[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 2 Molecular and Cellular Biology
The past decade has seen a revolution in two areas of cellular and molecular biology closely related to radiation therapy. The pathways involved in response to genomic stress, including mechanisms for sensing DNA damage and responding to cell cycle changes and DNA repair, have been elucidated by studies of model organisms, particularly the yeast Saccharomyces cerevisiae.1,2 In the second major area, understanding pathways of programmed cell death (PCD), which may play a major role in treatment response, studies of Caenorhabditis elegans were the most informative in demonstrating the significance of individual death genes.3 The challenge is to combine these two areas of knowledge to provide a rational understanding of how tumor cells can be made to enter the death pathways after processing DNA damage using the genomic stress response. Considerable technologic improvements also have enabled significant progress in translational work, including rational, in silico drug design and high-throughput screening of chemical libraries, which may permit the application of advances in molecular biologic knowledge to the development of drugs for use in combination with radiation therapy.
Essential Steps in Tumor Progression
Much evidence suggests that tumors are formed by stepwise progression of cells from a minimally altered state, where they are able to grow and form nodules or polyps (e.g., solid tumors) to a state of maximal transformation, characterized by multiply deviated cells that are capable of unlimited growth, manipulation of their local environment, invasion of surrounding tissues, and escape into the circulation to establish new colonies of secondary tumors, or metastases.4 Such phenotypic and genotypic progression involves an array of molecular and morphologic changes. In their landmark review, Hanahan and Weinberg5 suggested organizing these traits into six essential alterations in cell physiology: self-sufficiency in growth signals, insensitivity to growth inhibition, evasion of PCD, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis. Although the initial steps are believed to occur earlier in tumor progression, the exact order of occurrence varies among different types of malignant tumors, and some phenotypic transformations appear to require more than one molecular change.6,7 The occurrence in tumor cells of this series of radical, primarily genetic alterations in physiology is in part the result of an evolving instability of the tumor genome due to a breakdown in the DNA repair pathways that accompanies tumor progression.
In addition to progressing and acquiring enhanced malignant capabilities, most human tumors undergo further selections through exposure to various forms of cytotoxic therapy such as irradiation and chemotherapy, leading to the development of resistant phenotypes in the cells that survive the toxic insult.8 However, a new paradigm is emerging that may challenge our views of tumorigenisis. This is the concept of the cancer stem cell.9 According to this theory, tumors contain a small population of stem cells that divide slowly and give rise to more “differentiated” progeny that form the bulk of tumors. Tumor eradication would therefore involve killing this small, slowly dividing, and often treatment-resistant population. Studies in this area could revolutionize the cellular and molecular underpinnings of radiation biology.
All cancer treatments are aimed at reversing the hallmarks of neoplastic transformation and tipping the balance toward tumor cell death in both the bulk population and the stem cell fraction. Ionizing radiation kills cells almost exclusively through the generation of DNA double-strand breaks (DSBs).10 The evolving modification of the cellular phenotype during tumorigenesis leads to the development of resistance to therapy. Our major task is to explore the links among DSB response pathways, engagement of cell death mechanisms, and the outcome of radiation therapy.
Radiation Therapy and Radiation Biology
Ionizing radiation has long been known to be a potent modality in cancer therapy, killing cells and leading to tumor regression.11 Killing by ionizing radiation involves the oxygen-dependent generation of free radicals and subsequent damage to multiple molecular structures within the cell.10 The generation of DSBs is the dominant mechanism of direct cellular lethality. Indeed, it has been estimated that the existence of even one unrepaired DSB can lead to cell death.12
Within whole organisms, direct and indirect forms of lethality occur. Radiation can cause the death of hematopoietic stem cells in the bone marrow and stem cells in crypts of the small intestine, which leads to secondary damage caused by immunosuppression and infection, as the gastrointestinal lining is eroded.13,14 The challenge facing radiation therapy is to enhance cell killing within tumors while avoiding dose-related complications in organs containing rapidly renewing stem cell populations. Early studies using the clonogenic cell survival assay established the notion of optimizing radiobiologic parameters such as dose, fractionation, and cell cycle inhibition as a means of inhibiting reproductive cell death in critical normal tissues.15 The clonogenic cell survival assay is a useful measure of cell inactivation because the effects of ionizing radiation seen in vitro often mirror the responses of tumors in vivo.10 However, a weakness in the informative power of the method is that it does not allow for discrimination or elucidation of the various mechanisms leading to cell death. That kind of detailed information is essential for the successful combination of radiation therapy with other modalities, which may result in sensitization or protection, or both.
Irradiation activates a number of pathways that mediate reproductive death, including various forms of PCD (e.g., apoptosis, autophagy), replicative senescence, and necrosis, the default pathway that often dominates when other types of cell death are inhibited.16,17 Susceptibility to each of these forms of cell death changes over the course of tumor progression, and the tumor cell response to ionizing radiation is likely to reflect the aggregate of genetic changes that accompany tumor progression. The task of modern radiation biology is to examine mechanisms by which ionizing radiation gives rise to unrepaired DSBs and how such damage is coupled to the pathways of cell killing within the moving target of the evolving cancer cell.
DNA Double-Strand Break Response
All cells have evolved to live in an environment that is more or less mutagenic, and have evolved responses to permit survival when the genome is damaged.18 DNA may be damaged during the normal processes of DNA replication and segregation. Within tumor cells, these initial changes become progressively altered as cells compete for survival in the tumor milieu and are subjected to selection by various forms of cytotoxic therapy.19 The response to DSBs involves two principal components: arrest of cell proliferation to prevent replication and segregation of the damaged DNA and, when possible, repair of the lesion.19 A third component in the DSB response in multicellular organisms may be altruistic entry of irreversibly damaged cells into the PCD pathways.20,21 The latter effect is the desired response to cytotoxic therapy.
Mechanisms of Cell Cycle Arrest: The Cell Cycle and Checkpoints
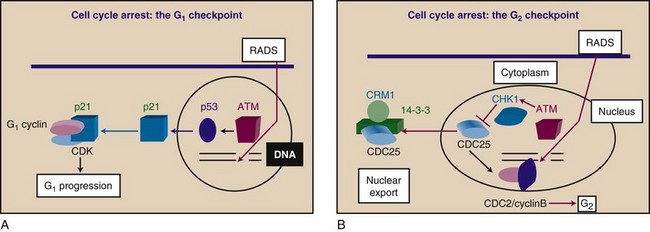
The cell cycle is a series of molecular programs that read out in an invariant order leading to duplication of the genome and other cellular components and permits division into two cells, each with a full set of chromosomes and the organelles required for life.22 Orderly, high-fidelity, complete duplication of the genome is required for cell cycle progression. This is achieved through a surveillance system that detects DNA damage or unreplicated DNA and imposes cell cycle arrest at designated checkpoints, blocking the forward progress of the cell cycle engine (Fig. 2-1). Because ionizing radiation produces severe damage to DNA, checkpoints are essential for facilitating repair and subsequent cell survival after radiation exposure, and provide an ultimate target for strategies aimed at sensitizing cells to radiation therapy.
Much accumulated evidence indicates that the cell cycle is driven by sequential activation of a series of protein kinases that determine the order and rate of the metabolic events required in each stage. These cell division kinases, or cyclin-dependent kinases (CDKs) are activated by binding proteins originally designated as cell division cycle (CDC) molecules, now known as cyclins.22 The complexes between cyclins and CDK molecules are master regulators that determine the rates of the individual component reactions that regulate each stage of the cell cycle.23 Phosphorylation of cell cycle effector proteins by the cyclin-CDK complexes acts as a switch, turning them on in orderly fashion. In mammalian cells, many cyclins regulate individual stages of the cell cycle, including G1 cyclins (cyclins D and E), S phase-specific cyclins, and G2-specific cyclins (cyclins A and B), which associate, respectively, with G1-specific (CDK2, CDK4, and CDK6), S-specific, and G2-specific (CDC2) cyclin-dependent kinases. The events of G1 phase (i.e., synthesis of enzymes and other molecules involved in DNA replication), S phase (i.e., DNA replication), and G2/M phase (i.e., chromosome condensation, disappearance of the nuclear envelope, and mitosis) are regulated by the transient accumulation and subsequent degradation of the cyclins.22,23,24 Switching off involves the targeted degradation of the cyclins by a protein destruction machine called the proteasome.24 Targeted destruction of the cyclins after they have carried out their function ensures unidirectional, irreversible progression around the cell cycle.23 The timing of the various cell cycle phases under normal conditions is related to the time required for cyclin-CDK complexes to reach appropriate, critical concentrations and for the phosphorylation of key substrates.23
Irradiation leads to prolongation of the cell cycle and arrests in G1, G2, and S phases because of checkpoint activation25 (see Fig. 2-1). Although this phenomenon was observed many years ago, it has only recently been understood at the biochemical and genetic levels. Arrest of cells at the G1 checkpoint has been particularly well studied (see Fig. 2-1A). G1 arrest is caused by the accumulation of a CDK inhibitory protein, CDKN1A (formerly designated p21 or Cip1), and this prevents key events that are required for transit through G1, including phosphorylation of the retinoblastoma (RB1) protein and activation of the E2F transcription factors (e.g., E2F1, E2F2, E2F4, E2F6) necessary for accumulation of enzymes required to traverse S phase.26,27 CDKN1A is induced at the transcriptional level by TP53 (also known as p53), which accumulates in irradiated cells and binds to the CDKN1A promoter, thereby activating transcription. TP53 accumulation depends on the activation of a DSB sensor molecule, the protein kinase ATM (ataxia-telangiectasia, mutated).26,27 The exact mechanism involved in sensing DSBs and activation of ATM is unclear, although it involves ATM autophosphorylation.28
The common mechanism for regulating the G1 and G2 checkpoints is inhibition of the CDK components of the interphase cell cycle engine and a block to phosphorylation and activation of effector molecules23,27 (see Fig. 2-1). A variation on this theme is seen at the mitotic spindle checkpoint. Progression through mitosis requires switching off CDK activity through targeted degradation of cyclin B.29 Arrest in M phase, when the mitotic spindle is compromised, involves the TP53-dependent stabilization of cyclin B. The mitotic spindle checkpoint guarantees that replication is followed by cell division, ensuring prevention of aneuploidy. Not surprisingly, the polyploidy nature of many tumor cells is related to TP53 inactivation.29
Most evidence indicates that ATM (and its homologs in other organisms) functions as a sensor for DNA damage through a complex series of interactions with the damaged DNA. This ultimately leads to ATM activation and downstream signaling cascades involving the kinases CHK1 and CHK2 (also called CHEK1 and CHEK2), which couple to the cell cycle engine at the level of individual cyclin-CDK complex molecules1,2,19,27,30 (see Fig. 2-1B). The molecular details of these processes are evolving rapidly, and referenced reviews can provide more detailed insight into the mechanisms of DNA damage, sensing, and checkpoint engagement.
TP53: Cellular Triage after Ionizing Radiation Exposure
TP53 is a nuclear phosphoprotein with sequence-specific DNA binding activity. It can function as both a transcriptional activator and repressor and plays a triage role in deciding whether to undergo cell cycle arrest and repair or to enter the pathways of PCD or replicative senescence31,32 (Fig. 2-2). When cells are exposed to ionizing radiation or chemotherapeutic agents, the levels of wild-type TP53 protein are increased. TP53 then transcriptionally activates a number of genes, most notably CDKN1A, the CDK inhibitor that mediates many of the properties of TP53.33 In addition to G1 arrest, genotoxic stress and ionizing radiation can induce TP53-dependent PCD pathways, including caspase-dependent apoptosis.33 TP53 can transcriptionally activate the proapoptotic BAX gene, induce synthesis of JUN kinase, and repress transcription of the antiapoptotic gene BCL2, suggesting that TP53 is a central mediator of PCD processes.31,34
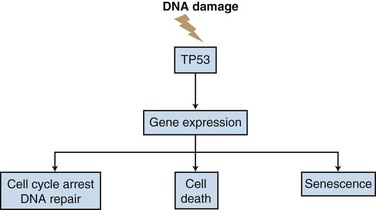
DNA damage in the presence of wild-type TP53 causes G1 arrest, followed by a period of DNA repair; if the damage is too great to be easily repaired, the cell is eliminated through TP53-dependent PCD pathways. However, approximately 50% of human tumors possess inactivating mutations in the TP53 gene.35 TP53 becomes inactivated in many tumors because of selection against its pro-apoptotic properties. The resultant loss of its central function as “sentinel of the genome” may be a type of collateral damage incurred in cells due to the selection advantage for survival accruing from the loss of TP53 apoptotic function.32,36 The loss of TP53 function is linked to poorer prognosis in malignant tumors such as lung, breast, colorectal, and hematopoietic tumors.37 Many tumor cell lines containing mutant TP53, including breast, glioma, and lymphoma cell lines, are more resistant to therapy than their wild-type TP53 counterparts.38,39
The loss of wild-type TP53 function in human malignant disease may be a key step in the progression of human cancer, and the TP53 status of cells may control the outcome of many tumor types in response to chemotherapy or radiation therapy.36 Although most of the other DSB response genes have molecular equivalents in yeast, TP53 does not. The TP53 gene appears to be required to determine the fate of damaged cells, which in mammalian cells may be PCD, a sacrifice that contributes to the well-being of the whole organism. TP53 monitors the degree of DNA damage and acts as a master switch, moving the cells from a state of cycle arrest and DNA repair to death or senescence pathways. Loss of TP53 in tumors compromises this critical surveillance and triage function. With the loss of this critical decision-point molecule, damaged and mutated tumor cells may survive to generate new and more malignant phenotypes (see Fig. 2-2).
ATM Gene: Master Regulator of the DNA Double-Strand Break Response
One of the many defects in ataxia-telangiectasia (AT) cells is increased chromosomal instability. Exposure to ionizing radiation also produces an increased number of chromosomal aberrations in AT cells compared with normal cells.40,41 This effect and the sequence similarity between ATM and the DNA repair protein DNA-PK suggested that AT cells might have deficient DNA repair. However, most studies have shown that AT cells do not have gross abnormalities in their ability to repair DNA damage. In general, it does not appear that the radiosensitivity of AT cells is caused by faulty DNA repair; it more likely results from an inability to detect the presence of DNA damage. Exposure to ionizing radiation causes normal cells to delay at the G1/S and G2/M transition phases of the cell cycle, and these checkpoints are thought to allow the cells to repair DNA damage before DNA synthesis or mitosis occurs.27 Both checkpoints are absent in AT cells, suggesting clues to the function of ATM.27
Although ATM and TP53 cooperate in radiation-induced apoptosis, there are ATM-independent pathways for the induction of TP53-dependent apoptosis, and many of the downstream effects of ATM are independent of TP53.27,28 Wild-type and knockout mice have been evaluated for acute and late toxicity after whole-body exposure to ionizing radiation. The ATM– and ATM/TP53-knockout mice had similar severe toxicity profiles, suggesting that TP53 does not play a role in acute radiation toxicity. Both TP53- and ATM-knockout mice preferentially developed lymphoid tumors, whereas ATM/TP53 double knockouts had an accelerated time to tumor formation and a broader spectrum of tumor types. Analysis of the acquired tumors in ATM-null/TP53-heterozygous mice revealed that three of seven had loss of the remaining TP53 allele. These studies showed that ATM and TP53 interact in a complex manner that is specific to cell type and outcome. This interaction most likely relies on a variety of other pathways (discussed later) and will require much additional work before a complete understanding of the ATM/TP53 relationship is obtained.
With isolation of the ATM gene it became possible to attempt correction of the cellular defects of the AT phenotype using gene transduction techniques. Several groups have reported that the introduction of ATM into AT cells resulted in reversal of AT defects.27,41 The transfection of full-length ATM into AT cells reversed DSBs, restored normal sensitivity to ionizing radiation, and decreased the number of chromosomal abnormalities. Transfection of full-length ATM also reversed the defective activation of CDKN1A (p21) and JUN kinase in response to ionizing radiation.42,43
Histones and Chromatin Structure
Native DNA exists in the cell in the form of chromatin complexed with a family of proteins called histones. Histones are involved in packaging DNA in the nucleus into a compact form. For gene transcription or DNA repair to occur, the histones are altered by post-translational modifications, most notably acetylation, that permit decondensation and access of factors to the DNA.44 The histone acetylase TIP60 is important both in this process and in the modification of ATM itself.26,45 For effective DNA repair, acetylation of histones by histone acetylases must occur to permit access of repair proteins to the sites of DSBs.46 DNA is then remodeled by ATP-dependent remodeling enzymes to permit repair molecules such as Mre11, RAD50, and NBS1 to access sites of DSBs.47,48 A novel histone, H2AX, found in low concentrations on chromatin, has been shown to play a crucial signaling role in the response to genomic stresses such as ionizing radiation. H2AX phosphorylation is one of the earliest events occurring after exposure to ionizing radiation, and ATM-dependent phosphorylation of H2AX may be involved in signaling to cell cycle checkpoints and DNA repair enzymes involved in recombination repair.49,50
Mechanisms of DNA Repair after Ionizing Radiation
Maintaining undamaged DNA is essential to cell survival. Cells have therefore evolved a wide range of mechanisms to halt cell cycle progression, survey DNA, and repair damage. Radiation causes a number of types of damage either by direct interaction with DNA or indirectly through effects on nearby water molecules and free radical generation. Types of damage include DNA base damage, damage to the deoxyribose sugar backbone, and physical breaks in one or both strands of the DNA. DNA damage induced by ionizing radiation tends to be clustered so that there is more than one damaged site in proximity along the double helix, known as locally multiply damaged sites.51
Among the less catastrophic forms of DNA damage induced by ionizing radiation are the singly damaged bases, which can be repaired by base excision repair. This is a process by which the damaged base is recognized and removed by an N-glycosylase, the apurinic or apyrimidinic (AP) site is cleaved by an AP endonuclease, a patch of DNA is excised, DNA is then resynthesized using the other strand as the template, and the repaired strand is ligated.18,52 In settings in which the base damage is not recognized by the N-glycosylase, another mechanism for repair exists, called nucleotide excision repair. In a manner somewhat similar to base excision repair, a damaged section of DNA is removed by incision and excision, a patch is resynthesized using the remaining strand, and the repaired strand is then ligated. Although no naturally occurring mammalian mutants have been identified that are defective in base excision repair, there are a number of different nucleotide excision repair mutants, which are exemplified by xeroderma pigmentosum and Cockayne’s syndrome.18 They are characterized by abnormalities in the repair of damage caused by ultraviolet light, although the clinical spectrum of these repair-deficiency syndromes varies. The abnormal genes are identified by finding the human gene that corrects rodent cell defects, called excision repair cross-complementing (ERCC) groups. For a comprehensive review on repair of single-strand breaks we recommend the article by Friedberg, Walker, and Siede.18
DNA Strand Breaks
Cellular lethality after radiation involves generation of DSBs.53,54,55 Misjoined or unrepaired DSBs lead to chromosome deletions, translocations, and acentric or dicentrics, with lethal consequences for the cell. As with the excision repair defects (e.g., ERCCs), there are several x-ray repair defects in the x-ray cross-complementation (XRCC) groups involved in the repair of DSBs. DNA single-strand repair is carried out in a manner similar to base damage repair, with the undamaged DNA strand serving as a template. DSB repair is more complicated because there is no adjacent, undamaged template available to form a template for repair of the broken strands. The ends of the broken DNA must be protected, and the damaged site must be reconstituted by the processes of homologous recombination and nonhomologous end joining (NHEJ).
DNA DSB repair has much in common with the recombinatorial processes involved in immunoglobulin and T-cell receptor gene rearrangement in the immune response. The XRCC groups that have been identified and are involved in DNA DSB repair include genes that produce DNA end-binding proteins XRCC6 (formerly designated KU70 or Ku-70) and XRCC5 (formerly designated KU80 or Ku-80) and a DNA-dependent protein kinase (DNA-PK). Defects in DNA repair genes are seen in severe combined immunodeficiency (SCID) mice, indicating the importance of recombination in the restoration of DNA integrity after DSBs and in the immune response.18,53
Double-Strand Break Damage and Repair
Experiments in yeast and human cells indicate that a single unrepaired DSB leads to cell death.53,55 However, such cells can detect such DSBs and repair them. DSB repair would be predicted to require three functional components: (1) a mechanism for detecting and gauging DNA damage, (2) a signal transduction system, and (3) an effector system for DNA repair. These components are discussed in the following paragraphs, commencing with the repair component. DSBs can be repaired by a number of mechanisms, but the most prevalent are homologous recombination and NHEJ. Human cells appear to differ from yeast in that while homologous recombination predominates in yeast, the opposite is true in mammalian cells.53,55
Genetic analysis has revealed the existence of a large number of genes that regulate DSB repair. One essential gene, involved in resistance to cell killing by ionizing radiation and the repair of DSB, is XRCC5, which encodes XRCC5, a protein that binds with high affinity to the ends of the double strands1,18 (Fig. 2-3A). XRCC5 functions in normal cellular processes that require DSB rejoining, most notably V(D)J rejoining in the immunoglobulin and T-cell receptor genes of immature B and T lymphocytes. XRCC5 exists in cells in a heterodimeric complex with XRCC6, the product of the XRCC6 gene. The XRCC5/XRCC6 heterodimer is required for the end-binding and repair functions.56 The XRCC (formerly designated KU) proteins recognize the ends of DSBs and protect them from further degradation before the onset of end-joining reactions required for DSB repair. An associated protein in the complex with important functions in DNA repair is DNA-PK, the product of the XRCC7 gene.56
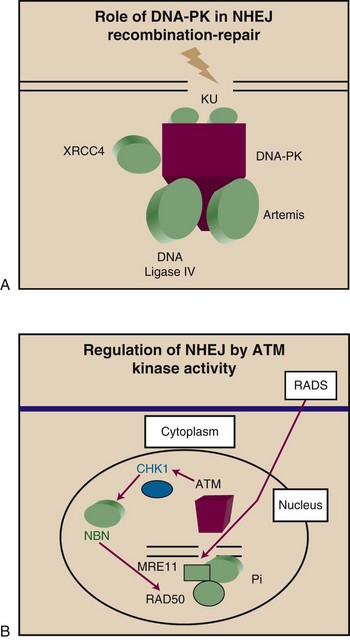
DNA-PK is a serine/threonine kinase involved in regulating the cellular response to DNA damage.56,57 DNA-PK is a member of the phosphatidylinositol-3-kinase gene family (PIK) that produce a group of high-molecular-weight proteins that contain a conserved kinase domain at the carboxyl-terminal end. PIK family proteins have been identified in yeast, in Drosophila, and in mammalian cells, and include the human ataxia-telangiectasia gene (XRCC7) for DNA-PK and the yeast genes TEL1 and MEC1.56,57 Homology with genes of other organisms can provide a clue to the function of the family of genes in mammalian systems. For example, the S. cerevisiae TEL1 gene is a homolog of the human ATM gene. TEL1 mutants have shortened telomeres and exhibit chromosome instability. In mice, the SCID mutation results in the loss of expression of functional DNA-PK.58 SCID mice are deficient in the repair of DNA DSBs, have faulty V(D)J recombination, and are sensitized to radiation.58 The binding of the XRCC complex (XRCC5 and XRCC6) to DNA-PK activates the serine/threonine kinase of the catalytic subunit of DNA-PK. The DNA-PK/XRCC complex is involved in recognition of DNA DSBs. In SCID cells, the catalytic subunit for DNA-PK is absent, and the loss of DNA-PK activity may account for the increased sensitivity to ionizing radiation, the inability to repair DNA DSBs, and the immune defects.
Genetic evidence indicates the existence of numerous other factors required for efficient DSB repair in human cells.18,53,55 Among the genes are homologs of the yeast RAD52 epistasis group, RAD50 and MRE11.26,59,60,61 RAD50 and MRE11, together with the product of the XRS2 gene, form a complex in S. cerevisiae that is involved in nonhomologous recombination (see Fig. 2-3B). RAD50 has been suggested to bind to DNA at the site of DSBs and mark them for repair or recombination. Such a signaling role for RAD50/MRE11 complexes is suggested by studies indicating the formation of nuclear foci containing these proteins in cells after irradiation.61 The RAD50/ MRE11 complexes also contain another important protein, NBN (nibrin; formerly NBS1 or p95), which is inactivated in the Nijmegen breakage syndrome.26,61 The RAD50/MRE11 foci fail to form in ATM-deficient cells, suggesting a role for this complex downstream of the ATM protein. DSB repair can also be carried out in human cells by homologous recombination, an alternative pathway involving a different group of genes, including RAD51 through RAD57. RAD51 is the eukaryotic homolog of the bacterial protein RecA and forms structures in meiotic chromosomes containing ATM, suggesting functional coupling between these proteins. The complex also contains TP53 and may be involved in coupling of DNA DSB formation to cell cycle arrest, as well as in carrying out recombination and repair.62
The tumor suppressor genes BRCA1 and BRCA2 also have function in DNA repair. Although these proteins have multiple functions, it has been shown that BRCA1 interacts with RAD51 and assists in cell cycle arrest through activation of CDKN1A, a gene that is normally activated by TP53.63 RAD51 is thus targeted by three major gene products: ATM, TP53, and BRCA1. Loss of RAD51 leads to cancer development, suggesting that this pathway of DNA repair and replication is crucial in protection of cells from genotoxic stress. That this RAD51 pathway of DSB repair is separate from the RAD50 end-joining pathway is indicated by findings that ionizing radiation causes the formation of nuclear foci containing RAD50 or RAD51 but never both complexes.64 However, further studies are required to understand the relative contributions of the two pathways to cell survival after exposure to ionizing radiation. As with the RAD50 pathway, understanding of mammalian recombination repair is still preliminary and is difficult to assemble into a fully coherent molecular pathway. BRCA2 mutant cancer cells are also deficient in DSB repair and have increased radiosensitivity.63 It is apparent that DNA damage recognition and repair are complex, involving interaction among molecules that can cause cell cycle arrest, apoptosis, and signal transduction. Defects in any of these pathways could lead to radiation sensitivity in normal tissues (and perhaps the corresponding tumor) or to the development of secondary mutations and the mutator phenotype, as with mismatch repair defects. For radiation therapy, the aim of course is to maximize accumulation of DSBs in tumor cells by targeting these pathways.
Pathways of Ionizing Radiation-Induced Programmed Cell Death
The ability to inhibit the pathways of cell death appears to be a key step in the origin of cancer cells, with the small percentage of cells that are able to evade PCD being the ones most capable of forming tumors.16 Multiple genetic alterations are involved in escape from PCD, most notably in the TP53 and BCL2 families.16 Radiation oncologists are faced with the undesirable situation in which tumor cells are selected for resistance to PCD while normal cells retain these altruistic pathways. Apoptosis and senescence are key PCD pathways with potential roles in cancer induction and resistance to therapy. Excellent reviews have been provided by Edinger and Thompson16 and Danial and Korsmeyer.65
Apoptosis is a normal physiologic process that permits altruistic death of cells during tissue remodeling or the elimination of T-cell populations after the reversal of viral infection.65 Apoptosis is characterized morphologically by cell shrinkage, membrane blebbing, chromatin condensation, and ultimate fragmentation of the cell into apoptotic bodies. Molecular characteristics include activation of a class of proteases (protein degrading enzymes) the caspases. These are involved in intracellular death signaling. In addition, activation of enzymes that degrade DNA (endonucleases) is seen. In the final stage of apoptosis, the cell corpses exhibit “eat me” signals, a process that involves alteration of the cell surface (including expression of annexin V binding sites on the plasma membrane), permitting neighboring cells to remove the apoptotic bodies without the induction of an inflammatory reaction. Apoptosis is mediated by changes in the outer mitochondrial membrane that lead to the release of death signals such as cytochrome c. BCL2 and related family members function as antiapoptotic factors by blocking prodeath changes in mitochondrial membrane potential or by antagonizing the proapoptotic factor BAX. The terminal stages involve DNA degradation, which cleaves DNA into 50- to 300-kb fragments and later into smaller fragments by cleavage of the exposed regions of DNA between nucleosomes.65
Replicative Senescence
In addition to vulnerability to overt killing, all somatic cells possess replicative checkpoints that place limits on the number of permitted cell divisions over the lifetime of the cell. For unlimited growth, cells must bypass crisis, the point at which the telomeres on chromosomes have shortened enough to prevent successful future cell divisions. TP53-sensitive (and CDKN1A-sensitive) expression of the enzyme telomerase in tumor cells is sufficient to bypass crisis and permit unlimited growth in some cells (see Fig. 2-2). In addition to crisis, cells undergo telomerase-independent forms of senescence, regulated in many cases by the retinoblastoma protein RB1 and the CDK inhibitory protein CDKN2A (formerly designated p16).66,67 However, TP53 appears to be the primary regulator of senescence and induces senescence downstream of DNA damage detected by ATM activation or other pathways.14 Few experiments have been carried out to examine the role of senescence in responses to ionizing radiation, although in one study, ionizing radiation induced premature senescence that occurred independently of ATM expression.68 A large increase in the volume of experiments dealing with this subject is anticipated.
Activation of Anabolic Signaling Pathways by Ionizing Radiation
One of the primary features of cancer is its autonomy in terms of growth signals. There are three operationally defined steps in growth signaling: (1) transmembrane receptor occupation by growth factors and transmembrane signaling, (2) stimulation of cytoplasmic cascades that amplify primary signals, and (3) activation of downstream effector proteins. Step 1 classically involves receptor dimerization and autophosphorylation of receptor tyrosine kinases (RTKs) or recruitment of nonreceptor tyrosine kinases (NRTKs). Such events occur commonly after ionizing radiation, and a generalized activation of the HER1 through HER4 RTKs occurs in irradiated cells69 (Fig. 2-4). Ionizing radiation can induce the release of growth factors such as epidermal growth factor (EGF, transforming growth factor–alpha) and paracrine activation of growth in adjacent cells.69 Increases in cellular phosphotyrosine levels were observed after exposure to x rays in a number of cells, and activation of NRTKs has been observed.69 Of particular importance in step 2 processes are members of the mitogen-activated protein kinase (MAPK) family, which play a crucial role in cell growth and survival. Ionizing radiation stimulates the activity of MAPK members that carry the mitogenic signal (e.g., MAPK1 [formerly ERK2], MAPK3 [formerly ERK1]).70 The paradigm for step 3 is the activation of factors that bind the promoters of mammalian immediate early genes such as FOS and EGR1 through receptor activation, tyrosine phosphorylation, and MAPK activation. Such transcription factors include serum response factor, ETS domain factors such as TCF62, ELK-1, activating protein-1 (AP1), and cyclic adenosine monophosphate-binding protein (i.e., cAMP response element binding [CREB]).71 Each of these factors is activated by ionizing radiation, and the promoter of at least one immediate early gene (EGR1) is induced by radiation treatment. Exposure to ionizing radiation may amplify the growth signals already active in cancer cells, suggesting that the RTK-MAPK pathway may be a fruitful target in the selection of radiation sensitizers.69,72
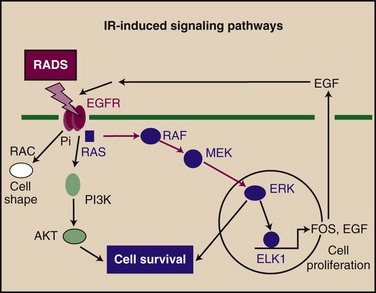
Radiation-induced signaling may be secondary to free radical generation. Radiation produces two main species that combine to kill cells in a clinical setting and that may also double as primary stress signals.10 These signals are reactive oxygen species (ROSs) and DSBs (discussed before). ROSs play a role in signal transduction after stimulation by platelet-derived growth factor (PDGF) and phorbol esters.70,73 ROSs produced by cellular stress, such as peroxide, superoxide, hydroxyl radicals, and nitrous oxide, may feed into this ROS-transduced pathway at a number of places.74 Cell kill by radiation is closely correlated with the accumulation of DSBs. The dictates of logic and much preliminary evidence strongly suggest a role for DSBs in the sensing and response to ionizing radiation.27 Cells evidently possess at least one system to sense and respond to radiation, the ATM family of protein kinases, which appear to be situated close to the primary event in DSB-induced signaling.27,53 ATM activation leads to phosphorylation of TP53, coupling DSB accumulation to cell cycle arrest in G1.30 Vectoral signaling is then coupled to the cyclic events of the cell division cycle. ATM belongs to the lipid kinase family that includes PIK and DNA-PK.27,40 PIK is activated by ionizing radiation and is required for cell survival during irradiation. DNA-PK is intimately involved in cell responses to radiation, and its ability to sense DSBs in combination with DNA end-binding proteins XRCC6 and XRCC5 suggests that it may be able to function independently of ATM in cell signaling after exposure to ionizing radiation.56
Radiation and the RNA World
Over recent years there has been a revolution in understanding the role of RNA in biology and cancer.75 In addition to its housekeeping function as mRNA and rRNA, novel RNAs have been shown recently to play key regulatory roles. Small interfering RNA and microRNA play negative regulatory roles in diminishing specific RNA levels and targeting structures in the 3′ and 5′ regions of mRNA.76,77 Large noncoding RNA plays a role in regulation of transcription, although its regulation of radiation responses is not known.78 Study of the roles of these molecules in the radiation response has begun.77,79
Tumor Microenvironment and Responses to Ionizing Radiation In Vivo
In addition to malignant cells, tumors contain normal cells and structural elements, including endothelial cells, fibroblasts, extracellular matrix molecules, inflammatory cells, and blood vessels. The tumor microenvironment is abnormal in being largely deficient in nutrients such as oxygen and glucose and abundant in the waste products of metabolism such as lactic acid and carbon dioxide.80,81 Therefore the concentrations of nutrients and cell-cell and cell-matrix contacts are abnormal and may be in a constant state of flux because of intermittent perfusion by the microvasvculature. The tumor microenvironment is a key determinant of the response to ionizing radiation. Hypoxic cells are markedly radioresistant because of the requirement for oxygen in the formation of the free radicals that mediate radiation-induced DSBs.10 Defining the tumor milieu and how it is regulated is therefore important in understanding the radiosensitivity of tumors.
Hypoxia, Microenvironment, and Radiation Response
The role of hypoxia in radioresistance has been studied for many years.10,82 Cell killing by radiation is decreased at intermediate and low oxygen conditions, and a number of studies have correlated low pretreatment tumor oxygenation with a worse disease outcome.83,84 Whether there is a direct cause-and-effect relationship between low oxygen concentration and outcome remains to be resolved, primarily through clinical trials that deliberately attempt to correct the poor oxygenation status of the tumors. In addition to directly inhibiting cell killing by ionizing radiation, hypoxia may amplify the mutator phenotype in the severely hypoxic cells in the central cores of tumors, increasing the rate of evolution of resistant clones and amplifying the malignant phenotype.85
Many genes are induced or repressed within the tumor microenvironment.86,87 The largely unregulated environment found in poorly vascularized tumors could be thought of as resembling the conditions under which many unicellular organisms grow. Survival for these organisms depends on their ability to withstand environmental stresses such as nutritional deprivation, temperature and pH changes, radiation exposure (e.g., ultraviolet light, x rays), and xenobiotics. Among the molecules induced by hypoxia are members of the unfolded protein response family stress proteins (e.g., glucose-regulated proteins [GRPs], redox enzymes such as heme oxygenase, metallothionien IIA, DT-diaphorase; transcription factors such as JUN, FOS, AP1, TP53, nuclear factor kappa B [NFκB], HIF1) and growth factors or cytokines (e.g., erythropoietin, vascular endothelial cell growth factor [VEGF], endothelial growth factor [EGF] receptor, interleukin-1 alpha [IL-1α]).86,87 HIF1 appears to be of key significance in the response of cells to the microenvironment at the molecular level because this transcription factor regulates expression of many of the factors that mediate angiogenesis.88
Tumor Angiogenesis
Another of the hallmarks of cancer is the ability of cancer cells to induce de novo angiogenesis in order to sustain growth.36,89,90 Tumors contain multiple angiogenic factors, including angiopoietin, acidic fibroblast growth factor (FGF), basic FGF, angiogenin, platelet-derived growth factor (PDGF), prostaglandins E1 and E2, transforming growth factor–beta, tumor necrosis factor–alpha, and VEGF (also known as vascular permeability factor). The VEGF family has received much attention as potential therapeutic targets. In addition, antiangiogenic factors are also expressed and include angiostatin, endostatin, interferon-alfa and interferon-beta, thrombospondin, and tissue inhibitor of metalloproteinase.90,91 The outcome for vascularization and tumor growth depends on a balance between the factors.
The use of antiangiogenic therapy with local radiation has been investigated in several laboratories.92,93 One important aspect for combining vascular targeting and radiation therapy and probably also for systemic agents is whether the antiangiogenic therapy increases or decreases tumor oxygenation. The relationship between angiogenesis, antiangiogenic therapy, and the more conventional cancer therapies remains to be elucidated but offers some exciting areas of investigation. The ability to suppress metastases and hold local tumor growth92,93 offered by vascular targeting may greatly increase the potential role for radiation therapy. However, if antiangiogenic therapy leads to decreased perfusion and/or hypoxia, the effects are likely to be confounding. Given the role of blood vessels in late radiation injury, the use of antiangiogenic therapy with radiation in clinical treatment will require careful study and assessment of long-term toxicities and late effects.
Targeting Molecular Pathways in the Radiation Response: Discovery of New Drugs
The accumulated data regarding the signaling circuitry underlying the response of cells to DSBs suggests an opportunity for the development of novel adjuvant therapies. Approaches based on small-molecule inhibitors of specific reactions, RNA interference (small interfering RNAs), and antisense and gene therapy are attractive and follow directly from the basic research available. There is much excitement regarding the potential use of kinase inhibitors in cancer treatment, based on the idea that they work catalytically, are present in small amounts, and often regulate key processes in the cell. The most popular paradigm is a kinase inhibitor, known as imatinib (Gleevec), which has greatly influenced the treatment of certain hematologic malignant diseases. Given that the cell cycle checkpoint component of the DNA DSB response is regulated largely at the post-transcriptional level by batteries of protein kinases and phosphatases, many inviting targets are available. Proof of principle that such an approach may work is suggested by studies using agents that inhibit the RAS signal transduction pathway, protein kinase C, or the PIK pathway, which were shown to enhance radiation-induced cell killing.* Members of the PIK family may be feasible targets for pharmacologic intervention in the clinic. Loss of function of PIK proteins is usually associated with increased radiosensitivity. Compounds that block the function of these proteins would greatly enhance the efficacy of agents that cause DNA damage, such as radiation therapy, and allow for the treatment of tumors that are particularly radioresistant. The fungal metabolite wortmannin is a specific inhibitor of the p110 PIK, now designated PIK3CD. Wortmannin forms a covalent adduct with Lys802 of PIK3CD, inactivating the kinase activity.101,102 This Lys802 is conserved in DNA-PK and AT proteins, suggesting that both would be sensitive to wortmannin. In vitro studies with a wide range of tumor-derived cells, including those of the breast, colon, and prostate, indicate that wortmannin is an effective radiosensitizer of human cells. Wortmannin can inhibit the kinase activity of DNA-PK in vitro and in vivo. DNA-PK also is involved in the activation of TP53 after irradiation of cells, and wortmannin suppresses the activation of TP53 after irradiation. Other studies have shown that wortmannin may exert its primary effect by inhibiting the repair of DNA strand breaks in cells.
In addition to drugs, the development of small interfering RNA approaches offers the promising possibility of “knocking down” selective genes that may mediate resistance.76 There has been much excitement regarding the use of targeted gene therapy to introduce genes of interest or their antisense partners within viral vectors. Some of the enthusiasm about these approaches has waned because of the potential toxicity of the viral vectors (particularly, adenovirus) and a number of other operational problems, such as accurately reaching the target site, achieving sufficient gene production in situ (i.e., drug, enzyme, or toxic molecule) to alter the radiation response, altering the radiation response sufficiently to show an impact on local tumor control, having a positive therapeutic ratio, and, ideally, having an impact on survival in addition to enhanced local control. Despite these problems, the approach remains popular, particularly for local expression of products that diffuse from cells, such as cytokines. With the advent of newer, safer vectors for delivery of agents, the approach holds promise.103
Earlier radiation modifiers in clinical use have been developed based on conventional radiation biology models, such as hypoxia (e.g., radiation sensitizers and enhancers, altered oxygen delivery), the competition model (e.g., thiol depletion and radioprotectors), and increasing susceptibility of DNA to radiation damage (e.g., halopyrimidines).101,102,104 Although such therapies were not based on the new molecular targets that have more recently been elucidated, much can be learned by studying the relationships between these therapies and the molecular processes. For example, it may be possible to understand how better to use the halopyrimidines in relation to cell cycle checkpoints, hypoxic sensitizers in relation to hypoxia-induced processes, and protectors in terms of specific DNA lesions or activity of repair enzymes.
As mentioned above, tumor cells exist in a unique microenvironment that is depleted of oxygen and glucose and rich in carbon dioxide and lactate. Drugs may be designed to prosper in this altered microenvironment and enhance treatment efficacy. Among these more conventional approaches, the bioreductive agents, such as mitomycin C and tirapazamine, require enzymatic activation that occurs preferentially in hypoxic environments.105
Normal Tissues
Radiation therapy depends for its selective treatment of cancer on tight control of delivery to tumors and avoidance of normal tissues. Most normal tissues that express wild-type TP53 are sensitive to TP53-induced PCD.106 Renewable tissues such as hematopoietic stem cells and crypt cells in the gastrointestinal tract may be particularly vulnerable.33 Indirect forms of lethality may occur in whole body irradiation, leading to the death of hematopoietic stem cells in the bone marrow, or depletion of mature tissues can lead to secondary damage caused by red blood cell depletion, immunosuppression, and gross infection as the gastrointestinal lining is lost.10,13,14
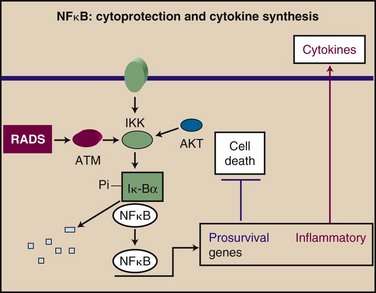
Additional changes occur that are thought to be the result of a persistent oxidative state in tissues such as lung, which may lead to chronic inflammatory changes and induction of cytokines and adhesion molecules.107,108 Changes may result from induction of the transcription factor NFκB by ionizing radiation in an ATM-dependent manner109,110 (Fig. 2-5). NFκB, in addition to its role in cytoprotection, plays a broader part in the acute inflammatory response through transcription of a wide range of proinflammatory cytokines and adhesion molecules.111 Induction of an inflammatory environment in normal tissues may be a side effect of activation of NFκB in a cytoprotective reaction (see Fig. 2-5).
Basic Concepts and Techniques of Radiation Sciences
Techniques and approaches to molecular and cellular biology in humans and other model organisms, largely developed in the 1960s and 1970s, continue to revolutionize knowledge of basic cancer biology and radiation sciences. The ability to inactivate genes by homologous recombination, which came into frequent use in the 1980s, has permitted the movement of this approach into animal models, allowing study of molecular genetics in vivo. This has permitted us to test the significance of the key genes in the cell cycle, DNA repair, and PCD, which mediate the sensitivity of cells to ionizing radiation and define their role in radiation response. Novel “big biology” approaches, including such genomic techniques as microarray screening, deep sequencing, and mass spectrometry permit discovery-based research with the potential for fundamental advances.112,113,114 Because of the progress in deciphering human and other genomes in the 1990s, a total readout of expression of all human genes can be undertaken. This has opened the way for identification of signature genes in human cancers that correspond to poor prognosis, treatment resistance, and metastasis. The impact of gene expression profiling on radiation biology is perhaps less than in general cancer biology, because most responses to genomic stress occur at the post-transcriptional level. The development of proteomics is being developed, based on advances in mass spectrometry, and this approach allied with deep sequencing may permit the rapid study of critical changes in post-translational modification, such as phosphorylation, acetylation, and ubiquitination, that mediate many regulatory responses to genomic stress. The increased speed and accuracy of drug development, based on robotic approaches to assaying chemical libraries and in silico design, should also increase the rate of drug discovery and hasten the translation of molecular findings into the clinic.
2 Zou L, Elledge SJ. Sensing and signaling DNA damage. Roles of Rad17 and Rad9 complexes in the cellular response to DNA damage. Harvey Lect. 2001;97:1-15.
3 Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans. Past, present and future. Trends Genet. 1998;14:410-416.
4 Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138-141.
5 Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70.
6 Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5:382-395.
8 Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy. Oncogene and non-oncogene addiction. Cell. 2009;136:823-837.
10 Hall EJ. Radiobiology for the radiobiologist. Philadelphia: Lippincott-Raven; 1993.
11 Fowler J. Biological foundations of radiotherapy. Amsterdam: Exerpta Medica; 1967.
12 Revell SH. Relationship between chromosome damage and cell death. New York: Alan Liss; 1983.
13 Dainiak N, Ricks RC. The evolving role of haematopoietic cell transplantation in radiation injury. Potentials and limitations. BJR Suppl. 2005;27:169-174.
15 Alper T, Gillies NE, Elkind MM. The sigmoid survival curve in radiobiology. Nature. 1960;186:1062-1063.
16 Edinger AL, Thompson CB. Death by design. Apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663-669.
17 Campisi J. Senescent cells, tumor suppression, and organismal aging. Good citizens, bad neighbors. Cell. 2005;120:513-522.
18 Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, DC: ASM Press; 1995.
23 Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221-234.
24 Lim HH, Surana U. Tome-1, wee1, and the onset of mitosis. Coupled destruction for timely entry. Mol Cell. 2003;11:845-846.
26 Iijima K, Ohara M, Seki R, Tauchi H. Dancing on damaged chromatin. Functions of ATM and the RAD50/MRE11/NBS1 complex in cellular responses to DNA damage. J Radiat Res (Tokyo). 2008;49:451-464.
29 Andreassen PR, Lohez OD, Margolis RL. G2 and spindle assembly checkpoint adaptation, and tetraploidy arrest. Implications for intrinsic and chemically induced genomic instability. Mutat Res. 2003;532:245-253.
31 Polyak K, Xia Y, Zweier JL, et al. A model for p53-induced apoptosis. Nature. 1997;389:300-305.
35 Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell. 1997;88:315-321.
36 Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030-9040.
42 Morgan SE, Lovly C, Pandita TK, et al. Fragments of ATM which have dominant-negative or complementing activity. Mol Cell Biol. 1997;17:2020-2029.
45 Robert F, Hardy S, Nagy Z, et al. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double-strand break repair. Mol Cell Biol. 2006;26:402-412.
47 Shim EY, Hong SJ, Oum JH, et al. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol. 2007;27:1602-1613.
50 Morrison AJ, Shen X. DNA repair in the context of chromatin. Cell Cycle. 2005;4:568-571.
51 Ward JF. Complexity of damage produced by ionizing radiation. Cold Spring Harb Symp Quant Biol. 2000;65:377-382.
53 Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst). 2004;3:781-796.
56 Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst). 2004;3:909-918.
59 Maser RS, Monsen KJ, Nelms BE, Petrini JH. Hmre11 and hrad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol. 1997;17:6087-6096.
62 Wray J, Liu J, Nickoloff JA, Shen Z. Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress. Cancer Res. 2008;68:2699-2707.
63 Scully R, Xie A, Nagaraju G. Molecular functions of BRCA1 in the DNA damage response. Cancer Biol Ther. 2004;3:521-527.
65 Danial NN, Korsmeyer SJ. Cell death. Critical control points. Cell. 2004;116:205-219.
69 Schmidt-Ullrich RK, Contessa JN, Lammering G, et al. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855-5865.
70 Stevenson MA, Pollock SS, Coleman CN, Calderwood SK. X-irradiation, phorbol esters, and H2O2 stimulate mitogen-activated protein kinase activity in NIH-3T3 cells through the formation of reactive oxygen intermediates. Cancer Res. 1994;54:12-15.
73 Chen Q, Olashaw N, Wu J. Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J Biol Chem. 1995;270:28499-28502.
77 Weidhaas JB, Babar I, Nallur SM, et al. Micrornas as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111-11116.
79 Jaklevic B, Uyetake L, Wichmann A, et al. Modulation of ionizing radiation-induced apoptosis by bantam microrna in Drosophila. Dev Biol. 2008;320:122-130.
80 Gullino PM. The internal milieu of tumors. Prog Exp Tumor Res. 1966;8:1-25.
82 Weinmann M, Welz S, Bamberg M. Hypoxic radiosensitizers and hypoxic cytotoxins in radiation oncology. Curr Med Chem Anti-Cancer Agents. 2003;3:364-374.
83 Hockel M, Schlenger K, Aral B, et al. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509-4515.
85 Bindra RS, Glazer PM. Genetic instability and the tumor microenvironment. Towards the concept of microenvironment-induced mutagenesis. Mutat Res. 2005;569:75-85.
88 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425-437.
89 Folkman J. Angiogenesis and apoptosis. Semin Cancer Biol. 2003;13:159-167.
96 Dutta PR, Maity A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007;254:165-177.
99 Jaboin JJ, Shinohara ET, Moretti L, et al. The role of mtor inhibition in augmenting radiation induced autophagy. Technol Cancer Res Treat. 2007;6:443-447.
105 Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-447.
107 Epperly MW, Guo H, Shields D, et al. Correlation of ionizing irradiation-induced late pulmonary fibrosis with long-term bone marrow culture fibroblast progenitor cell biology in mice homozygous deletion recombinant negative for endothelial cell adhesion molecules. In Vivo. 2004;18:1-14.
108 Robbins ME, Zhao W. Chronic oxidative stress and radiation-induced late normal tissue injury. A review. Int J Radiat Biol. 2004;80:251-259.
110 Piret B, Schoonbroodt S, Piette J. The ATM protein is required for sustained activation of NF-kappaB following DNA damage. Oncogene. 1999;18:2261-2271.
112 Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863-14868.
1 Sanchez Y, Bachant J, Wang H, et al. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166-1171.
2 Zou L, Elledge SJ. Sensing and signaling DNA damage. Roles of Rad17 and Rad9 complexes in the cellular response to DNA damage. Harvey Lect. 2001;97:1-15.
3 Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans. Past, present and future. Trends Genet. 1998;14:410-416.
4 Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138-141.
5 Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70.
6 Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5:382-395.
7 Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027-1038.
8 Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy. Oncogene and non-oncogene addiction. Cell. 2009;136:823-837.
9 Leonard JM, Ye H, Wetmore C, Karnitz LM. Sonic Hedgehog signaling impairs ionizing radiation-induced checkpoint activation and induces genomic instability. J Cell Biol. 2008;183:385-391.
10 Hall EJ. Radiobiology for the radiobiologist. Philadelphia: Lippincott-Raven; 1993.
11 Fowler J. Biological foundations of radiotherapy. Amsterdam: Exerpta Medica; 1967.
12 Revell SH. Relationship between chromosome damage and cell death. New York: Alan Liss; 1983.
13 Dainiak N, Ricks RC. The evolving role of haematopoietic cell transplantation in radiation injury. Potentials and limitations. BJR Suppl. 2005;27:169-174.
14 Dubois A, Walker RI. Prospects for management of gastrointestinal injury associated with the acute radiation syndrome. Gastroenterology. 1988;95:500-507.
15 Alper T, Gillies NE, Elkind MM. The sigmoid survival curve in radiobiology. Nature. 1960;186:1062-1063.
16 Edinger AL, Thompson CB. Death by design. Apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663-669.
17 Campisi J. Senescent cells, tumor suppression, and organismal aging. Good citizens, bad neighbors. Cell. 2005;120:513-522.
18 Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, DC: ASM Press; 1995.
19 Mcgowan CH, Russell P. The DNA damage response. Sensing and signaling. Curr Opin Cell Biol. 2004;16:629-633.
20 Stergiou L, Hengartner MO. Death and more. DNA damage response pathways in the nematode C. elegans. Cell Death Differ. 2004;11:21-28.
21 Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39-85.
22 Murray AW, Kirschner MW. What controls the cell cycle? Sci Am. 1991;264:56-63.
23 Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221-234.
24 Lim HH, Surana U. Tome-1, wee1, and the onset of mitosis. Coupled destruction for timely entry. Mol Cell. 2003;11:845-846.
25 Rudoltz MS, Kao G, Blank KR, et al. Molecular biology of the cell cycle. Potential for therapeutic applications in radiation oncology. Semin Radiat Oncol. 1996;6:284-294.
26 Iijima K, Ohara M, Seki R, Tauchi H. Dancing on damaged chromatin. Functions of ATM and the RAD50/MRE11/NBS1 complex in cellular responses to DNA damage. J Radiat Res (Tokyo). 2008;49:451-464.
27 Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316-323.
28 Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499-506.
29 Andreassen PR, Lohez OD, Margolis RL. G2 and spindle assembly checkpoint adaptation, and tetraploidy arrest. Implications for intrinsic and chemically induced genomic instability. Mutat Res. 2003;532:245-253.
30 Morgan SE, Kastan MB. P53 and ATM: cell cycle, cell death, and cancer. Adv Cancer Res. 1997;71:1-25.
31 Polyak K, Xia Y, Zweier JL, et al. A model for p53-induced apoptosis. Nature. 1997;389:300-305.
32 Schmitt CA, Fridman JS, Yang M, et al. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002;1:289-298.
33 Komarova EA, Christov K, Faerman AI, Gudkov AV. Different impact of p53 and p21 on the radiation response of mouse tissues. Oncogene. 2000;19:3791-3798.
34 Fei P, El-Deiry WS. P53 and radiation responses. Oncogene. 2003;22:5774-5783.
35 Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell. 1997;88:315-321.
36 Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030-9040.
37 Wahl GM, Carr AM. The evolution of diverse biological responses to DNA damage. Insights from yeast and p53. Nat Cell Biol. 2001;3:E277-286.
38 Feki A, Irminger-Finger I. Mutational spectrum of p53 mutations in primary breast and ovarian tumors. Crit Rev Oncol Hematol. 2004;52:103-116.
39 Munro AJ, Lain S, Lane DP. P53 abnormalities and outcomes in colorectal cancer. A systematic review. Br J Cancer. 2005;92:434-444.
40 Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177-202.
41 Ting NS, Lee WH. The DNA double-strand break response pathway. Becoming more brcaish than ever. DNA Repair (Amst). 2004;3:935-944.
42 Morgan SE, Lovly C, Pandita TK, et al. Fragments of ATM which have dominant-negative or complementing activity. Mol Cell Biol. 1997;17:2020-2029.
43 Zhang N, Chen P, Khanna KK, et al. Isolation of full-length ATM cdna and correction of the ataxia-telangiectasia cellular phenotype. Proc Natl Acad Sci U S A. 1997;94:8021-8026.
44 Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol. 1999;119:1-16.
45 Robert F, Hardy S, Nagy Z, et al. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double-strand break repair. Mol Cell Biol. 2006;26:402-412.
46 Tamburini BA, Tyler JK. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol. 2005;25:4903-4913.
47 Shim EY, Hong SJ, Oum JH, et al. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol. 2007;27:1602-1613.
48 Shim EY, Ma JL, Oum JH, et al. The yeast chromatin remodeler RSC complex facilitates end joining repair of DNA double-strand breaks. Mol Cell Biol. 2005;25:3934-3944.
49 Morrison AJ, Highland J, Krogan NJ, et al. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767-775.
50 Morrison AJ, Shen X. DNA repair in the context of chromatin. Cell Cycle. 2005;4:568-571.
51 Ward JF. Complexity of damage produced by ionizing radiation. Cold Spring Harb Symp Quant Biol. 2000;65:377-382.
52 Leadon SA. Repair of DNA damage produced by ionizing radiation: A minireview. Semin Radiat Oncol. 1996;6:295-305.
53 Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst). 2004;3:781-796.
54 Collis SJ, Deweese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24:949-961.
55 Lisby M, Rothstein R. DNA repair. Keeping it together. Curr Biol. 2004;14:R994-996.
56 Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst). 2004;3:909-918.
57 Abraham RT. PI 3-kinase related kinases. ‘Big’ players in stress-induced signaling pathways. DNA Repair (Amst). 2004;3:883-887.
58 Jackson SP, Jeggo PA. DNA double-strand break repair and V(D)J recombination: involvement of DNA-PK. Trends Biochem Sci. 1995;20:412-415.
59 Maser RS, Monsen KJ, Nelms BE, Petrini JH. Hmre11 and hrad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol. 1997;17:6087-6096.
60 Petrini JH. S-phase functions of the Mre11 complex. Cold Spring Harb Symp Quant Biol. 2000;65:405-411.
61 Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458-462.
62 Wray J, Liu J, Nickoloff JA, Shen Z. Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress. Cancer Res. 2008;68:2699-2707.
63 Scully R, Xie A, Nagaraju G. Molecular functions of BRCA1 in the DNA damage response. Cancer Biol Ther. 2004;3:521-527.
64 Yuan SS, Chang HL, Lee EY. Ionizing radiation-induced Rad51 nuclear focus formation is cell cycle-regulated and defective in both ATM(-/-) and c-Abl(-/-) cells. Mutat Res. 2003;525:85-92.
65 Danial NN, Korsmeyer SJ. Cell death. Critical control points. Cell. 2004;116:205-219.
66 Chen W, Kang J, Xia J, et al. P53-related apoptosis resistance and tumor suppression activity in UVB-induced premature senescent human skin fibroblasts. Int J Mol Med. 2008;21:645-653.
67 Fujita K, Mondal AM, Horikawa I, et al. P53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat Cell Biol. 2009;11:1135-1142.
68 Naka K, Tachibana A, Ikeda K, Motoyama N. Stress-induced premature senescence in htert-expressing ataxia telangiectasia fibroblasts. J Biol Chem. 2004;279:2030-2037.
69 Schmidt-Ullrich RK, Contessa JN, Lammering G, et al. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855-5865.
70 Stevenson MA, Pollock SS, Coleman CN, Calderwood SK. X-irradiation, phorbol esters, and H2O2 stimulate mitogen-activated protein kinase activity in NIH-3T3 cells through the formation of reactive oxygen intermediates. Cancer Res. 1994;54:12-15.
71 Zhang HM, Li L, Papadopoulou N, et al. Mitogen-induced recruitment of ERK and MSK to SRE promoter complexes by ternary complex factor Elk-1. Nucleic Acids Res. 2008;36:2594-2607.
72 Lammering G, Hewit TH, Valerie K, et al. Anti-erbb receptor strategy as a gene therapeutic intervention to improve radiotherapy in malignant human tumours. Int J Radiat Biol. 2003;79:561-568.
73 Chen Q, Olashaw N, Wu J. Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J Biol Chem. 1995;270:28499-28502.
74 Schmidt-Ullrich RK, Dent P, Grant S, et al. Signal transduction and cellular radiation responses. Radiat Res. 2000;153:245-257.
75 Sharp PA. The centrality of RNA. Cell. 2009;136:577-580.
76 Ventura A, Jacks T. Micrornas and cancer. Short rnas go a long way. Cell. 2009;136:586-591.
77 Weidhaas JB, Babar I, Nallur SM, et al. Micrornas as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111-11116.
78 Mercer TR, Dinger ME, Mattick JS. Long non-coding rnas. Insights into functions. Nat Rev Genet. 2009;10:155-159.
79 Jaklevic B, Uyetake L, Wichmann A, et al. Modulation of ionizing radiation-induced apoptosis by bantam microrna in Drosophila. Dev Biol. 2008;320:122-130.
80 Gullino PM. The internal milieu of tumors. Prog Exp Tumor Res. 1966;8:1-25.
81 Gullino PM. Tumor pathophysiology. Rhe perfusion model. Antibiot Chemother. 1980;28:35-42.
82 Weinmann M, Welz S, Bamberg M. Hypoxic radiosensitizers and hypoxic cytotoxins in radiation oncology. Curr Med Chem Anti-Cancer Agents. 2003;3:364-374.
83 Hockel M, Schlenger K, Aral B, et al. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509-4515.
84 Vaupel P, Mayer A, Hockel M. Tumor hypoxia and malignant progression. Methods Enzymol. 2004;381:335-354.
85 Bindra RS, Glazer PM. Genetic instability and the tumor microenvironment. Towards the concept of microenvironment-induced mutagenesis. Mutat Res. 2005;569:75-85.
86 Pugh CW, Ratcliffe PJ. The von Hippel-Lindau tumor suppressor, hypoxia-inducible factor-1 (HIF-1) degradation, and cancer pathogenesis. Semin Cancer Biol. 2003;13:83-89.
87 Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med. 2002;8:S62-S67.
88 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425-437.
89 Folkman J. Angiogenesis and apoptosis. Semin Cancer Biol. 2003;13:159-167.
90 Gupta MK, Qin RY. Mechanism and its regulation of tumor-induced angiogenesis. World J Gastroenterol. 2003;9:1144-1155.
91 Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15-18.
92 Chan LW, Camphausen K. Angiogenic tumor markers, antiangiogenic agents and radiation therapy. Expert Rev Anticancer Ther. 2003;3:357-366.
93 Koukourakis MI. Tumour angiogenesis and response to radiotherapy. Anticancer Res. 2001;21:4285-4300.
94 Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin Cancer Res. 1997;3:1149-1156.
95 Dudek AZ, Zwolak P, Jasinski P, et al. Protein kinase C-beta inhibitor enzastaurin (LY317615.HCI) enhances radiation control of murine breast cancer in an orthotopic model of bone metastasis. Invest New Drugs. 2008;26:13-24.
96 Dutta PR, Maity A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007;254:165-177.
97 Fujiwara K, Iwado E, Mills GB, et al. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int J Oncol. 2007;31:753-760.
98 Gustafson DL, Frederick B, Merz AL, Raben D. Dose scheduling of the dual VEGFR and EGFR tyrosine kinase inhibitor vandetanib (ZD6474, Zactima) in combination with radiotherapy in EGFR-positive and EGFR-null human head and neck tumor xenografts. Cancer Chemother Pharmacol. 2008;61:179-188.
99 Jaboin JJ, Shinohara ET, Moretti L, et al. The role of mtor inhibition in augmenting radiation induced autophagy. Technol Cancer Res Treat. 2007;6:443-447.
100 Park JK, Jung HY, Park SH, et al. Combination of PTEN and gamma-ionizing radiation enhances cell death and G(2)/M arrest through regulation of AKT activity and p21 induction in non-small-cell lung cancer cells. Int J Radiat Oncol Biol Phys. 2008;70:1552-1560.
101 Kubota N, Okada S, Inada T, et al. Wortmannin sensitizes human glioblastoma cell lines carrying mutant and wild type TP53 gene to radiation. Cancer Lett. 2000;161:141-147.
102 Losada R, Rivero MT, Slijepcevic P, et al. Effect of Wortmannin on the repair profiles of DNA double-strand breaks in the whole genome and in interstitial telomeric sequences of Chinese hamster cells. Mutat Res. 2005;570:119-128.
103 Millington M, Arndt A, Boyd M, et al. Towards a clinically relevant lentiviral transduction protocol for primary human CD34 hematopoietic stem/progenitor cells. PLoS ONE. 2009;4:e6461.
104 Coleman CN. Chemical sensitizers and protectors. Int J Radiat Oncol Biol Phys. 1998;42:781-783.
105 Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-447.
106 Rosen EM, Fan S, Rockwell S, Goldberg ID. The molecular and cellular basis of radiosensitivity. Implications for understanding how normal tissues and tumors respond to therapeutic radiation. Cancer Invest. 1999;17:56-72.
107 Epperly MW, Guo H, Shields D, et al. Correlation of ionizing irradiation-induced late pulmonary fibrosis with long-term bone marrow culture fibroblast progenitor cell biology in mice homozygous deletion recombinant negative for endothelial cell adhesion molecules. In Vivo. 2004;18:1-14.
108 Robbins ME, Zhao W. Chronic oxidative stress and radiation-induced late normal tissue injury. A review. Int J Radiat Biol. 2004;80:251-259.
109 Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565-576.
110 Piret B, Schoonbroodt S, Piette J. The ATM protein is required for sustained activation of NF-kappaB following DNA damage. Oncogene. 1999;18:2261-2271.
111 Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81-S96.
112 Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863-14868.
113 Peterson BK, Hare EE, Iyer VN, et al. Big genomes facilitate the comparative identification of regulatory elements. PLoS ONE. 2009;4:e4688.
114 Scherf U, Ross DT, Waltham M, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24:236-244.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 2 Molecular and Cellular Biology
The past decade has seen a revolution in two areas of cellular and molecular biology closely related to radiation therapy. The pathways involved in response to genomic stress, including mechanisms for sensing DNA damage and responding to cell cycle changes and DNA repair, have been elucidated by studies of model organisms, particularly the yeast Saccharomyces cerevisiae.1,2 In the second major area, understanding pathways of programmed cell death (PCD), which may play a major role in treatment response, studies of Caenorhabditis elegans were the most informative in demonstrating the significance of individual death genes.3 The challenge is to combine these two areas of knowledge to provide a rational understanding of how tumor cells can be made to enter the death pathways after processing DNA damage using the genomic stress response. Considerable technologic improvements also have enabled significant progress in translational work, including rational, in silico drug design and high-throughput screening of chemical libraries, which may permit the application of advances in molecular biologic knowledge to the development of drugs for use in combination with radiation therapy.
Essential Steps in Tumor Progression
Much evidence suggests that tumors are formed by stepwise progression of cells from a minimally altered state, where they are able to grow and form nodules or polyps (e.g., solid tumors) to a state of maximal transformation, characterized by multiply deviated cells that are capable of unlimited growth, manipulation of their local environment, invasion of surrounding tissues, and escape into the circulation to establish new colonies of secondary tumors, or metastases.4 Such phenotypic and genotypic progression involves an array of molecular and morphologic changes. In their landmark review, Hanahan and Weinberg5 suggested organizing these traits into six essential alterations in cell physiology: self-sufficiency in growth signals, insensitivity to growth inhibition, evasion of PCD, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis. Although the initial steps are believed to occur earlier in tumor progression, the exact order of occurrence varies among different types of malignant tumors, and some phenotypic transformations appear to require more than one molecular change.6,7 The occurrence in tumor cells of this series of radical, primarily genetic alterations in physiology is in part the result of an evolving instability of the tumor genome due to a breakdown in the DNA repair pathways that accompanies tumor progression.
In addition to progressing and acquiring enhanced malignant capabilities, most human tumors undergo further selections through exposure to various forms of cytotoxic therapy such as irradiation and chemotherapy, leading to the development of resistant phenotypes in the cells that survive the toxic insult.8 However, a new paradigm is emerging that may challenge our views of tumorigenisis. This is the concept of the cancer stem cell.9 According to this theory, tumors contain a small population of stem cells that divide slowly and give rise to more “differentiated” progeny that form the bulk of tumors. Tumor eradication would therefore involve killing this small, slowly dividing, and often treatment-resistant population. Studies in this area could revolutionize the cellular and molecular underpinnings of radiation biology.
All cancer treatments are aimed at reversing the hallmarks of neoplastic transformation and tipping the balance toward tumor cell death in both the bulk population and the stem cell fraction. Ionizing radiation kills cells almost exclusively through the generation of DNA double-strand breaks (DSBs).10 The evolving modification of the cellular phenotype during tumorigenesis leads to the development of resistance to therapy. Our major task is to explore the links among DSB response pathways, engagement of cell death mechanisms, and the outcome of radiation therapy.
Radiation Therapy and Radiation Biology
Ionizing radiation has long been known to be a potent modality in cancer therapy, killing cells and leading to tumor regression.11 Killing by ionizing radiation involves the oxygen-dependent generation of free radicals and subsequent damage to multiple molecular structures within the cell.10 The generation of DSBs is the dominant mechanism of direct cellular lethality. Indeed, it has been estimated that the existence of even one unrepaired DSB can lead to cell death.12
Within whole organisms, direct and indirect forms of lethality occur. Radiation can cause the death of hematopoietic stem cells in the bone marrow and stem cells in crypts of the small intestine, which leads to secondary damage caused by immunosuppression and infection, as the gastrointestinal lining is eroded.13,14 The challenge facing radiation therapy is to enhance cell killing within tumors while avoiding dose-related complications in organs containing rapidly renewing stem cell populations. Early studies using the clonogenic cell survival assay established the notion of optimizing radiobiologic parameters such as dose, fractionation, and cell cycle inhibition as a means of inhibiting reproductive cell death in critical normal tissues.15 The clonogenic cell survival assay is a useful measure of cell inactivation because the effects of ionizing radiation seen in vitro often mirror the responses of tumors in vivo.10 However, a weakness in the informative power of the method is that it does not allow for discrimination or elucidation of the various mechanisms leading to cell death. That kind of detailed information is essential for the successful combination of radiation therapy with other modalities, which may result in sensitization or protection, or both.
Irradiation activates a number of pathways that mediate reproductive death, including various forms of PCD (e.g., apoptosis, autophagy), replicative senescence, and necrosis, the default pathway that often dominates when other types of cell death are inhibited.16,17 Susceptibility to each of these forms of cell death changes over the course of tumor progression, and the tumor cell response to ionizing radiation is likely to reflect the aggregate of genetic changes that accompany tumor progression. The task of modern radiation biology is to examine mechanisms by which ionizing radiation gives rise to unrepaired DSBs and how such damage is coupled to the pathways of cell killing within the moving target of the evolving cancer cell.
DNA Double-Strand Break Response
All cells have evolved to live in an environment that is more or less mutagenic, and have evolved responses to permit survival when the genome is damaged.18 DNA may be damaged during the normal processes of DNA replication and segregation. Within tumor cells, these initial changes become progressively altered as cells compete for survival in the tumor milieu and are subjected to selection by various forms of cytotoxic therapy.19 The response to DSBs involves two principal components: arrest of cell proliferation to prevent replication and segregation of the damaged DNA and, when possible, repair of the lesion.19 A third component in the DSB response in multicellular organisms may be altruistic entry of irreversibly damaged cells into the PCD pathways.20,21 The latter effect is the desired response to cytotoxic therapy.
Mechanisms of Cell Cycle Arrest: The Cell Cycle and Checkpoints
The cell cycle is a series of molecular programs that read out in an invariant order leading to duplication of the genome and other cellular components and permits division into two cells, each with a full set of chromosomes and the organelles required for life.22 Orderly, high-fidelity, complete duplication of the genome is required for cell cycle progression. This is achieved through a surveillance system that detects DNA damage or unreplicated DNA and imposes cell cycle arrest at designated checkpoints, blocking the forward progress of the cell cycle engine (Fig. 2-1). Because ionizing radiation produces severe damage to DNA, checkpoints are essential for facilitating repair and subsequent cell survival after radiation exposure, and provide an ultimate target for strategies aimed at sensitizing cells to radiation therapy.
Much accumulated evidence indicates that the cell cycle is driven by sequential activation of a series of protein kinases that determine the order and rate of the metabolic events required in each stage. These cell division kinases, or cyclin-dependent kinases (CDKs) are activated by binding proteins originally designated as cell division cycle (CDC) molecules, now known as cyclins.22 The complexes between cyclins and CDK molecules are master regulators that determine the rates of the individual component reactions that regulate each stage of the cell cycle.23 Phosphorylation of cell cycle effector proteins by the cyclin-CDK complexes acts as a switch, turning them on in orderly fashion. In mammalian cells, many cyclins regulate individual stages of the cell cycle, including G1 cyclins (cyclins D and E), S phase-specific cyclins, and G2-specific cyclins (cyclins A and B), which associate, respectively, with G1-specific (CDK2, CDK4, and CDK6), S-specific, and G2-specific (CDC2) cyclin-dependent kinases. The events of G1 phase (i.e., synthesis of enzymes and other molecules involved in DNA replication), S phase (i.e., DNA replication), and G2/M phase (i.e., chromosome condensation, disappearance of the nuclear envelope, and mitosis) are regulated by the transient accumulation and subsequent degradation of the cyclins.22,23,24 Switching off involves the targeted degradation of the cyclins by a protein destruction machine called the proteasome.24 Targeted destruction of the cyclins after they have carried out their function ensures unidirectional, irreversible progression around the cell cycle.23 The timing of the various cell cycle phases under normal conditions is related to the time required for cyclin-CDK complexes to reach appropriate, critical concentrations and for the phosphorylation of key substrates.23
Irradiation leads to prolongation of the cell cycle and arrests in G1, G2, and S phases because of checkpoint activation25 (see Fig. 2-1). Although this phenomenon was observed many years ago, it has only recently been understood at the biochemical and genetic levels. Arrest of cells at the G1 checkpoint has been particularly well studied (see Fig. 2-1A). G1 arrest is caused by the accumulation of a CDK inhibitory protein, CDKN1A (formerly designated p21 or Cip1), and this prevents key events that are required for transit through G1, including phosphorylation of the retinoblastoma (RB1) protein and activation of the E2F transcription factors (e.g., E2F1, E2F2, E2F4, E2F6) necessary for accumulation of enzymes required to traverse S phase.26,27 CDKN1A is induced at the transcriptional level by TP53 (also known as p53), which accumulates in irradiated cells and binds to the CDKN1A promoter, thereby activating transcription. TP53 accumulation depends on the activation of a DSB sensor molecule, the protein kinase ATM (ataxia-telangiectasia, mutated).26,27 The exact mechanism involved in sensing DSBs and activation of ATM is unclear, although it involves ATM autophosphorylation.28
The common mechanism for regulating the G1 and G2 checkpoints is inhibition of the CDK components of the interphase cell cycle engine and a block to phosphorylation and activation of effector molecules23,27 (see Fig. 2-1). A variation on this theme is seen at the mitotic spindle checkpoint. Progression through mitosis requires switching off CDK activity through targeted degradation of cyclin B.29 Arrest in M phase, when the mitotic spindle is compromised, involves the TP53-dependent stabilization of cyclin B. The mitotic spindle checkpoint guarantees that replication is followed by cell division, ensuring prevention of aneuploidy. Not surprisingly, the polyploidy nature of many tumor cells is related to TP53 inactivation.29
Most evidence indicates that ATM (and its homologs in other organisms) functions as a sensor for DNA damage through a complex series of interactions with the damaged DNA. This ultimately leads to ATM activation and downstream signaling cascades involving the kinases CHK1 and CHK2 (also called CHEK1 and CHEK2), which couple to the cell cycle engine at the level of individual cyclin-CDK complex molecules1,2,19,27,30 (see Fig. 2-1B). The molecular details of these processes are evolving rapidly, and referenced reviews can provide more detailed insight into the mechanisms of DNA damage, sensing, and checkpoint engagement.
TP53: Cellular Triage after Ionizing Radiation Exposure
TP53 is a nuclear phosphoprotein with sequence-specific DNA binding activity. It can function as both a transcriptional activator and repressor and plays a triage role in deciding whether to undergo cell cycle arrest and repair or to enter the pathways of PCD or replicative senescence31,32 (Fig. 2-2). When cells are exposed to ionizing radiation or chemotherapeutic agents, the levels of wild-type TP53 protein are increased. TP53 then transcriptionally activates a number of genes, most notably CDKN1A, the CDK inhibitor that mediates many of the properties of TP53.33 In addition to G1 arrest, genotoxic stress and ionizing radiation can induce TP53-dependent PCD pathways, including caspase-dependent apoptosis.33 TP53 can transcriptionally activate the proapoptotic BAX gene, induce synthesis of JUN kinase, and repress transcription of the antiapoptotic gene BCL2, suggesting that TP53 is a central mediator of PCD processes.31,34
DNA damage in the presence of wild-type TP53 causes G1 arrest, followed by a period of DNA repair; if the damage is too great to be easily repaired, the cell is eliminated through TP53-dependent PCD pathways. However, approximately 50% of human tumors possess inactivating mutations in the TP53 gene.35 TP53 becomes inactivated in many tumors because of selection against its pro-apoptotic properties. The resultant loss of its central function as “sentinel of the genome” may be a type of collateral damage incurred in cells due to the selection advantage for survival accruing from the loss of TP53 apoptotic function.32,36 The loss of TP53 function is linked to poorer prognosis in malignant tumors such as lung, breast, colorectal, and hematopoietic tumors.37 Many tumor cell lines containing mutant TP53, including breast, glioma, and lymphoma cell lines, are more resistant to therapy than their wild-type TP53 counterparts.38,39
The loss of wild-type TP53 function in human malignant disease may be a key step in the progression of human cancer, and the TP53 status of cells may control the outcome of many tumor types in response to chemotherapy or radiation therapy.36 Although most of the other DSB response genes have molecular equivalents in yeast, TP53 does not. The TP53 gene appears to be required to determine the fate of damaged cells, which in mammalian cells may be PCD, a sacrifice that contributes to the well-being of the whole organism. TP53 monitors the degree of DNA damage and acts as a master switch, moving the cells from a state of cycle arrest and DNA repair to death or senescence pathways. Loss of TP53 in tumors compromises this critical surveillance and triage function. With the loss of this critical decision-point molecule, damaged and mutated tumor cells may survive to generate new and more malignant phenotypes (see Fig. 2-2).
ATM Gene: Master Regulator of the DNA Double-Strand Break Response
One of the many defects in ataxia-telangiectasia (AT) cells is increased chromosomal instability. Exposure to ionizing radiation also produces an increased number of chromosomal aberrations in AT cells compared with normal cells.40,41 This effect and the sequence similarity between ATM and the DNA repair protein DNA-PK suggested that AT cells might have deficient DNA repair. However, most studies have shown that AT cells do not have gross abnormalities in their ability to repair DNA damage. In general, it does not appear that the radiosensitivity of AT cells is caused by faulty DNA repair; it more likely results from an inability to detect the presence of DNA damage. Exposure to ionizing radiation causes normal cells to delay at the G1/S and G2/M transition phases of the cell cycle, and these checkpoints are thought to allow the cells to repair DNA damage before DNA synthesis or mitosis occurs.27 Both checkpoints are absent in AT cells, suggesting clues to the function of ATM.27
Although ATM and TP53 cooperate in radiation-induced apoptosis, there are ATM-independent pathways for the induction of TP53-dependent apoptosis, and many of the downstream effects of ATM are independent of TP53.27,28 Wild-type and knockout mice have been evaluated for acute and late toxicity after whole-body exposure to ionizing radiation. The ATM– and ATM/TP53-knockout mice had similar severe toxicity profiles, suggesting that TP53 does not play a role in acute radiation toxicity. Both TP53- and ATM-knockout mice preferentially developed lymphoid tumors, whereas ATM/TP53 double knockouts had an accelerated time to tumor formation and a broader spectrum of tumor types. Analysis of the acquired tumors in ATM-null/TP53-heterozygous mice revealed that three of seven had loss of the remaining TP53 allele. These studies showed that ATM and TP53 interact in a complex manner that is specific to cell type and outcome. This interaction most likely relies on a variety of other pathways (discussed later) and will require much additional work before a complete understanding of the ATM/TP53 relationship is obtained.
With isolation of the ATM gene it became possible to attempt correction of the cellular defects of the AT phenotype using gene transduction techniques. Several groups have reported that the introduction of ATM into AT cells resulted in reversal of AT defects.27,41 The transfection of full-length ATM into AT cells reversed DSBs, restored normal sensitivity to ionizing radiation, and decreased the number of chromosomal abnormalities. Transfection of full-length ATM also reversed the defective activation of CDKN1A (p21) and JUN kinase in response to ionizing radiation.42,43
Histones and Chromatin Structure
Native DNA exists in the cell in the form of chromatin complexed with a family of proteins called histones. Histones are involved in packaging DNA in the nucleus into a compact form. For gene transcription or DNA repair to occur, the histones are altered by post-translational modifications, most notably acetylation, that permit decondensation and access of factors to the DNA.44 The histone acetylase TIP60 is important both in this process and in the modification of ATM itself.26,45 For effective DNA repair, acetylation of histones by histone acetylases must occur to permit access of repair proteins to the sites of DSBs.46 DNA is then remodeled by ATP-dependent remodeling enzymes to permit repair molecules such as Mre11, RAD50, and NBS1 to access sites of DSBs.47,48 A novel histone, H2AX, found in low concentrations on chromatin, has been shown to play a crucial signaling role in the response to genomic stresses such as ionizing radiation. H2AX phosphorylation is one of the earliest events occurring after exposure to ionizing radiation, and ATM-dependent phosphorylation of H2AX may be involved in signaling to cell cycle checkpoints and DNA repair enzymes involved in recombination repair.49,50
Mechanisms of DNA Repair after Ionizing Radiation
Maintaining undamaged DNA is essential to cell survival. Cells have therefore evolved a wide range of mechanisms to halt cell cycle progression, survey DNA, and repair damage. Radiation causes a number of types of damage either by direct interaction with DNA or indirectly through effects on nearby water molecules and free radical generation. Types of damage include DNA base damage, damage to the deoxyribose sugar backbone, and physical breaks in one or both strands of the DNA. DNA damage induced by ionizing radiation tends to be clustered so that there is more than one damaged site in proximity along the double helix, known as locally multiply damaged sites.51
Among the less catastrophic forms of DNA damage induced by ionizing radiation are the singly damaged bases, which can be repaired by base excision repair. This is a process by which the damaged base is recognized and removed by an N-glycosylase, the apurinic or apyrimidinic (AP) site is cleaved by an AP endonuclease, a patch of DNA is excised, DNA is then resynthesized using the other strand as the template, and the repaired strand is ligated.18,52 In settings in which the base damage is not recognized by the N-glycosylase, another mechanism for repair exists, called nucleotide excision repair. In a manner somewhat similar to base excision repair, a damaged section of DNA is removed by incision and excision, a patch is resynthesized using the remaining strand, and the repaired strand is then ligated. Although no naturally occurring mammalian mutants have been identified that are defective in base excision repair, there are a number of different nucleotide excision repair mutants, which are exemplified by xeroderma pigmentosum and Cockayne’s syndrome.18 They are characterized by abnormalities in the repair of damage caused by ultraviolet light, although the clinical spectrum of these repair-deficiency syndromes varies. The abnormal genes are identified by finding the human gene that corrects rodent cell defects, called excision repair cross-complementing (ERCC) groups. For a comprehensive review on repair of single-strand breaks we recommend the article by Friedberg, Walker, and Siede.18
DNA Strand Breaks
Cellular lethality after radiation involves generation of DSBs.53,54,55 Misjoined or unrepaired DSBs lead to chromosome deletions, translocations, and acentric or dicentrics, with lethal consequences for the cell. As with the excision repair defects (e.g., ERCCs), there are several x-ray repair defects in the x-ray cross-complementation (XRCC) groups involved in the repair of DSBs. DNA single-strand repair is carried out in a manner similar to base damage repair, with the undamaged DNA strand serving as a template. DSB repair is more complicated because there is no adjacent, undamaged template available to form a template for repair of the broken strands. The ends of the broken DNA must be protected, and the damaged site must be reconstituted by the processes of homologous recombination and nonhomologous end joining (NHEJ).
DNA DSB repair has much in common with the recombinatorial processes involved in immunoglobulin and T-cell receptor gene rearrangement in the immune response. The XRCC groups that have been identified and are involved in DNA DSB repair include genes that produce DNA end-binding proteins XRCC6 (formerly designated KU70 or Ku-70) and XRCC5 (formerly designated KU80 or Ku-80) and a DNA-dependent protein kinase (DNA-PK). Defects in DNA repair genes are seen in severe combined immunodeficiency (SCID) mice, indicating the importance of recombination in the restoration of DNA integrity after DSBs and in the immune response.18,53
Double-Strand Break Damage and Repair
Experiments in yeast and human cells indicate that a single unrepaired DSB leads to cell death.53,55 However, such cells can detect such DSBs and repair them. DSB repair would be predicted to require three functional components: (1) a mechanism for detecting and gauging DNA damage, (2) a signal transduction system, and (3) an effector system for DNA repair. These components are discussed in the following paragraphs, commencing with the repair component. DSBs can be repaired by a number of mechanisms, but the most prevalent are homologous recombination and NHEJ. Human cells appear to differ from yeast in that while homologous recombination predominates in yeast, the opposite is true in mammalian cells.53,55
Genetic analysis has revealed the existence of a large number of genes that regulate DSB repair. One essential gene, involved in resistance to cell killing by ionizing radiation and the repair of DSB, is XRCC5, which encodes XRCC5, a protein that binds with high affinity to the ends of the double strands1,18 (Fig. 2-3A). XRCC5 functions in normal cellular processes that require DSB rejoining, most notably V(D)J rejoining in the immunoglobulin and T-cell receptor genes of immature B and T lymphocytes. XRCC5 exists in cells in a heterodimeric complex with XRCC6, the product of the XRCC6 gene. The XRCC5/XRCC6 heterodimer is required for the end-binding and repair functions.56 The XRCC (formerly designated KU) proteins recognize the ends of DSBs and protect them from further degradation before the onset of end-joining reactions required for DSB repair. An associated protein in the complex with important functions in DNA repair is DNA-PK, the product of the XRCC7 gene.56
