confirmed by multiple studies. As cytogenetic analysis requires short-term culture with attendant selection, it remained unknown whether the findings were representative of the majority of the meningioma tumor cells in a patient. In 1987, molecular genetic methods studying DNA from homogenized tumor tissue using polymorphic DNA markers provided evidence that loss of chromosome 22 occurred frequently in the vast majority, if not all of the cells in patients’ tumors.15–18
MOLECULAR GENETICS
Identification of the NF2 Gene as a Tumor Suppressor Gene in Meningiomas
With the establishment of molecular methods, it was rapidly determined that approximately 50% of WHO grade I meningiomas have monosomy chromosome 22, with a further 10% showing interstitial allelic losses that involve band 22q12.15,17–19 The incidence of loss of alleles on chromosome 22 was observed to vary between meningioma types.20 A recent study of 126 sporadic tumors using array-CGH reported losses to be most common in fibroblastic meningiomas (>80%) followed by transitional meningiomas (>60%), but relatively infrequent in meningothelial meningiomas (<40%).21 The figures for the other and less common variants are less well established.
In parallel with such studies, the hunt for the NF2 gene showed linkage to the same 22q12 region. In 1993 the NF2 gene was identified22,23 and with the clinically well established association of meningiomas with the NF2 syndrome, mutations of the retained NF2 allele were rapidly shown in the majority of meningiomas with loss of one copy of chromosome 22. Thus NF2 was identified as a tumor suppressor gene in meningiomas.20,24,25 In meningiomas, the loss of this tumor suppressor protein most commonly occurs by loss of one NF2 gene allele and mutation of the retained allele. Loss of the protein products of a tumor suppressor gene may occur due to inhibition of expression by promotor-methylation of a wild-type gene copy. However, in meningiomas there is little evidence to support the methylation of the NF2 promoter as an alternative mechanism.21
As there is a close relationship between the loss of alleles on chromosome 22 (encompassing one NF2 gene) and the frequency of mutation of the retained NF2 gene, the incidence of loss of both wild-type NF2 genes varies in the different subtypes of meningioma in a similar manner. Fibroblastic meningiomas have the highest incidence of NF2 mutations (>50%) while mutations in the meningothelial variant are as low as 18%.21 Loss of both wild type NF2 genes has also been shown to occur in the uncommon childhood meningiomas.26 In line with these genetic data, immunocytochemistry for a protein product of the NF2 gene—most commonly known as merlin—has been found frequently in meningothelial tumors but is commonly absent in many of the other variants.27–30
Meningiomas are not the only tumors with loss of both copies of wild-type NF2. As expected this occurs in the majority of schwannomas31,32 as well in approximately 60% of spinal ependymomas and malignant mesotheliomas.33,34 Other tumors in which mutations have been found include malignant melanomas, and thyroid carcinomas. The status of the NF2 gene in other tumor types remains poorly documented.
Thus, at least 30% of all meningiomas and the vast majority of meningothelial meningiomas do not lose alleles on 22q, nor do they have mutations or aberrant methylation of the NF2 gene. The genes involved in the oncogenesis of these meningiomas remain uncertain. As NF2 codes for a member of the 4.1 superfamily of genes other members have been investigated to some degree. Loss of heterozygosity studies have indicated losses of one copy of the DAL1 gene (also known as EPB41L3; 4.1B) at 18p11.32 and its expression in meningiomas.35,36 It is not clear how frequently this occurs, with widely differing values reported in different studies, while immunocytochemically assessed loss of expression of the protein product, has been documented in up to 76% of all meningiomas.30,35,37 However, no or very few definite mutations of retained alleles of DAL1 have been found;38 nor has any evidence for promoter methylation been reported. The EPB41 gene (1p36; coding the 4.1R protein) has also been studied and no convincing evidence for its involvement has been presented.39 No copy number abnormalities have been noted in the region of the other family members ezrin and radixin and screening for mutations in moesin has proved negative.37
Interstitial deletions of 22q not involving the NF2 locus have been interpreted to indicate another meningioma tumor suppressor gene located on this chromosomal arm. Several candidate genes have been investigated including the SMARCB1/INI1 gene (no wild-type in atypical teratoid / rhaboid tumors) and the MN1 gene, and single cases with abnormalities have been identified.40,41 A homozygous deletion at 22q12 in a sporadic meningioma resulted in the identification of a β-adaptin gene (AP1B1 or BAM22) in the deleted region. Loss of expression was found in 9/70 meningiomas, 7 of which had no detectable abnormalities of the NF2 gene. However, no further indications that this might constitute another tumor suppressor gene for meningioma on 22q have been presented.42,43
The NF2 Gene and Its Transcripts and Protein Products
The NF2 tumor suppressor gene at 22q12.2 is composed of 17 exons and covers a genomic region of 95 kb (Fig. 8-1). It encodes transcripts with at least eight well-documented alternatively spliced open reading frames. There are two dominant splice variants. The first and longest open reading frame found in transcripts is 1785 bases with the splicing out of exon 16 and encodes a protein of 595 residues, merlin (also referred to as isoform 1). The second has a slightly shorter open reading frame and splices in exon 16, altering the 3′ sequence, and encodes a protein of 590 residues (see Fig. 8-1). The other commonly expressed splice variants lack exon 2, exon 3, or both, with the remaining isoforms expressed at low levels.44 Most investigations have focused on isoform 1 and while there have been some studies of isoform 2, almost nothing is known of the function of the proteins the other splice variants code for. Transcripts of approximately 6.1 kb and 2.7 kb are expressed ubiquitously but in a ratio that is tissue specific.44 Some tissues also expressed a transcript of approximately 3.9 kb. There is also some evidence that initiation of transcription can occur at several start sites, affecting the length of the 5′ untranslated sequences while the 3′ untranslated sequences in the large transcripts consist of thousands of bases.44 The full-length splice variants encode a deduced protein with approximately 47% identity to several members of the 4.1/ERM family of proteins believed to be involved in linking cytoskeletal components to cell membrane proteins including moesin, ezrin, and radixin: thus the name merlin.23
FIGURE 8-1 Overview of the location, the genetic makeup, and the two dominant splice variants of the NF2 gene as presently understood. (Data from Ensembl release 49, March 2008. http://www.ensembl.org/index.html). A and B, The location of the NF2
[/not-level-membership-for-neurosurgery-category]
