Chapter 486 Molecular and Cellular Biology of Cancer
Genes Involved in Oncogenesis
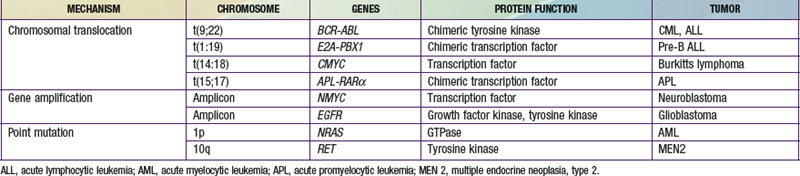
The three main mechanisms by which proto-oncogenes can be activated include amplification, point mutations, and translocation (Table 486-1). MYC, which codes for a protein that regulates transcription, is an example of a proto-oncogene that is activated by amplification. Patients with neuroblastoma in which the MYC gene is amplified 10-300-fold have a poorer outcome. Point mutations can also activate proto-oncogenes. The NRAS proto-oncogene codes for a guanine-nucleotide-binding protein with guanosine triphosphatase activity that is important in signal transduction and is mutated in 25-30% of acute nonmyelogenous leukemias, resulting in a constitutively active protein. The RET protein is a transmembrane tyrosine kinase receptor that is important in signal transduction. A point mutation in the RET gene results in the constitutive activation of a tyrosine kinase, as found in multiple neoplasia syndromes and familial thyroid carcinoma.
Syndromes Predisposing to Cancer
Several syndromes are associated with an increased risk of developing malignancies, which can be characterized by different mechanisms (Table 486-2). One mechanism involves the inactivation of tumor suppressor genes such as RB in familial retinoblastoma. Interestingly, patients with retinoblastoma in which one of the alleles is inactivated throughout all of the patient’s cells are also at a very high risk for developing osteosarcoma. A familial syndrome, Li-Fraumeni syndrome, in which one mutant P53 allele is inherited, has also been described in patients who develop sarcomas, leukemias, and cancers of the breast, bone, lung, and brain. Neurofibromatosis is a condition characterized by the proliferation cells of neural crest origin, leading to neurofibromas. These patients are at a higher risk of developing malignant schwannomas and pheochromocytomas. Neurofibromatosis is often inherited in an autosomal dominant fashion, although 50% of the cases present without a family history and occur secondary to the high rate of spontaneous mutations of the NF1 gene.
Table 486-2 FAMILIAL OR GENETIC SUSCEPTIBILITY TO MALIGNANCY
| DISORDER | TUMOR/CANCER | COMMENT |
|---|---|---|
| CHROMOSOMAL SYNDROMES | ||
| Chromosome 11p-(deletion) with sporadic aniridia | Wilms tumor | Associated with genitourinary anomalies, mental retardation, WT1 gene |
| Chromosomal 13q-(deletion) | Retinoblastoma, sarcoma | Associated with mental retardation, skeletal malformations; autosomal dominant (bilateral) or sporadic new mutations, RB1 gene |
| Trisomy 21 | Lymphocytic or nonlymphocytic leukemia, especially megakaryocytic leukemia; transient leukemoid reaction | Risk of ALL is increased 20%; risk of AML is 400%; patients have an increased sensitivity to chemotherapy |
| Klinefelter syndrome (47, XXY) | Breast cancer, extragonadal germ cell tumors | |
| Trisomy 8 | Preleukemia | |
| Noonan Syndrome | JMML | Autosomal dominant; mutations in PTPN11 gene |
| Monosomy 5 or 7 | Myelodysplastic syndrome | Recurrent infections may precede neoplasia |
| CHROMOSOMAL INSTABILITY | ||
| Xeroderma pigmentosum | Basal cell and squamous cell carcinomas; melanoma | Autosomal recessive; failure to repair UV damaged DNA. Mutations in XP gene on chromosome 3p25 |
| Fanconi anemia | Leukemia, myelodysplasic syndrome, liver neoplasias, rare head and neck tumors, GI and GU cancers | Autosomal recessive; chromosome fragility; positive diepoxybutane test result. Mutations in FANCX gene family. |
| Bloom syndrome | Leukemia, lymphoma, and solid tumors | Autosomal recessive; increase sister chromatid exchange; mutations in BLM gene; member of the RecQ helicase gene |
| Ataxia-teleangiectasia | Lymphoma, leukemia, less commonly central nervous system and non-neural solid tumors | Autosomal recessive; sensitive to x-irradiation, radiomimetic drugs; mutation in ATM tumor suppressor gene |
| Dysplastic nevus syndrome | Melanoma | Autosomal dominant; some cases associated with mutations in CDKN2A gene |
| Rothmund-Thompson syndrome | Osteosarcoma; skin cancers | Autosomal recessive; mutation in RecQ helicase gene family |
| Werner’s syndrome (premature aging) | Soft tissue sarcomas | Autosomal recessive; mutation in the WRN gene; member of the RecQ helicase gene family |
| IMMUNODEFICIENCY SYNDROMES | ||
| Wiskott-Aldrich syndrome | Lymphoma, leukemia | X-linked recessive; WAS gene mutation (Xp11.22-23); WASp protein functions in signal transduction associated with cytoskeletal actin filament rearrangement |
| X-linked immunodeficiency (Duncan syndrome) | Lymphoproliferative disorder | X- linked; Epstein-Barr viral infection can result in fatal outcome; mutation in SH2D1A gene locus |
| X-linked agammaglobulinemia (Bruton’s disease) | Lymphoma, leukemia | X-linked; mutation in BKT gene resulting in absence of mature B cells |
| Severe combined immunodeficiency | Leukemia, lymphoma | X-linked; mutations in ADA gene |
| OTHERS | ||
| Neurofibromatosis 1 | Neurofibroma, optic glioma, acoustic neuroma, astrocytoma, meningioma, pheochromoctoma, sarcoma | Autosomal dominant, mutation in tumor suppressor gene, NF1 |
| Neurofibromatosis 2 | Bilateral acoustic neuromas, meningiomas | Autosomal dominant; mutation in tumor suppressor gene, NF2 |
| Tuberous sclerosis | Fibroangiomatous nevi, myocardial rhabdomyoma | Autosomal dominant |
| Gorlin-Goltz syndrome (Nevus basal cell carcinoma syndrome) | Multiple basal cell carcinomas; medulloblastoma | Autosomal dominant; mutation in PTCH gene |
| Li-Fraumeni syndrome | Bone, soft tissue sarcoma, breast | Mutation of P53 tumor-suppressor gene, autosomal dominant |
| Retinoblastoma | Sarcoma | Autosomal recessive; Increased risk of secondary malignancy 10-20 yr later, mutation in RB tumor suppressor gene |
| Hemihypertrophy ± Beckwith syndrome | Wilms tumor, hepatoblastoma, adrenal carcinoma | WT1 gene. 25% develop tumor, most in first 5 yr of life |
| von Hippel-Landau disease | Hemangioblastoma of the cerebellum and retina, pheochromocytoma, renal cancer | Autosomal dominant, mutation of tumor-suppressor gene, VHL gene |
| Multiple endocrine neoplasia syndrome, type 1 (Wermer syndrome) | Parathyroid, pancreatic islet, and pituitary tumors | Autosomal dominant; mutation in PYGM tumor suppressor gene |
| Multiple endocrine neoplasia syndrome, type 2A (Sipple syndrome) | Medullary carcinoma of the thyroid, hyperparathyroidism, pheochromocytoma | Autosomal dominant; mutations in Cys rich regions of the RET gene activates this proto-oncogene; RET codes for a tyrosine kinase; monitor calcitonin and calcium levels |
| Multiple endocrine neoplasia type 2B (multiple mucosal neuroma syndrome) | Mucosal neuroma, pheochromocytoma, medullary thyroid carcinoma, Marfan habitus; neuropathy | Autosomal dominant; mutation in catalytic site (codon 883 or 914) activates proto-oncogene; RET codes for a tyrosine kinase |
| Familial adenomatous polyposis | Colorectal, thyroid carcinoma, duodenal and periampullar carcinomas; pediatric hepatoblastoma | Autosomal dominant; mutation in APC gene |
| Familial juvenile polyposis | Colorectal carcinoma | Autosomal dominant; mutation in SMAD4 gene |
| Hereditary nonpolyposis colon cancer (Lynch syndrome, NHPCC) | Colon cancer | Autosomal dominant; mutation in mismatch repair genes; hMSH2, hMLH1, PMS1, PMS2, hMSH6, hMSG3 |
| Turcot syndrome | Pediatric brain tumors and increased risk of colon carcinoma and polyps | Mutation in the APC gene |
| Familial adenomatous polyposis coli | Adenocarcinoma of colon | Autosomal dominant, APC gene |
| Gardner syndrome | Adenocarcinoma of colon, skull and soft tissue tumors | Autosomal dominant, APC gene |
| Peutz-Jeghers syndrome | Gastrointestinal carcinoma, ovarian neoplasia | Autosomal dominant, LKB1 gene codes for a Ser/Thr kinase that regulates cell cycle, metabolism, cell polarity |
| Hemochromatosis | Hepatocellular carcinoma | Autosomal dominant; malignancy associated with cirrhotic liver |
| Glycogen storage disease 1 (von Gierke Disease) | Hepatocellular carcinoma | Autosomal recessive; malignancy associated with cirrhotic liver Mutation in glucose-6-phosphatase or glucose 6-phosphatase translocase genes |
| Tyrosinemia, galactosemia | Hepatocellular carcinoma | Autosomal recessive; tumor associated with cirrhotic liver |
| BRCA1 and BRCA2 | Breast, ovarian | DNA repair defect. |
ALL, acute lymphocytic leukemia; AML, acute myelocytic leukemia; GI, gastrointestinal; GU, genitourinary; JMML, juvenile myelomonocytic leukemia; NHPCC, nonhereditary polyposis colon cancer.
Bjornsson HT, Sigurdsson MI, Fallin MD, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877-2883.
Blackburn E. Telomeres and telomerase; their mechanism of action and the effects of altering their functions. FEBS Lett. 2005;579:859-862.
Butel JS. Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis. 2000;21:405-426.
Dong LM, Potter JD, White E, et al. Genetic susceptibility to cancer. JAMA. 2008;299:2423-2436.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148-1158.
Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008;359:2143-2153.
Reinhart B, Chaillet JR. Genomic imprinting: cis-acting sequences and regional control. Int Rev Cytol. 2005;243:173-213.