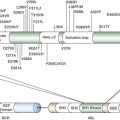
ATM
BRAF
BRD3
CBL
CTNNB1
EGFR
FBXW7
FGFR3
GOPC
KEAP1
KIAA0427
KRAS
NF1
PIK3CA
PPP2R1A
PTEN
RB1
RBM10
SETD2
SMAD4
SMARCA4
STK11
TP53
U2AF1
APC
BCL11A
BCL2L1
BRAF
CDK6
CDKN2A
CREBBP
CSMD1
DDR2
EGFR
EYS
FAM123B
FBXW7
FGFR1
FOXP1
HLA-A
HRAS
KEAP1
MLL2
MUC16
NF1
NFE2L2
NOTCH1
PIK3CA
PTEN
RB1
REL
SMAD4
SMARCA4
TNFAIP3
TP53
TSC1
VGLL4
WHSC1L1
WWOX
BCLAF1
C17orf108
CDYL
CNTNAP2
COL22A1
COL4A2
DIP2C
ELAVL2
GRIK3
GRM8
KHSRP
KIF21A
PLSCR4
RASSF8
RB1
RIMS2
RUNX1T1
SATB2
TMEM132D
TP53
ZDBF2
Genes in bold are present in more than one histological subtype.
Data generated through of analysis of 183 lung adenocarcinomas 24 ; TCGA project of 178 previously untreated, stage I-IV primary lung squamous cell carcinoma 9 ; and 34 primary SCLC tumors and 17 SCLC cell lines. 25
Identification of Novel Pathways
Identification of Therapeutic Targets
Lessons Learned and Future Directions
Genome-Wide Functional (siRNA, shRNA Library) Screening
Epigenetic Changes in Lung Carcinogenesis
Methylation and Histone Modification
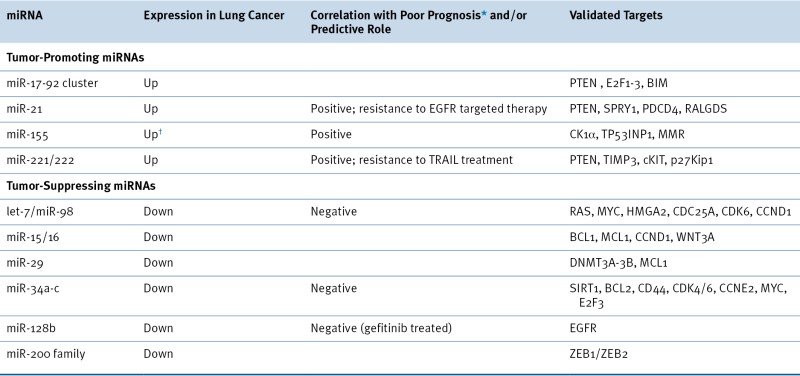
microRNA-Mediated Regulation
Table 32-3
DNA Methylation as a Biomarker in Lung Cancer
| Early Detection | Prognostic | Predictive |
| APC CDH13 DAPK1 DNMT1 FHIT GATA5 GSTP1 MAGEA1 MAGEB2 MGMT p16 PAX5-b RARβ2 RASSF1A RASSF5 RUNX3 TCF21 |
APC CDH1 CDH13 CXCL12 DAPK1 DLEC1 EPB41L3 (DAL-1) ESR1 FHIT IGFBP-3 MGMT MLH1 MSH2 p16 PYCARD (ASC) PTEN RASSF1A RRAD RUNX3 SPARC TIMP3 TMS1 TSLC1 WIF1 |
SFN (14-3-3 sigma) |
Data summarized from the following reviews: References 26–28.
Table 32-4
Targeted Therapies Approved in Clinical Trial or in Preclinical Study for Lung Cancer
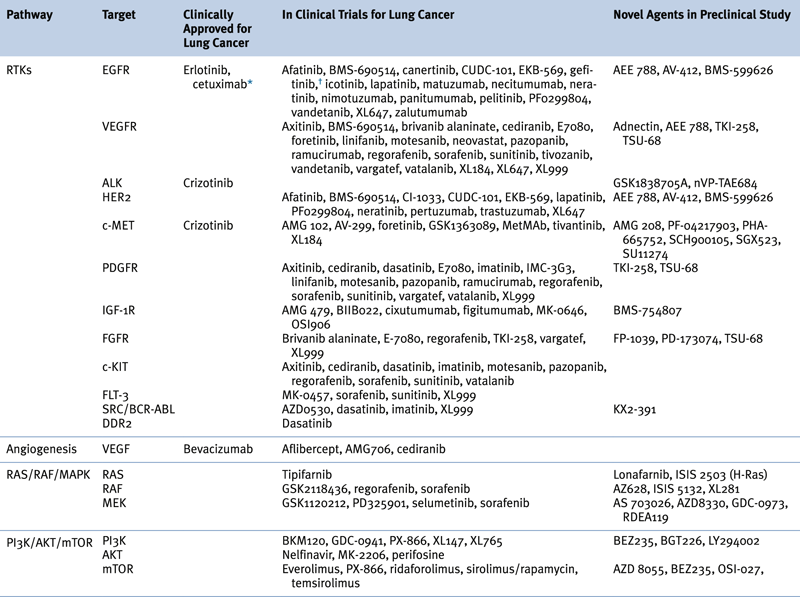
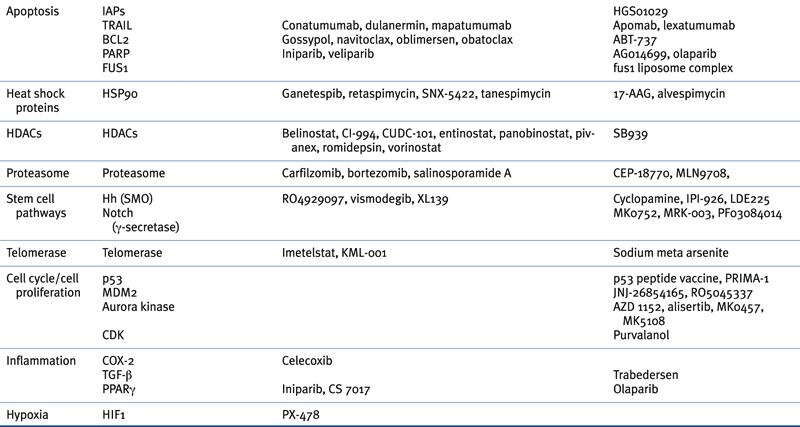
∗ Although not currently U.S. FDA-approved for non–small-cell lung cancer, cetuximab is a recommended treatment in several practice guidelines, including those of the American Society of Clinical Oncology (ASCO) and the National Comprehensive Cancer Network (NCCN).
† Previously approved in the United States and still approved elsewhere.
Adapted from Reference 29.
Oncogenes, Tumor Suppressor Genes, and Signaling Pathways in Lung Cancer
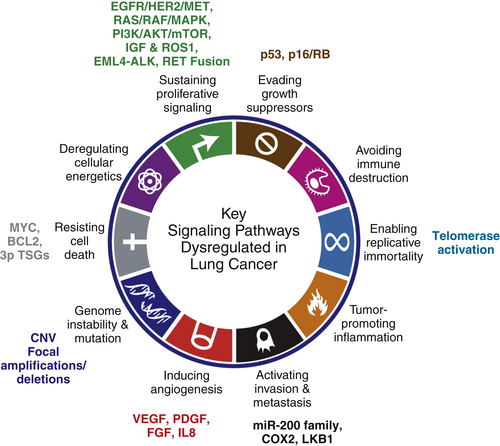
Hallmark: Sustaining Proliferative Signaling
EGFR/HER2/MET Signaling
EGFR
ERBB2 (HER2)
MET
RAS/RAF/MAPK Pathway
PI3K/AKT/mTOR Pathway
STK11 (LKB1)
Insulin Growth Factor (IGF) Pathway and ROS1
Other Fusion Proteins: EML4-ALK and RET
EML4-ALK
RET
Hallmark: Resisting Cell Death and Evading Growth Suppressors
MYC
The 3P Tumor Suppressor Genes: Regulators of Apoptosis
The p53 Pathway
The p16INK4a-RB Pathway
Hallmark: Enabling Replicative Immortality
Hallmark: Inducing Angiogenesis
Hallmark: Activation Invasion and Metastasis
Lung Cancer Stem Cells
Lineage-Dependent Oncogenes: SOX2 and NKX2-1 (TITF1)
Preclinical Model Systems for Lung Cancer
Translation of Molecular Data to the Clinic: Rationale-Based Targeted Therapy
Acknowledgments
1. Cancer statistics . CA Cancer J Clin . 2012 ; 2012 ( 62 ) : 10 – 29 .
2. The clonal evolution of tumor cell populations . Science . 1976 ; 194 : 23 – 28 .
3. Clinical implications and utility of field cancerization . Cancer Cell Int . 2007 ; 7 : 2 .
4. Hallmarks of cancer: the next generation . Cell . 2011 ; 144 : 646 – 674 .
5. Global cancer statistics . CA Cancer J Clin . 2002 ; 2005 ( 55 ) : 74 – 108 .
6. Chipping away at the lung cancer genome . Nat Med . 2012 ; 18 : 349 – 351 .
7. Genomic medicine in non-small cell lung cancer: paving the path to personalized care . Respirology . 2011 ; 16 : 257 – 263 .
8. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study . Nat Med . 2008 ; 14 : 822 – 827 .
9. Comprehensive genomic characterization of squamous cell lung cancers . Nature . 2012 .
10. Identification of driver mutations in tumor specimens from 1000 patients with lung adenocarcinoma: The NCI’s lung cancer mutation consortium (LCMC) . J Clin Oncol . 2011 : 29 .
11. The concept of synthetic lethality in the context of anticancer therapy . Nature reviews. Cancer . 2005 ; 5 : 689 – 698 .
12. Molecular biology of lung cancer: clinical implications . Clin Chest Med . 2011 ; 32 : 703 – 740 .
13. Cancer. Addiction to oncogenes–the Achilles heel of cancer . Science . 2002 ; 297 : 63 – 64 .
14. Transcriptional regulation and transformation by Myc proteins . Nat Rev Mol Cell Biol . 2005 ; 6 : 635 – 645 .
15. Epithelial-mesenchymal transitions in development and disease . Cell . 2009 ; 139 : 871 – 890 .
16. Clinicopathological significance of Sip1-associated epithelial mesenchymal transition in non-small cell lung cancer progression . Anticancer Res . 2009 ; 29 : 4099 – 4106 .
17. Cancer stem cells in lung cancer: Evidence and controversies . Respirology . 2013 ; 18 : 757 – 764 .
18. Lineage dependency and lineage-survival oncogenes in human cancer . Nat Rev Cancer . 2006 ; 6 : 593 – 602 .
19. REFERENCE DELETED IN PROOFS
20. SnapShot: Lung cancer models . Cell . 2012 ; 149 246-246 e1 .
21. Lung cancer in never smokers–a different disease . Nat Rev Cancer . 2007 ; 7 : 778 – 790 .
22. Lung cancer in never smokers: molecular profiles and therapeutic implications . Clin Cancer Res . 2009 ; 15 : 5646 – 5661 .
23. Lung cancer in never smokers: a review . J Clin Oncol . 2007 ; 25 : 561 – 570 .
24. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing . Cell . 2012 ; 150 : 1107 – 1120 .
25. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer . Nat Genet. 2012 ; 44 : 1111 – 1116 .
26. Aberrant methylation in non-small cell lung cancer . Surg Today . 2010 ; 40 : 602 – 607 .
27. Lung cancer: from single-gene methylation to methylome profiling . Cancer Metastasis Rev . 2010 ; 29 : 95 – 107 .
28. Genetic and epigenetic changes in lung carcinoma and their clinical implications . Mod Pathol . 2011 ; 24 : 932 – 943 .
29. Targeted therapies for lung cancer: clinical experience and novel agents . Cancer J . 2011 ; 17 : 512 – 527 .
30. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy . Nat Rev Cancer . 2011 ; 11 : 849 – 864 .
31. Mechanistic Roles of Noncoding RNAs in Lung Cancer Biology and Their Clinical Implications . Genet Res Int . 2012 ; 2012 : 737416 .
32. microRNAs and lung cancer: tumors and 22-mers . Cancer Metastasis Rev . 2010 ; 29 : 109 – 122 .
33. Clinical implications of microRNAs in lung cancer . Semin Oncol . 2011 ; 38 : 776 – 780 .
34. Involvement of microRNAs in lung cancer biology and therapy . Transl Res . 2011 ; 157 : 20020 – 20028 .