[level-membership-for-cardiovascular-category]
Chapter 3 Molecular and Cellular Basis of Cardiac Electrophysiology
Basic Concepts
Membrane Potential and Conduction
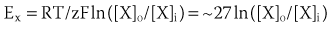
where R is the gas constant, T is temperature, F is Faraday’s constant, and z is the valence of the ionic species. If the resting cardiac myocyte is assumed to have an intracellular [K+] of ~150 mmol/L and an extracellular [K+] of 4 mmol/L, then the Nernst potential for K+ (EK) is roughly –90 mV. The Nernst potential represents the voltage at which the osmotic tendency for K+ to flow across the membrane is exactly balanced by the electrical tendency to flow in the opposite direction, which results in a net zero ionic flux and thus no net current flow. Accordingly, the K+ Nernst potential is sometimes referred to as the zero current potential, at which no K+ current would flow through open K channels. By contrast, the Nernst potential for Na+ in the resting cell is approximately +60 mV, indicating that if a pathway for Na+ to enter the cell were present, Na+ would enter the cell to move the membrane voltage (mV) toward the Nernst potential for Na+. Indeed, this is precisely what happens when Na channels are open and initiate phase 0 of the action potential. The Nernst potential for potassium is, in fact, very close to the resting membrane potential of the ventricular myocyte, indicating that the resting heart cell membrane is highly permeable to K+. Actual resting potentials are less negative than EK due to small conductances of other ionic species with less negative Nernst potentials. In the course of the cardiac cycle, the cell membrane becomes permeable to different ionic species, and these changes in permeability determine time-dependent changes in the membrane potential, with each ion striving to move the membrane voltage to its Nernst potential and inscribing regionally specific action potentials (Figure 3-1).
Passive Membrane Properties and Cable Theory
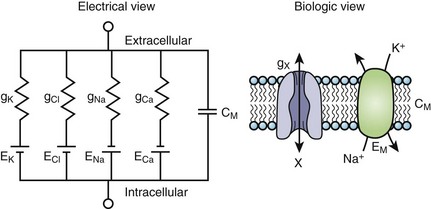
The cardiac cell membrane can be modeled as a circuit comprising variable resistors (ion channels) in parallel with a capacitor (lipid bilayer), an RC circuit (Figure 3-2). The flow of current across the membrane will alter the charge on the capacitor (and therefore the membrane potential) and change the membrane resistance. The flow of current occurs not only across but obviously along the inside and outside of the cell membrane from cell to cell as well, that is, current propagates.
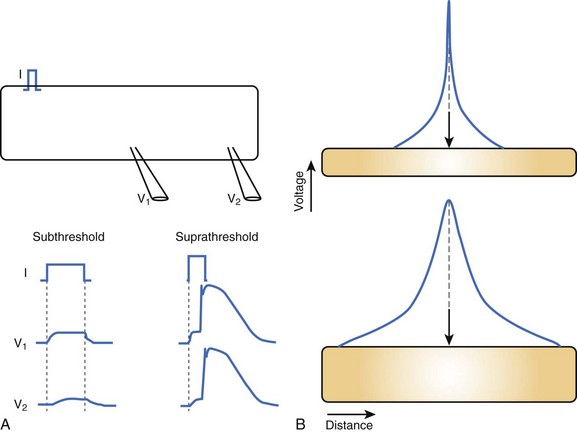
Cable theory, originally developed to understand current flow in trans-oceanic telegraphic cables, can be used to model passive current flow and propagation in a cardiac muscle fiber. In their simplest formulation, the cable equations define the distribution of voltage along a continuous, uniform cable of infinite length stimulated by a point source. The predictions of cable theory are that (1) a change in voltage exhibits a characteristic decay along the cable defined by the space constant (distance over which voltage decays to 1/e of the value at the site of injection) (Figure 3-3, A); and (2) there is an inverse relationship between resistance to current flow (both transmembrane and intercellular) and the cable diameter (Figure 3-3, B). That is, the space constant is directly proportional to the cable diameter, and thus, greater lengths of the cable are influenced by the same current injection into a thick, rather than a thin, cable.
Substantial anatomic and biophysical limitations exist when applying the cable theory description of conduction to cardiac muscle. Anatomically, the shape of the heart is complex, and at any level of integration above a single muscle fiber, it does not resemble a cable. Conduction through the myocardium is not continuous; instead, myocardial cells are connected by gap junction channels that create a non-uniformity of intercellular resistance. Macroscopic discontinuities such as fibrous tissue and blood vessels also significantly perturb the cable view of conduction in the myocardium. Finally, the cardiac cell membrane does not just comprise RC circuits; instead, when stimulated to the threshold, it will generate action potentials (see Figure 3-3, A). Despite the limitations of cable theory, it serves as the foundation for several important concepts in impulse propagation and was used to demonstrate the electrical nature of conduction in the heart.1
While this discussion implicitly treats the activation wave in one dimension, the behavior of propagating waves in the heart is more complex. The complexity can be appreciated if one considers propagation in two dimensions. In this circumstance, the shape of the wavefront is a major determinant of the efficiency of propagation. A convex wavefront, as might be observed after point stimulation, creates a large sink around a smaller activating source. This mismatch reduces conduction velocity and the safety factor for propagation. Conversely, a concave activation front produces a source-sink mismatch that favors the source; this results in a high safety factor and more rapid impulse transmission. Thus, not only do source-sink characteristics influence propagation, they also influence the curvature of the wave front (see Figure 3-4).
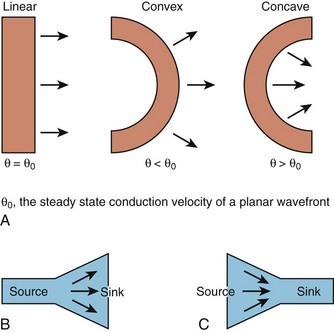
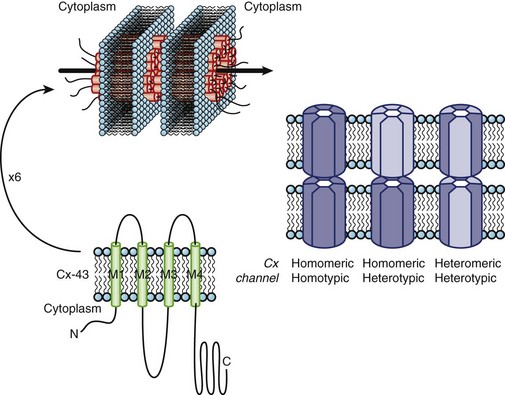
Directionally different conduction velocity is a characteristic feature of cardiac muscle known as anisotropic conduction. Anisotropic conduction has its basis in the structure of the myocyte and cardiac tissue; myocytes are rod shaped and are organized in bundles that are oriented along the long axis of the cell. Transmurally, the axes of these bundles undergo significant changes in orientation through the ventricular wall (~120 degrees maximal deviation).2 Communication among myocytes occurs via gap junction channels that are non-uniformly distributed over the surface of the heart cell, with larger numbers of channel poised to propagate the impulse longitudinally, rather than transversely, to the long axis of the muscle fiber.3 The implications of anisotropy for conduction in the longitudinal and transverse direction under pathologic conditions are controversial. In the context of uniform depression of conduction, as might exist with antiarrhythmic drug treatment, propagation in the transverse direction is preserved compared with conduction in the longitudinal direction. However, when cellular uncoupling occurs, such as in ischemia, longitudinal propagation may exhibit a higher safety factor than does transverse conduction.
Major Breakthroughs: Voltage Clamp, Molecular Cloning
A stimulus of sufficient magnitude applied to a myocyte (or any excitable cell) elicits a typical change in membrane potential known as the action potential. The ionic current basis of the action potential was confirmed and quantitatively studied using the voltage clamp developed in the middle of the twentieth century.4 Voltage clamping is a technique whereby the experimenter controls the transmembrane voltage and measures the current at that defined voltage. Much of what we know about ionic currents in myocytes comes from voltage clamp experiments and a more recently developed type of voltage clamp called the patch clamp.5 A variant of the patch clamp technique permits the measurement of ionic currents through single ion channels.
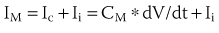
Molecular Basis of Cardiac Action Potentials
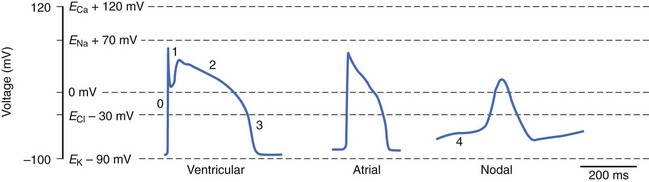
Cardiac myocytes possess a characteristically long action potential (200 to 400 ms, see Figure 3-1), compared with neurons or skeletal muscle cells (1 to 5 ms). The action potential profile is sculpted by the orchestrated activity of multiple ionic currents, each with its distinctive time- and voltage-dependent amplitudes. The currents, in turn, are carried by complex transmembrane proteins that passively conduct ions down their electrochemical gradients through selective pores (ion channels), actively transport ions against their electrochemical gradients (pumps, transporters), or electrogenically exchange ionic species (exchangers).
Action potentials in the heart are regionally distinct. The regional variability in cardiac action potentials is the result of differences in the numbers and types of ion channel proteins expressed by different cell types in the heart. Further, unique sets of ionic currents are active in pacemaking and muscle cells, and the relative contributions of these currents may vary in the same cell type in different regions of the heart.6
Ion Channels and Transporters: Molecular Building Blocks of the Action Potential
Currents that underlie the action potential are carried by complex, multi-subunit transmembrane glycoproteins called ion channels (Table 3-1). These channels open and close in response to a number of biologic stimuli, including a change in voltage, ligand binding (directly to the channel or to a G-protein–coupled receptor), and mechanical deformation. Other ion-motive transmembrane proteins such as exchangers and transporters make important contributions to cellular excitability in the heart. Ion pumps establish and maintain the ionic gradients across the cell membrane that permit current flow through ion channels. If pumps, transporters, or exchangers are not electrically neutral (e.g., 3 Na+ for 1 Ca2+), they are termed electrogenic and can further influence electrical signaling in the heart.
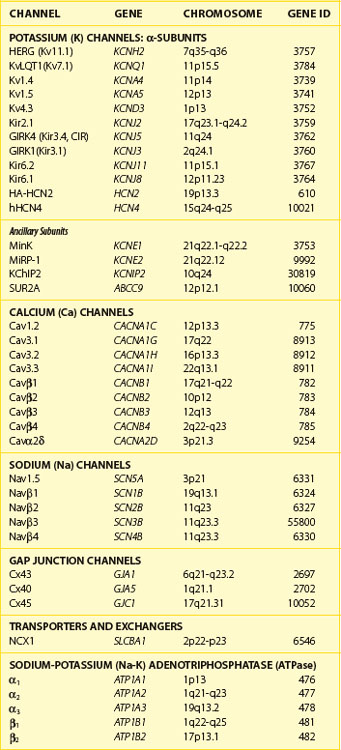
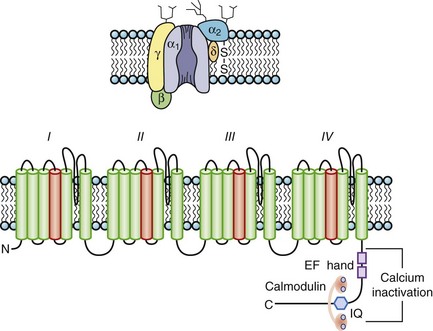
The most abundant superfamily of ion channels expressed in the heart consists of voltage-gated ion channels. Various structural themes are common to all voltage-dependent ion channels. First, the architecture is modular, consisting either of four homologous subunits (in K channels, see Figures 3-7 and 3-8) or of four internally homologous domains (in Na and Ca channels) (see Figures 3-5 and 3-6). Second, proteins wrap around a central pore (see Figure 3-8). The pore-lining (P segment) regions exhibit exquisite conservation within a given channel family of like selectivity (e.g., jellyfish, eel, fruit fly, and human Na channels have very similar P segments), but not among families with different selectivities. Third, the general strategy for activation gating (opening and closing in response to changes in the membrane voltage) is highly conserved: The fourth transmembrane segment (S4), typically studded with positively charged residues, lies within the membrane field and moves in response to depolarization, thus opening the channel. Fourth, most ion channel complexes include not only the pore-forming proteins (α-subunits) but also auxiliary subunits (e.g., β-subunits) that modify channel function.
Sodium Channels
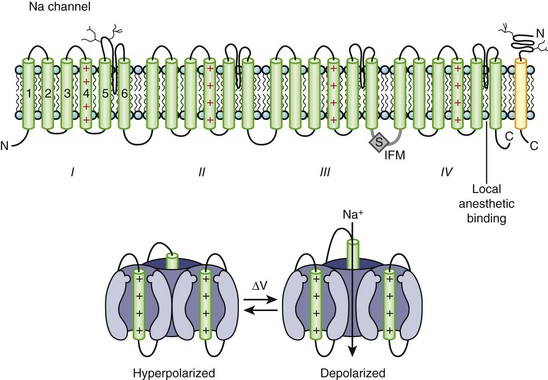
Na channels were the first ion channels to be cloned and have their sequence determined.7 In humans, more than 10 distinct Na channel genes have been cloned from excitable tissues, with striking homology to the complementary DNA (cDNA) cloned from eel electroplax. The cardiac Na channel gene (SCN5A) resides on the short arm of chromosome 3 (3p21) (see Table 3-1). The Na channel complex is composed of several subunits, but only the α-subunit is required for function. Figure 3-5 shows that the α-subunit consists of four internally homologous domains (labeled I to IV), each of which contains six transmembrane segments. The four domains fold together so as to create a central pore, whose structural constituents determine the selectivity and conductance properties of the Na channel.
One of the seminal contributions of Hodgkin and Huxley was the notion that Na channels occupy several “states” (which are now viewed as different conformations of the protein) in the process of opening (activation); yet another set of conformations is entered when the channels close during maintained depolarization (inactivation).4 The m gates that underlie activation and the h gate that mediates inactivation were postulated to have intrinsic voltage dependence and to function independently.8 While some of the implicit structural predictions of that formulation have withstood the test of time, others have not. For example, the four S4 segments are now widely acknowledged to serve as activation voltage “sensors.” In the process of activation, several charged residues in each S4 segment physically traverse the membrane (see Figure 3-5, bottom panel). The contributions of each S4 segment to activation are markedly asymmetrical; some of the charged residues play a much more prominent role than do others in “homologous” positions.9 Other studies have revealed that activation is coupled with inactivation. Indeed, the time course of current decay during maintained depolarization predominantly reflects the voltage dependence of activation, although single-channel inactivation itself does vary with voltage (particularly in cardiac Na channels). If the S4s are the sensors, where are the activation “gates” themselves? This crucial question still remains unresolved. However, according to experimental evidence, S6 is the leading contender for the physical activation gate.
Inactivation of Na channels is as arcane a process as activation. Not only is there loose coupling to activation, but there are multiple inactivation processes. One common approach to distinguishing inactivated states is to determine the rate at which they recover the ability to activate: Repriming from the traditional “fast” inactivation occurs over tens of milliseconds, while recovery from “slow” inactivation may need tens of seconds or longer. Fast inactivation is at least partly mediated by the cytoplasmic linker between domains III and IV (the crucial residues are labeled IFM in Figure 3-5), which may function as a hinged lid, docking onto a receptor formed by amino acids in the S4-S5 linkers of domains III and IV. This notion is consistent with observations that fast inactivation can be disrupted by internal proteases. Nevertheless, it is increasingly clear that mutations scattered widely throughout the channel affect inactivation gating. The structural determinants of slow inactivation are less well localized than those of fast inactivation. Mutations in the P region of domain I affect both activation gating and slow inactivation, while various widely scattered disease mutations identified in paramyotonia congenita and other skeletal myopathies suppress slow inactivation of the Na channel.
Four different β-subunits (NaVβ1 to 4, SCN1B-SCN4B) have been isolated, and all appear to be single membrane–spanning domain (type I topology) proteins with a large extracellular V-shaped immunoglobulin (Ig) fold often found in cell adhesion molecules and small carboxyl terminal cytoplasmic domain.10 The effects of the particular β-subunit on the kinetics and voltage dependence of the α-subunit vary depending on the particular β-subunit and the cell expression system. Despite the expression of β1 mRNA, using subtype-specific antisera, the functional role of β-subunits in cardiac Na currents is still being debated. Several pieces of evidence are consistent with a role for β-subunit(s) in heart cells. First, β-subunits are found in cardiac myocytes, although no NaVβ1 is found in association with the α-subunit protein from the rat heart. Second, NaVβ1 variably modulates the function of NaV1.5 in heterologous expression systems, including the sensitivity of the channel to blockade by antiarrhythmic drugs and free fatty acids. Finally, mutations in β-subunits have been directly or indirectly implicated in heritable cardiac arrhythmias.
In contrast to PKA, protein kinase C (PKC) alters the function of all of the mammalian Na channel isoforms. The PKC effect is largely attributable to phosphorylation of a highly conserved serine in the III-IV linker (see Figure 3-5). Conventional PKC isoforms reduce the maximal conductance of the channels and alter gating in an isoform-specific fashion. The macroscopic current decay of neuronal channels is uniformly slowed by PKC, which suggests destabilization of the inactivated state. Cardiac channels exhibit a hyperpolarizing shift in the steady-state availability curve consistent with an enhancement of inactivation from closed states.
Alteration of ion channel function is an important pathophysiologic mechanism of various familial diseases of muscle and brain and of inherited arrhythmias. The Na channelopathies were among the first molecularly characterized human ion channel diseases.11 Rare allelic variants in SCN5A have been linked to inherited ventricular arrhythmias,12 conduction system disease, and sudden infant death syndrome. Complex electrophysiological phenotypes have been associated with mutations in both α- and β-subunits of Na channels. Rare variants or mutations have been associated with sudden death in women (in a population-based study) and with atrial fibrillation. More common acquired forms of long QT syndrome (LQTS), generally associated with drug ingestion, have been linked to common variants of SCN5A. Finally, disease-causing mutations in the Na channel have been associated with alterations in drug blockade. The highly variant phenotypes of these arrhythmic syndromes are, in part, explained by the variable effects of the mutations on channel subunit function or expression.
Calcium Channels: L-type
The pore-forming subunit (α1) of the calcium (Ca) channel is built on the same structural framework as the Na channel.13 As is the case with the Na channel, a number of genes encode surface membrane Ca channel α1-subunits. The predominant sarcolemmal Ca channels in the heart are the L-type (large and long-lasting) and T-type (tiny and transient) Ca channels (Table 3-2). The cardiac L-type Ca channel (CaV1.2) is a multi-subunit transmembrane protein composed of α1C (α11.2) (165 kDa), β (55 kDa), and α2 (130 kDa) to δ (32 kDa) subunits. Three genes are known to encode L-type Ca channel α1-subunits (CaVα11.x to α13.x), and the CaVα11.2 is the gene expressed in the heart (see Table 3-1). Distinct splice variants of the CaVα11.2 gene have been described, and these contribute to the diversity of the cardiac L-type Ca channel function. Similar to the α-subunit of the Na channel, the S5-S6 linkers (P segments) of the α1-subunit of the Ca channel form the ion-selective pore (Figure 3-6). However, unlike Na channels, each P segment contributes a glutamic acid to a cluster that serve to bind Ca2+ in the channel pore. The β-subunit is completely cytoplasmic and noncovalently binds to the α1C subunit, modifying its function and contributing to appropriate membrane trafficking of the channel complex. Although β2a has been proposed to be the major L-type Ca channel β-subunit, splice variants of all β-subunits β1 to β4 are expressed in the mammalian heart in a complex spatial and temporal pattern. As many as five genes have been suggested to encode the α2-δ subunit: These gene products undergo post-translational processing to produce the mature extracellular α2-subunit linked by a disulfide bond to the transmembrane δ-subunit. In heterologous expression systems, α2– to δ-subunits enhance expression of Ca channels and hasten current activation and deactivation in the presence of α1– and β-subunits.
| L-TYPE | T-TYPE | |
|---|---|---|
| Pore-forming α-subunit | α1C | α1H |
| Auxiliary subunits | β, α2-δ | ? |
| Permeability | Ba2+ > Ca2+ | Ba2+ ≅ Ca2+ |
| Activation threshold | >–30 mV | >–60 mV |
| Inactivation threshold | >–40 mV | >–90 mV |
| Inactivation | ||
| Rate | Slow | Fast |
| Calcium-dependent | Yes | No |
| Voltage-sensitive | Yes | Yes |
| Recovery | Fast | Slow |
| Localization in heart | All | Nodal > Purkinje > atria |
| Blocker sensitivity | ||
| Dihydropyridines | ++++ | + |
| Phenylalkylamines | ++++ | + |
| Benzothiazepines | ++++ | + |
| Tetralols | ++ | +++ |
| Ni2+ | + | +++ |
| Cd2+ | +++ | + |
Ba, Barium; Cd, cadmium; Ni, nickel.
Ca channels inactivate by both Ca2+-dependent inactivation (CDI) and voltage-dependent inactivation (VDI) processes. CDI and VDI are regulated by channel phosphorylation and β-subunits, which suggests shared structural mechanisms that perhaps involve the I-II domain linker. The C-terminus contains peptide sequences that bind calmodulin (CaM) and Ca2+; these are an IQ motif named for the signature amino acids (isoleucine and glutamine) in the sequence and a helix-loop-helix structural domain, referred to as an EF hand, that mediate CDI. CaM is permanently tethered to the channel complex and serves as a Ca2+ sensor for the L-type channel. The Ca2+-CaM complex facilitates the interaction of CaM with the IQ motif resulting in the occlusion of the inner mouth of the Ca channel pore and terminating the inward Ca2+ flux despite continued depolarization. As cytoplasmic Ca2+ concentration falls, calmodulin unbinds Ca2+ and the IQ motif, thus relieving Ca2+-dependent inactivation. Spatial discrimination of Ca2+ concentration by CaM may involve regions in the N-terminus of the channel. A Ca2+-binding EF hand motif in the carboxyl terminus of α1 appears to be necessary to confer Ca2+-dependent inactivation to the CaVα11.2-subunit, although not through a direct binding of Ca2+ (see Figure 3-6, bottom). The I-II interdomain linker has been demonstrated as the site of β-subunit binding as well as a critical structural determinant of VDI. Mutations in this linker have been associated with the highly arrhythmogenic Timothy syndrome.14
Calcium Channels: T-type
The other major Ca channel present in the sarcolemma of heart (and prominently in vascular smooth muscle) cells is the T-type channel. The T-type channel has a biophysical fingerprint that is distinct from that of the L-type channel, opening at more negative voltages, inactivating more rapidly, and having a lower conductance than the L-type channel (for reviews, see references 15 and 16). The distribution of the T-type current is more restricted in the heart than is that of the L-type current. The T-type current has been recorded in the SA node, AV node, atrium, and Purkinje cells but not in the normal adult ventricle (see Table 3-2). The T-type current plays a prominent role in phase 4 diastolic depolarization and the action potential upstroke of pacemaking cells. T-type currents play a role in the developing heart and in pathologic remodeling in some species, but this current has not been detected in normal or diseased human ventricular myocytes. Three genes encoding the T-type Ca current α1-subunits have been cloned. The CaVα13.1 (CACNA1G) cDNA was isolated from neuronal tissue and was the first of this new class of Ca channels cloned. The gene resides on human chromosome 17q22 (see Table 3-1), with a predicted topology similar to that of other Ca channel cDNAs.16 The gene encoding CaVα13.2 (I) expressed in human heart resides on chromosome 16p13.3. The gene encoding CaVα3.3 (CACNA1I) on chromosome 22q13 first appeared as part of the output of the Human Genome Project.17 Expression of the CaVα13 cDNAs produces currents with the biophysical hallmarks of the T-type Ca current. Interestingly, CaVα13.2 lacks a consensus β-subunit–binding motif in the I-II linker of the channel and expresses robustly without the need for other subunits in contrast to L-type channels. The T-type channel genes lack an EF hand or IQ motifs, which suggests modes of inactivation distinct from the L-type channel.
Potassium Channels
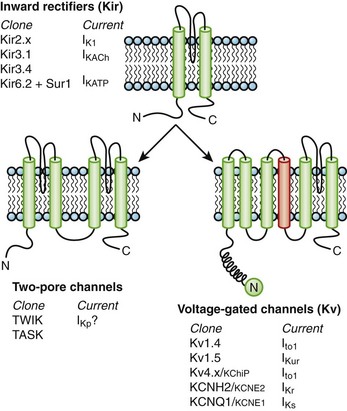
Currents through K channels are the major repolarizing currents in the heart, but the relative importance of any specific channel varies regionally in the heart. K channels are the most diverse subfamily of channel proteins comprising molecules with three distinct molecular architectures (Figure 3-7). The inward rectifier currents (IK1, IKACh, IKAdo), designated Kir, are encoded by a K channel that is evolutionarily the most primitive and comprises only two membrane-spanning repeats (analogous to S5 and S6) and a pore, or P, segment. The latter contains the K channel signature sequence (TVGYGDM) that underlies the K+-selective permeability of the channel. The first K channel gene isolated was from Drosophilia melongaster. This mutant fruit fly was called Shaker because of its response to ether anesthesia. The gene that caused the Shaker phenotype was isolated by positional cloning and encoded a voltage-dependent K (Kv) channel.18 Since the original cloning of the Shaker K channel (Kv1.x), a number of K channel genes in the same or closely related gene families have been isolated (Kv1.x to Kv11.x). The voltage-dependent K channels that have been identified in the mammalian heart are shown in Figure 3-7. The voltage-dependent K channels, which are structurally similar to a single domain of the Na channel or the Ca channel, are composed of six membrane-spanning segments, including a highly basic S4 segment. The cytoplasmic half of the S6 membrane–spanning repeat appears to mediate drug blockade of voltage-gated K channels, analogous to regions of the Na channel that bind local anesthetics. Similar to Kir channels, Kv channels must tetramerize to form the intact channel and are typically associated with ancillary subunits. Within a subfamily of K channels (e.g., Kv1.x), subunits may hetero-multimerize, but it is believed that assembly does not occur across subfamilies. It seems likely that two rounds of gene duplication generated Ca and Na channels from the less complex Kv structure. It is possible that a more straightforward gene duplication of an inward rectifier channel produced the third type of K channel, the two-pore K+-selective channel (see Figure 3-7).
The cDNAs that encode the α-subunits of the K channel are sufficient to generate K+-selective currents, but a number of ancillary subunits that modify channel function (Kvβ, KCNE, KChIP) have been identified. A family of related proteins (Kvβ1 to Kvβ3) modulates the function of Kv channels. The β-subunits bind to the amino terminus of Kv α-subunits that modify their function in an isoform-specific fashion (for review see reference 19). The crystal structure of Kvβ2 complexed with the amino terminus of Kv1.1 has been solved,20 and it has been suggested that it functions as an oxidoreductase. Indeed, Kvβ1.2 has been shown to confer oxygen sensitivity to Kv4.2 channels. A recently described, unrelated family of proteins, KChIPs, that contain Ca2+-binding EF hand motifs modulate the function of the members of the Kv4 family, which suggests the possibility that more than one type of ancillary subunit can interact with Kv4 channels. The molecular details of the interaction of KChIP and Kv4 subunits are still being studied (Figure 3-8, left).
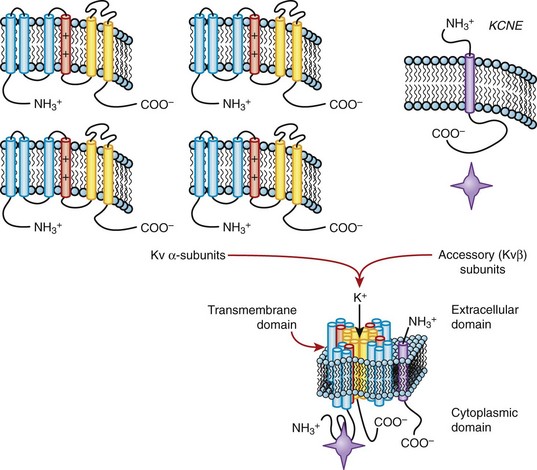
Still other K channel ancillary subunits are transmembrane proteins that are predicted to have the ability to not only alter channel gating but, in some cases, also influence channel pore properties. Most relevant to the heart are the gene products of KCNE1 (minK) and KCNE2 (MiRP-1), which are thought to coassemble with KvLQT1 (KCNQ1)21,22 and HERG (KCNH2) to form the two components of the delayed rectifier current IKs and IKr, respectively (see Figure 3-8, right, and see below). α-Subunits, which by themselves are not functional (e.g., Kv9.x), may modulate the function of other Kv α–encoded channels.
In response to a depolarizing voltage pulse, K channels, like other voltage-gated ion channels, undergo a series of conformational changes that alter function. The S4 membrane–spanning repeats are critical components of the activation gating machinery. Many K channels (like Na and Ca channels) close in the face of continued depolarization; that is, they inactivate. The molecular basis of inactivation, however, is mechanistically heterogeneous. The first type of inactivation to be understood in molecular detail in K channels validated a scheme, referred to as the ball-and-chain mechanism, proposed by Armstrong23 In an elegant series of experiments, Aldrich and coworkers demonstrated the “ball” role of the amino terminus of the Shaker K channel in the inactivation process that they called N-type (because it involves the amino terminus).24,25 After channel activation by a depolarizing stimulus, the amino terminus binds to and plugs the cytoplasmic mouth of the channel pore, thus terminating the K+ flux (see Figure 3-7, bottom). Channels that have the amino terminus removed fail to undergo this type of inactivation, but it can be restored if a peptide that resembles the amino terminal ball is added to the cytoplasm of the cell.25 A second form of inactivation (carboxyl-terminal, or C-type) involves the outer mouth of the channel pore and amino acid residues in S6 and in the P segment. It has been suggested that C-type inactivation of the channel protein resembles the closing of a camera shutter; that is, it involves constriction of the outer pore of the channel.
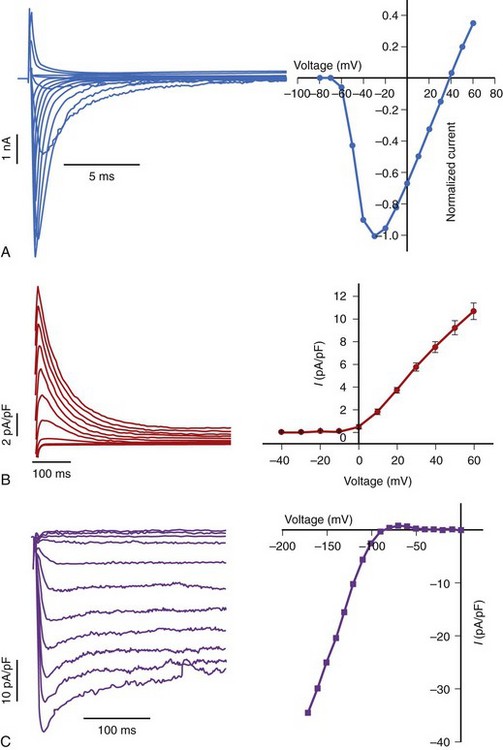
K channels serve multiple roles in the maintenance of normal cardiac electrophysiology. Of the multiple subtypes (voltage-gated, inward-rectifier, twin-pore), voltage-dependent K channels underlie both the transient outward (sometimes called A-type) current and the delayed rectifier current in the heart. The transient outward K current activates and inactivates rapidly and is a critical determinant of phase 1 repolarization of the ventricular action potential (see Figure 3-1). The two components of the transient outward current in the heart are a Ca2+-independent K current (Ito1) and a Ca2+-dependent current (Ito2). The latter is a K current in some species and a chloride (Cl) current in others. The channels that encode cardiac Ito1 not only vary among species but may vary regionally in the ventricle as well. Kv1.4 is a minor, but important, component of Ito1 in some species, including humans. However, in the human ventricle, Ito1 is primarily encoded by Kv4.3, which recovers from inactivation much faster than do homomeric Kv1.4 channels. Indeed, the Kv1.4 channel recovers so slowly (2 to 3 seconds) that it cannot be a significant component of the cardiac Ito1 at physiologic heart rates. However, it is possible that hetero-multimerization of Kv1.4 with other Kv1 family genes and/or coassembly with β-subunits could alter the kinetics of the current. Another argument against Kv1.4 as the major component of cardiac Ito1 is the insensitivity of expressed Kv1.4 to blockade by low-dose flecainide (10 µM), whereas expressed Kv4 and native cardiac Ito1 are both flecainide sensitive. The Kv4 family of genes is expressed in relative abundance in the mammalian heart—Kv4.3 in larger mammalian ventricles, such as dog and human, and Kv4.2 in rodent ventricle. Data suggest that Kv1.4 mRNA and protein are also present in mammalian ventricular myocytes and that a physiologic correlate of Kv1.4-based Ito1 may be a slowly recovering transient outward current in the subendocardium of the human ventricle.
The delayed rectifier K current (IK) plays a major role in terminating repolarization in the cells of large mammalian hearts. IK is a composite current made up of a rapid component (IKr) and a slow component (IKs). Definition of the genetics of LQTS clarified the molecular basis of both components of the delayed rectifier. MinK (KCNE1) was initially considered a “minimal K channel” that encoded a current that resembled IKs. Subsequently, positional cloning identified the disease gene in chromosome-11 linked LQTS as KvLQT126 (Kv7.1), but the current encoded by KCNQ1 was a functional orphan, not resembling any known cardiac K current. However, the co-expression of KCNQ1 and KCNE1 generated a current with a much closer resemblance to native IKs than to either of the subunits expressed alone.21,22 An alternatively spliced variant of KCNQ1 is expressed in the heart and exerts a dominant negative effect on IKs in vitro; thus, native IKs may be regulated, in part, by the extent of such alternative splicing.
Long QT genetics identified KCNQ1 as the gene underlying IKs, and HERG (Kv11.1) encoded by KCNH2 underlying IKr.27 IKr exhibits a number of unusual physiologic properties, which when disrupted (by mutations in KCNH2, by hypokalemia, or by drug blockade) disrupt normal repolarization. With depolarizations to progressively more positive potentials, activating IKr actually decreases. This “inward rectification” is a manifestation of the very rapid inactivation that HERG channels undergo once they are open. The extent of this fast inactivation increases at positive potentials and with lower extracellular K+. The latter explains the decrease in IKr (causing action potential and QT prolongation) observed in hypokalemia. Further, when the action potential enters phase 3, channels recover from inactivation, transitioning rapidly to an open (conducting) state before closing relatively slowly. Thus, as the action potential begins to repolarize, IKr increases markedly, further accelerating repolarization. HERG channels are blocked by many drugs, including methansulfonanilide drugs such as dofetilide and sotalol. As with KVLQT1, HERG may coassemble with other proteins to produce native IKr. Database mining for homologs of KCNE1 uncovered a related gene, MiRP-1 (encoded by KCNE2), in the same locus on chromosome 21 that encodes a topologically similar, small polypeptide with an extracellular amino terminus, a single transmembrane domain, and a cytoplasmic carboxy tail (see Figure 3-8). When MiRP-1 is co-expressed with HERG voltage-dependent gating, single-channel conductance, regulation by K+o, and biphasic blockade by methansulfonanilides are all modified. However, the role of MiRP-1 in native cardiac IKr remains uncertain. HERG exists in alternatively spliced forms, but the role that different splice variants play in generating the native current is uncertain. As with KCNH2 and KCNQ1, mutations in KCNE1 and KCNE2 have been linked to LQTS.
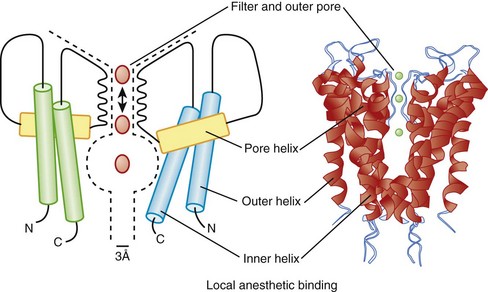
Another major class of K channel genes expressed in the heart encodes inwardly rectifying currents. The term inward rectification is used to describe the fact that these channels pass current more readily into cells than out of them (Figure 3-9). All inward rectifiers share a similar topology, with only two membrane-spanning repeats and a pore loop, and they must tetramerize to form the intact channel. In 1998, a major advance in ion channel biology occurred with the determination of the structure of a bacterial inward rectifier channel from Streptomyces lividans, called KcsA.28 The structure is remarkable in that it accounts for a number of the physical principles that underlie K+-selective permeation.29 The crystal structure demonstrated that the linker between the two membrane-spanning domains (P segments) form the outer mouth of the channel and that the K channel signature sequence forms the selectivity filter. High rates of ion flux are maintained despite the relatively avid binding of K+ due to the presence of the two K+ ions in the selectivity region that repel each other. The second membrane-spanning repeat, analogous to the S6 of Kv channels, forms much of the inner mouth of the channel, where antiarrhythmic drug binding is expected to occur (Figure 3-10). Models of other K channels have since been generated by structural or homology approaches. A KCNQ1 structural model has been proposed, based on homology to other K channel structures, and this, in turn, has been used to identify the key structural features of interactions between KCNQ1 and KCNE1.
The other inward rectifiers in the heart exhibit specialized functions, as in response to neurohormones or metabolic stress. The Kir3 family of inward rectifier channels underlies the K current that is coupled to the M2 muscarinic (IKACh) or A1 adenosine receptors (IKAdo) in nodal cells and atria. IKACh (IKAdo) is a heteromultimer of the products of two different genes in the Kir3 family, initially referred to as GIRK (G-protein inwardly rectifying K channel, Kir3.1) and CIR (cardiac inward rectifier, Kir3.4) (KCNJ3 and KCNJ5). Kir3.1 and Kir3.4 tetramerize in a 2 : 2 ratio to form the IKACh channel protein, which encodes a current that is directly activated by the β-subunit and γ-subunit of an inhibitory G-protein (Figure 3-11, left). IKACh is the primary mediator of the negative chronotropic and dromotropic effects of parasympathetic activation in the heart.
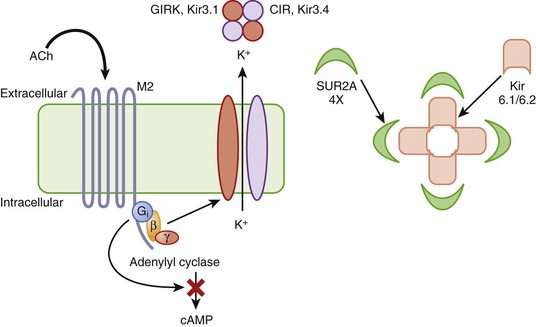
Another inward rectifier, IKATP, links electrical signaling to the metabolic state of the myocyte. Changes in the activity of IKATP profoundly influence the electrophysiology of the heart in ischemia and play a key role in the endogenous cellular mechanism that limits the injurious effect of myocardial ischemia known as ischemic preconditioning.30 IKATP is believed to be a hetero-multimeric channel complex comprising a tetrameric assembly of Kir6.2 channels (KCNJ8 and KCNJ11) at its core surrounded by four sulfonylurea receptor subunits (SUR2A, Figure 3-11, right). SUR2A, encoded by ABCC9, is an ATP-binding cassette (ABC) protein that imparts sensitivity to sulfonylureas and K channel openers such as pinacidil and chromakalim to the channel complex.
If “Funny” or Pacemaker Current
If is a current that contributes to diastolic depolarization in pacemaking cells in the heart. The current is found in many cell types, but its features are variable. For example, If is present in ventricular myocytes, but its activation voltage is so negative that it is not likely to be of physiologic significance.31 If activates slowly on hyperpolarization and deactivates rapidly with depolarization. If supports a mixed monovalent cation (Na+ and K+) current with a reversal potential of –20 to –30 mV. The current is highly regulated: β-Adrenergic stimulation increases If and hastens diastolic depolarization. A family of genes topologically similar to voltage-dependent K channels and related to cyclic nucleotide–gated channels in photoreceptors in the retina appears to encode If. A number of hyperpolarization-activated cyclic nucleotide–gated channels (HA-CNG) have been cloned from the heart, and several exhibit the general features of If in cardiac pacemaking cells. It has been suggested that If itself is a composite current with fast and slow components encoded by HCN2 and HCN4, respectively. Support for If as the pacemaker current in the heart also comes from a genetic model of bradycardia in zebrafish with a dramatically reduced If.
Electrogenic Transporters
Na+-Ca2+ Exchanger
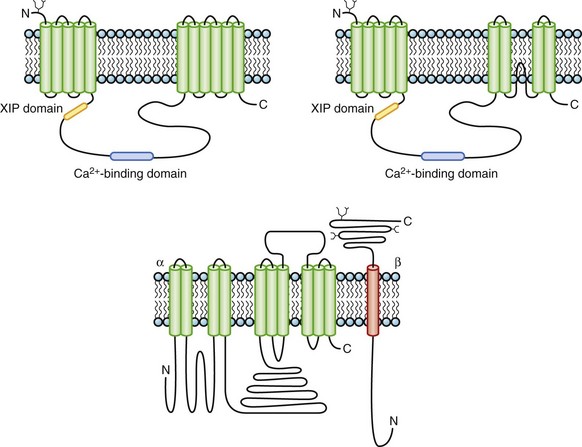
The Na+-Ca2+ exchanger is an electrogenic ion transporter that exchanges three Na+ ions for one Ca2+. The highest levels of exchange activity have been observed in the heart. The cardiac NCX, a transmembrane glycoprotein, was originally proposed to have 11 or 12 transmembrane repeats based on hydropathy analysis. More recent mutagenesis data challenge the original topologic models and instead suggest that there may be only nine transmembrane segments32 (Figure 3-12). NCX contains two membrane-spanning domains, with the first five transmembrane segments being separated from the remainder by a large cytoplasmic loop that makes up about half of the molecule. The intracellular loop contains domains that bind Ca2+ and the endogenous NCX inhibitory domain, XIP.
Na+-K+-ATPase
The Na+-K+-ATPase, or Na pump, is responsible for establishing and maintaining major ionic gradients across the cell membrane. The Na pump belongs to the widely distributed class of P-type ATPases that are responsible for transporting a number of cations. The P-type designation of this family of enzymes refers to the formation of a phosphorylated aspartyl intermediate during the catalytic cycle. The Na+-K+-ATPase hydrolyzes a molecule of ATP to transport two K+ into the cell and three Na+ out and is thereby electrogenic, generating a time-independent outward current. The Na+-K+-ATPase is oligomeric and consists of α-, β- and, possibly, γ-subunits. There are four different α- and three distinct β-isoforms (for review see reference 38). The evidence that the γ-subunit is part of the complex comes from photoaffinity labeling with ouabain derivatives and immunoprecipitation studies. The γ-subunit belongs to a family of small membrane-spanning proteins, including phospholemman, which support ionic fluxes.
Na+-K+-ATPase isoforms exhibit tissue-specific distributions. The α1β1 isoform is broadly distributed, α2-containing isoforms are preferentially expressed in the heart, skeletal muscle, adipocytes, and brain, α3 is predominantly a brain isoform, and α4 is found in abundance in the testis. The structural diversity of the Na+-K+-ATPase comes from variations in α- and β-genes, splice variants of α-subunits, and the promiscuity of subunit associations, which are all themes that also underlie the diversity of ion channels, particularly K channels. The α-subunit is catalytic and binds digitalis glycosides in the extracellular linker between the first and second membrane-spanning regions (see Figure 3-12, bottom). α1-, α2-, and α3-subunits are found in the human heart. In the rat, α3-subunits bind glycosides three orders of magnitude greater affinity than do α1-containing pumps. However, in humans, the binding affinities of the α subunits are far less variable. The β-subunits are essential for normal pump function and influence the Na+ and K+ affinities of the α-subunits and also serve as chaperones ensuring the proper trafficking of the α-subunit to the sarcolemma. Only β2 appears to be present in significant quantities in the human heart.
Molecular Basis of Activation and Recovery of the Heart
The aggregate activity of If and diminished IK1 slowly depolarizes the nodal cell until ~–40 mV when Ca currents are activated, hastening the rate of rise of the action potential. First, the transient T-type Ca current (ICa,T) is activated, driving the membrane potential toward ECa; then, the longer-lasting, dihydropyridine-sensitive L-type Ca current (ICa,L) is activated. Simultaneously, the more slowly activating outward K currents (delayed rectifier, IK) are activated, hindering the movement of the membrane potential toward ECa. Ultimately, Ca currents inactivate, and the membrane potential moves back toward EK, turning off IK and activating If and then starting the cycle again. Current continues to flow through the electrogenic Na+-Ca2+ exchanger throughout the cycle, and the magnitude and direction of this current depend on the membrane potential as well as intracellular Ca2+ and Na+ concentrations. It has recently been demonstrated that cyclic variations of submembrane Ca2+ concentration drive the activation of the Na+-Ca2+ exchanger during diastole to act in concert with ion channels to confer pacemaking activity on SA node cells, a phenomenon known as the calcium clock.33a
As in the case of the nodal cell action potential, the highly orchestrated activity of a number of ionic currents inscribes the muscle cell action potential. A prototypical action potential from atrial and ventricular myocytes with a schematic of the trajectory of the underlying ionic currents is shown in Figure 3-13. The action potential is divided into five phases: (1) Phase 0 is rapid upstroke; (2) phase 1 is early repolarization; (3) phase 2 is the plateau; (4) phase 3 is late repolarization; and (5) phase 4 is the resting potential or, in the case of a nodal action potential, diastolic depolarization (see Figure 3-1). Unlike in the case of nodal cells, a true resting potential can be defined in cardiac muscle cells, and it is ~90 mV, close to EK; thus, at rest, cardiac muscle cells are mostly permeable to K+ due to the activity of IK1.
Cellular and Molecular Basis of Cardiac Electrophysiology
Excitability and Propagation
Propagation of a wave of excitation in a homogeneous cable-like medium is continuous and obeys the laws of cable theory (see Passive Membrane Properties and Cable Theory). In such a preparation, the maximal upstroke velocity of the action potential (dV/dt)max is an indirect measure of depolarizing ionic current and conduction velocity.34 A continuous cable model is a structural oversimplification of all cardiac tissue with the possible exception of normal papillary muscles. Continuous propagation of excitation waves is not characteristic of cardiac tissue. Due to the structural and functional complexities of the myocardium, discontinuous conduction (see below) is the rule.
A feedback exists between network properties (cell-to-cell coupling via gap junctions) and active membrane properties (ionic currents) in propagation in cardiac tissue preparations.3 Under conditions of normal cellular coupling, fluctuations in local conduction velocity, action potential shape, and ionic current flow are small. However, with cellular uncoupling—such as that which accompanies ischemia—the interaction between intercellular conduction and active membrane properties assumes greater significance. In cardiac muscle, the Na current is the main determinant of membrane depolarization and local circuit current. When cells are uncoupled, discontinuity of conduction increases, the delay between activation of cells increases, and the Na current may sufficiently inactivate such that the currents active during the plateau (i.e., L-type Ca current) of the action potential become essential for driving excitatory current through gap junctions. In both experimental models and computer simulations, blocking the L-type Ca current was shown to reduce the safety factor for conduction and to lower the intercellular resistance that produces conduction blockade. Cellular uncoupling and discontinuous conduction has important implications for safety factors for the propagation of the impulse. With moderate cell-to-cell uncoupling in simple models of propagation, conduction is slower but has a higher safety factor. However, with more significant uncoupling, the transmitted current is so small that insufficient Na current is recruited to initiate an action potential.
Repolarization and Refractory Periods
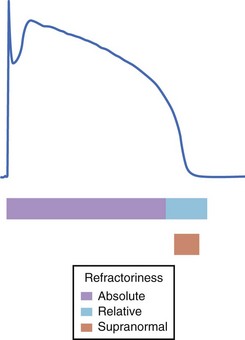
Refractoriness of tissue, a consequence of the long duration of the cardiac action potential, allows only gradual recovery of excitability. Refractoriness is essential to the normal mechanical function of the heart, as it permits relaxation of cardiac muscle prior to the next activation. Refractoriness of cardiac muscle is classified as either absolute or relative: The former occurs immediately after phase 0 and during the plateau and no stimulus, regardless of its strength, can re-excite the cell; the latter occurs during phase 3, when the cell is excitable but the stimulus strength for activation exceeds that at rest (Figure 3-14). The molecular basis of refractoriness is the lack of availability of depolarizing current (Na current in muscle) because repolarization to negative potentials is required for channels to recover from fast inactivation and thus be available to pass the excitatory current. The duration of refractoriness of any cardiac tissue thus depends on the complement of ion channels (and, in particular, depolarizing currents) expressed. When the depolarizing current becomes available to activate, outward currents (typically delayed rectifier K currents) increase the stimulus strength required to reach the threshold, making the tissue relatively refractory (compared with the rested state).
Cellular and Molecular Mechanisms Contributing to Cardiac Arrhythmias
Cardiac arrhythmias result from abnormalities of impulse generation, conduction, or both. It is, however, difficult to establish an underlying mechanism for many clinical arrhythmias. Criteria such as initiation and termination with pacing and entrainment are used in the clinical electrophysiology laboratory to make the diagnosis of re-entry in some cases. Even fewer specific tools are available to diagnose non–re-entrant arrhythmias. It is clear that molecular changes in the heart predispose to the development of abnormalities of cardiac rhythm. However, an exclusively molecular approach to understanding the mechanisms of arrhythmia is limited by failure to include the cellular and network properties of the heart. We will attempt to place in context the role of cellular and molecular changes in the development of clinically significant rhythm disturbances. A summary of the cellular and molecular changes that underlie prototypical arrhythmias and their putative mechanisms is provided in Table 3-3.
Alterations in Impulse Initiation: Automaticity
Spontaneous (phase 4) diastolic depolarization underlies the property of automaticity, which is characteristic of cells in SA and AV nodes, the His-Purkinje system, the coronary sinus, and, possibly, pulmonary veins. Phase 4 depolarization results from the concerted action of a number of ionic currents, but the relative importance of these currents remains controversial (Figure 3-15). The inwardly rectifying K current (IK1) maintains the resting membrane potential and resists depolarization; thus, the activity of other currents (e.g., Ca currents) or a reduction of IK1 (and other K conductances) must occur to permit the cell to reach the threshold for the firing of an action potential. If may play a particularly prominent role in the normal automaticity of Purkinje fibers, although this hypothesis is not without controversy. Deactivation of IK is another mechanism allowing depolarizing currents to move the membrane potential toward the threshold. Ca currents, both the T-type and the L-type, figure prominently in diastolic depolarization and in the upstroke of the action potential in nodal tissue and latent atrial pacemakers. A number of other time-independent currents may play a role in diastolic depolarization and pacemaking activity, including currents through the electrogenic Na+-K+-ATPase and the Na+-Ca2+ exchanger and background currents.
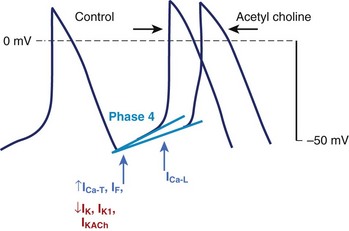
The rate of phase 4 depolarization and, therefore, the firing rate of pacemaker cells are dynamically regulated. Prominent among the factors that modulate phase 4 is the tone of the autonomic nervous system. The negative chronotropic effect of activation of the parasympathetic nervous system is the result of the release of acetylcholine that binds to muscarinic receptors, releasing G-protein βγ-subunits that activate a potassium current (IKACh) in nodal and atrial cells (see Figure 3-11). The resultant increase in K+ conductance opposes membrane depolarization, slowing the rate of rise of phase 4 of the action potential. Agonist activation of muscarinic receptors also antagonizes activation of the sympathetic nervous system through inhibition of adenylyl cyclase, reducing cAMP and inhibiting protein kinase A (PKA). Conversely, augmentation of the tone of the sympathetic nervous system increases myocardial catecholamine concentrations, which activate both α- and β-receptors. The effect of β1 adrenergic stimulation predominates in pacemaking cells, increasing the L-type Ca current and shifting the voltage dependence of If to more positive potentials, thus augmenting the slope of phase 4 and increasing the rate of SA node firing. L-type Ca current density is increased by PKA-mediated phosphorylation, which results in an increase in the rate of rise of phase 4 and the upstroke velocity of the action potential in nodal cells. The positive chronotropic effect of β-adrenergic stimulation has been related to increased subsarcolemmal Ca2+ release through the ryanodine receptor (RyR2) accelerating the rate of diastolic depolarization. Enhanced sympathetic nervous system activity can dramatically increase the rate of firing of SA nodal cells producing sinus tachycardia, with rates in excess of 200 beats/min. In contrast, the increased rate of firing of Purkinje cells is more limited, rarely producing ventricular tachyarrhythmia in excess of 120 beats/min.
After-Depolarizations and Triggered Automaticity
Triggered automaticity or activity refers to impulse initiation that is dependent on after-depolarizations (Figure 3-16). After-depolarizations are membrane voltage oscillations that occur during EADs or following DADs an action potential.35
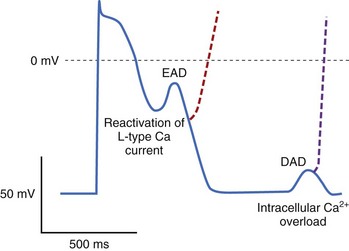
Mutations in the cardiac ryanodine receptor (RYR2), the SR Ca2+ release channel in the heart, have been identified in kindreds with the syndrome of catecholamine-stimulated polymorphic ventricular tachycardia (CPVT) and ventricular fibrillation with short QT intervals. It seems likely that perturbed [Ca2+]i changes in the relationship between SR Ca2+ load and the threshold for Ca2+ release, and thus perhaps DADs, contribute to the arrhythmias characteristic of this syndrome. Indeed, murine models of CPVT demonstrate adrenergically mediated arrhythmias, with DADs in vitro. It is likely that some ventricular tachycardias that complicate digitalis intoxication are initiated by triggered activity. It has also been suggested that DADs underlie some forms of idiopathic ventricular tachycardia, particularly from the right ventricular outflow tract (see Table 3-3). In a recent study, flecainide has been demonstrated to inhibit Ca2+ from RyRs in a murine model of CPVT, and preliminary data suggest efficacy in patients with refractory symptoms.
A fundamental condition that underlies the development of EADs is action potential prolongation, which is manifest on surface electrocardiogram as QT prolongation. Hypokalemia, hypomagnesemia, bradycardia, and drugs can cause predisposition to the formation of EADs, with drugs being the most common cause.36 Antiarrhythmics with class IA and III actions produce action potential and QT prolongation intended to be therapeutic but frequently cause proarrhythmia. Noncardiac drugs such as some phenothiazines, some nonsedating antihistamines, and some antibiotics can also prolong the duration of the action potential and cause predisposition to EAD-mediated triggered arrhythmias. Decreased [K+]o paradoxically decreases some membrane K currents (particularly IKr) in the ventricular myocyte, which explains why hypokalemia causes prolongation of the action potential and EADs. Indeed, K infusions in patients with congenital LQTS and drug-induced QT prolongation have been shown to reduce the QT interval.36a
EAD-mediated triggered activity likely contributes to the initiation of the characteristic polymorphic ventricular tachycardia—torsades de pointes—seen in patients with congenital and acquired forms of LQTS.36b In addition, exaggerated dispersion of repolarization is also likely to play a role in both the initiation and the maintenance of torsades de pointes by generating a substrate for functional re-entry (see below). Acquired prolongation of the QT interval most often is the result of drug therapy or electrolyte disturbances, as noted previously. However, structural heart diseases such as cardiac hypertrophy and cardiac failure may also delay ventricular repolarization (so-called electrical remodeling) and cause predisposition to arrhythmias related to abnormalities of repolarization.37,38 The abnormalities of repolarization in cardiac hypertrophy and cardiac failure are often magnified by concomitant drug therapy or electrolyte disturbances.
Abnormal Impulse Conduction: Re-entry
The most common arrhythmia mechanism is re-entry. Re-entry is as much a property of the networks of myocytes as it is a property of individual heart cells. Fundamentally, re-entry is circulation of an activation wave around an inexcitable obstacle. Thus, the requirements for re-entry are two electrophysiologically dissimilar pathways for impulse propagation around an inexcitable region such that unidirectional blockade occurs in one of the pathways and a region of excitable tissue exists at the head of the propagating wavefront.39 Structural and electrophysiological properties of the heart may contribute to the development of the inexcitable obstacle and of unidirectional blockade. The complex geometry of muscle bundles in the heart and spatial heterogeneity of cellular coupling or other active membrane properties (i.e., ionic currents) appear to be critical.
A key feature in classifying re-entrant arrhythmias, particularly for therapy, is the presence and size of an excitable gap. An excitable gap exists when the tachycardia circuit is longer than the tachycardia wavelength (λ = conduction velocity × refractory period), allowing appropriately timed stimuli to reset the propagation in the circuit. Re-entrant arrhythmias may exist in the heart in the absence of an excitable gap and with a tachycardia wavelength nearly the same size as the path length. In this case, the wavefront propagates through partially refractory tissue with no anatomic obstacle and no fully excitable gap. This is referred to as leading circle re-entry,40 which is a form of functional re-entry (re-entry that depends on functional properties of the tissue). Unlike excitable gap re-entry, no fixed anatomic circuit exists in leading circle re-entry, and it may, therefore, not be possible to disrupt the tachycardia with pacing or destruction of a part of the circuit. Furthermore, the circuit in leading circle re-entry tends to be less stable than that in excitable gap re-entrant arrhythmias, with large variations in cycle length and predilection to termination. Atrial flutter represents an example of a re-entrant tachycardia with a large excitable gap not always due to an anatomic constraint but to functional blockade (reflecting the special properties of the crista terminalis discussed above). Experimental data and computer simulations have highlighted the shortcomings of tenets on leading circle re-entry and suggest that spiral waves may better explain some forms of functional re-entry.
1 Weidmann S. Effect of current flow on the membrane potential of cardiac muscle. J Physiol (Lond). 1951;115:227-236.
2 LeGrice IJ, Smaill BH, Chai LZ, et al. Laminar structure of the heart: Ventricular myocyte arrangement and connective tissue architecture in the dog. Am J Physiol. 1995;269(2 Pt 2):H571-H582.
3 Spach MS, Heidlage JF. The stochastic nature of cardiac propagation at a microscopic level. Electrical description of myocardial architecture and its application to conduction. Circ Res. 1995;76(3):366-380.
4 Hodgkin AL, Huxley AF, Katz B. Ionic currents underlying activity in the axon of the squid. Arch Sci Physiol. 1949;3:129-150.
5 Hamill OP, Marty A, Neher E, et al. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391(2):85-100.
6 Antzelevitch C, Sicouri S, Litovsky SH, et al. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res. 1991;69(6):1427-1449.
7 Noda M, Shimizu S, Tanabe T, et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121-127.
8 Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500-544.
9 Stühmer W, Conti F, Suzuki H, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339(6226):597-603.
10 Isom L, De Jongh K, Patton D, et al. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256(5058):839-842.
11 Ptacek LJ, George ALJr., Griggs RC, et al. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991;67(5):1021-1027.
12 Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80(5):805-811.
13 Tanabe T, Takeshima H, Mikami A, et al. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature. 1987;328(6128):313-318.
14 Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19-31.
15 Vassort G, Talavera K, Alvarez JL. Role of T-type Ca2+ channels in the heart. Cell Calcium. 2006;40(2):205-220.
16 Perez-Reyes E. Molecular characterization of T-type calcium channels. Cell Calcium. 2006;40(2):89-96.
17 Dunham I, Shimizu N, Roe BA, et al. The DNA sequence of human chromosome 22. Nature. 1999;402(6761):489-495.
18 Tempel BL, Papazian DM, Schwarz TL, et al. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science. 1987;237(4816):770-775.
19 Snyders DJ. Structure and function of cardiac potassium channels. Cardiovasc Res. 1999;42(2):377-390.
20 Gulbis JM, Zhou M, Mann S, et al. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289(5476):123-127.
21 Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80-83.
22 Barhanin J, Lesage F, Guillemare E, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78-80.
23 Armstrong CM. Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J Gen Physiol. 1969;54(5):553-575.
24 Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250(4980):533-538.
25 Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250(4980):568-571.
26 Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17-23.
27 Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795-803.
28 Doyle DA, Cabral JM, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69-77.
29 Hille B, Armstrong CM, MacKinnon R. Ion channels: From idea to reality. Nat Med. 1999;5(10):1105-1109.
30 O’Rourke B. Myocardial K(ATP) channels in preconditioning. Circ Res. 2000;87(10):845-855.
31 DiFrancesco D. Cardiac pacemaker: 15 years of “new” interpretation. Acta Cardiol. 1995;50(6):413-427.
32 Nicoll DA, Ottolia M, Lu L, et al. A new topological model of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem. 1999;274(2):910-917.
33 Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am J Physiol. 1998;275(5 Pt 2):F633-F650.
33a Lakatta EG, Maltsev VA, Vinogradova TM. A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res. 2010;106:659-673.
34 Hodgkin AL. A note on conduction velocity. J Physiol (Lond). 1954;125:221-224.
35 Cranefield PF. Action potentials, afterpotentials, and arrhythmias. Circ Res. 1977;41(4):415-423.
36 Roden D, Lazzara R, Rosen M, et al. Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. Circulation. 1996;94(8):1996-2012.
36a Compton SJ, Lux RL, Ramsey MR, et al. Genetically defined therapy of inherited long-QT syndrome. Correction of abnormal repolarization by potassium. Circulation. 1996;94:1018-1022.
36b Choy AM, Lang CC, Chomsky CM, et al. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation. 1997;96:2149-2154.
37 Tomaselli GF, Beuckelmann DJ, Calkins HG, et al. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90(5):2534-2539.
38 Nattel S, Maguy A, Le Bouter S, et al. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87(2):425-456.
39 Mines GR. On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can. 1914;IV:43-52.
40 Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of trachycardia. Circ Res. 1973;33(1):54-62.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 3 Molecular and Cellular Basis of Cardiac Electrophysiology
Basic Concepts
Membrane Potential and Conduction
where R is the gas constant, T is temperature, F is Faraday’s constant, and z is the valence of the ionic species. If the resting cardiac myocyte is assumed to have an intracellular [K+] of ~150 mmol/L and an extracellular [K+] of 4 mmol/L, then the Nernst potential for K+ (EK) is roughly –90 mV. The Nernst potential represents the voltage at which the osmotic tendency for K+ to flow across the membrane is exactly balanced by the electrical tendency to flow in the opposite direction, which results in a net zero ionic flux and thus no net current flow. Accordingly, the K+ Nernst potential is sometimes referred to as the zero current potential, at which no K+ current would flow through open K channels. By contrast, the Nernst potential for Na+ in the resting cell is approximately +60 mV, indicating that if a pathway for Na+ to enter the cell were present, Na+ would enter the cell to move the membrane voltage (mV) toward the Nernst potential for Na+. Indeed, this is precisely what happens when Na channels are open and initiate phase 0 of the action potential. The Nernst potential for potassium is, in fact, very close to the resting membrane potential of the ventricular myocyte, indicating that the resting heart cell membrane is highly permeable to K+. Actual resting potentials are less negative than EK due to small conductances of other ionic species with less negative Nernst potentials. In the course of the cardiac cycle, the cell membrane becomes permeable to different ionic species, and these changes in permeability determine time-dependent changes in the membrane potential, with each ion striving to move the membrane voltage to its Nernst potential and inscribing regionally specific action potentials (Figure 3-1).
Passive Membrane Properties and Cable Theory
The cardiac cell membrane can be modeled as a circuit comprising variable resistors (ion channels) in parallel with a capacitor (lipid bilayer), an RC circuit (Figure 3-2). The flow of current across the membrane will alter the charge on the capacitor (and therefore the membrane potential) and change the membrane resistance. The flow of current occurs not only across but obviously along the inside and outside of the cell membrane from cell to cell as well, that is, current propagates.
Cable theory, originally developed to understand current flow in trans-oceanic telegraphic cables, can be used to model passive current flow and propagation in a cardiac muscle fiber. In their simplest formulation, the cable equations define the distribution of voltage along a continuous, uniform cable of infinite length stimulated by a point source. The predictions of cable theory are that (1) a change in voltage exhibits a characteristic decay along the cable defined by the space constant (distance over which voltage decays to 1/e of the value at the site of injection) (Figure 3-3, A); and (2) there is an inverse relationship between resistance to current flow (both transmembrane and intercellular) and the cable diameter (Figure 3-3, B). That is, the space constant is directly proportional to the cable diameter, and thus, greater lengths of the cable are influenced by the same current injection into a thick, rather than a thin, cable.
Substantial anatomic and biophysical limitations exist when applying the cable theory description of conduction to cardiac muscle. Anatomically, the shape of the heart is complex, and at any level of integration above a single muscle fiber, it does not resemble a cable. Conduction through the myocardium is not continuous; instead, myocardial cells are connected by gap junction channels that create a non-uniformity of intercellular resistance. Macroscopic discontinuities such as fibrous tissue and blood vessels also significantly perturb the cable view of conduction in the myocardium. Finally, the cardiac cell membrane does not just comprise RC circuits; instead, when stimulated to the threshold, it will generate action potentials (see Figure 3-3, A). Despite the limitations of cable theory, it serves as the foundation for several important concepts in impulse propagation and was used to demonstrate the electrical nature of conduction in the heart.1
While this discussion implicitly treats the activation wave in one dimension, the behavior of propagating waves in the heart is more complex. The complexity can be appreciated if one considers propagation in two dimensions. In this circumstance, the shape of the wavefront is a major determinant of the efficiency of propagation. A convex wavefront, as might be observed after point stimulation, creates a large sink around a smaller activating source. This mismatch reduces conduction velocity and the safety factor for propagation. Conversely, a concave activation front produces a source-sink mismatch that favors the source; this results in a high safety factor and more rapid impulse transmission. Thus, not only do source-sink characteristics influence propagation, they also influence the curvature of the wave front (see Figure 3-4).
Directionally different conduction velocity is a characteristic feature of cardiac muscle known as anisotropic conduction. Anisotropic conduction has its basis in the structure of the myocyte and cardiac tissue; myocytes are rod shaped and are organized in bundles that are oriented along the long axis of the cell. Transmurally, the axes of these bundles undergo significant changes in orientation through the ventricular wall (~120 degrees maximal deviation).2 Communication among myocytes occurs via gap junction channels that are non-uniformly distributed over the surface of the heart cell, with larger numbers of channel poised to propagate the impulse longitudinally, rather than transversely, to the long axis of the muscle fiber.3 The implications of anisotropy for conduction in the longitudinal and transverse direction under pathologic conditions are controversial. In the context of uniform depression of conduction, as might exist with antiarrhythmic drug treatment, propagation in the transverse direction is preserved compared with conduction in the longitudinal direction. However, when cellular uncoupling occurs, such as in ischemia, longitudinal propagation may exhibit a higher safety factor than does transverse conduction.
Major Breakthroughs: Voltage Clamp, Molecular Cloning
A stimulus of sufficient magnitude applied to a myocyte (or any excitable cell) elicits a typical change in membrane potential known as the action potential. The ionic current basis of the action potential was confirmed and quantitatively studied using the voltage clamp developed in the middle of the twentieth century.4 Voltage clamping is a technique whereby the experimenter controls the transmembrane voltage and measures the current at that defined voltage. Much of what we know about ionic currents in myocytes comes from voltage clamp experiments and a more recently developed type of voltage clamp called the patch clamp.5 A variant of the patch clamp technique permits the measurement of ionic currents through single ion channels.
Molecular Basis of Cardiac Action Potentials
Cardiac myocytes possess a characteristically long action potential (200 to 400 ms, see Figure 3-1), compared with neurons or skeletal muscle cells (1 to 5 ms). The action potential profile is sculpted by the orchestrated activity of multiple ionic currents, each with its distinctive time- and voltage-dependent amplitudes. The currents, in turn, are carried by complex transmembrane proteins that passively conduct ions down their electrochemical gradients through selective pores (ion channels), actively transport ions against their electrochemical gradients (pumps, transporters), or electrogenically exchange ionic species (exchangers).
Action potentials in the heart are regionally distinct. The regional variability in cardiac action potentials is the result of differences in the numbers and types of ion channel proteins expressed by different cell types in the heart. Further, unique sets of ionic currents are active in pacemaking and muscle cells, and the relative contributions of these currents may vary in the same cell type in different regions of the heart.6
Ion Channels and Transporters: Molecular Building Blocks of the Action Potential
Currents that underlie the action potential are carried by complex, multi-subunit transmembrane glycoproteins called ion channels (Table 3-1). These channels open and close in response to a number of biologic stimuli, including a change in voltage, ligand binding (directly to the channel or to a G-protein–coupled receptor), and mechanical deformation. Other ion-motive transmembrane proteins such as exchangers and transporters make important contributions to cellular excitability in the heart. Ion pumps establish and maintain the ionic gradients across the cell membrane that permit current flow through ion channels. If pumps, transporters, or exchangers are not electrically neutral (e.g., 3 Na+ for 1 Ca2+), they are termed electrogenic and can further influence electrical signaling in the heart.
The most abundant superfamily of ion channels expressed in the heart consists of voltage-gated ion channels. Various structural themes are common to all voltage-dependent ion channels. First, the architecture is modular, consisting either of four homologous subunits (in K channels, see Figures 3-7 and 3-8) or of four internally homologous domains (in Na and Ca channels) (see Figures 3-5 and 3-6). Second, proteins wrap around a central pore (see Figure 3-8). The pore-lining (P segment) regions exhibit exquisite conservation within a given channel family of like selectivity (e.g., jellyfish, eel, fruit fly, and human Na channels have very similar P segments), but not among families with different selectivities. Third, the general strategy for activation gating (opening and closing in response to changes in the membrane voltage) is highly conserved: The fourth transmembrane segment (S4), typically studded with positively charged residues, lies within the membrane field and moves in response to depolarization, thus opening the channel. Fourth, most ion channel complexes include not only the pore-forming proteins (α-subunits) but also auxiliary subunits (e.g., β-subunits) that modify channel function.
Sodium Channels
Na channels were the first ion channels to be cloned and have their sequence determined.7 In humans, more than 10 distinct Na channel genes have been cloned from excitable tissues, with striking homology to the complementary DNA (cDNA) cloned from eel electroplax. The cardiac Na channel gene (SCN5A) resides on the short arm of chromosome 3 (3p21) (see Table 3-1). The Na channel complex is composed of several subunits, but only the α-subunit is required for function. Figure 3-5 shows that the α-subunit consists of four internally homologous domains (labeled I to IV), each of which contains six transmembrane segments. The four domains fold together so as to create a central pore, whose structural constituents determine the selectivity and conductance properties of the Na channel.
One of the seminal contributions of Hodgkin and Huxley was the notion that Na channels occupy several “states” (which are now viewed as different conformations of the protein) in the process of opening (activation); yet another set of conformations is entered when the channels close during maintained depolarization (inactivation).4 The m gates that underlie activation and the h gate that mediates inactivation were postulated to have intrinsic voltage dependence and to function independently.8 While some of the implicit structural predictions of that formulation have withstood the test of time, others have not. For example, the four S4 segments are now widely acknowledged to serve as activation voltage “sensors.” In the process of activation, several charged residues in each S4 segment physically traverse the membrane (see Figure 3-5, bottom panel). The contributions of each S4 segment to activation are markedly asymmetrical; some of the charged residues play a much more prominent role than do others in “homologous” positions.9 Other studies have revealed that activation is coupled with inactivation. Indeed, the time course of current decay during maintained depolarization predominantly reflects the voltage dependence of activation, although single-channel inactivation itself does vary with voltage (particularly in cardiac Na channels). If the S4s are the sensors, where are the activation “gates” themselves? This crucial question still remains unresolved. However, according to experimental evidence, S6 is the leading contender for the physical activation gate.
Inactivation of Na channels is as arcane a process as activation. Not only is there loose coupling to activation, but there are multiple inactivation processes. One common approach to distinguishing inactivated states is to determine the rate at which they recover the ability to activate: Repriming from the traditional “fast” inactivation occurs over tens of milliseconds, while recovery from “slow” inactivation may need tens of seconds or longer. Fast inactivation is at least partly mediated by the cytoplasmic linker between domains III and IV (the crucial residues are labeled IFM in Figure 3-5), which may function as a hinged lid, docking onto a receptor formed by amino acids in the S4-S5 linkers of domains III and IV. This notion is consistent with observations that fast inactivation can be disrupted by internal proteases. Nevertheless, it is increasingly clear that mutations scattered widely throughout the channel affect inactivation gating. The structural determinants of slow inactivation are less well localized than those of fast inactivation. Mutations in the P region of domain I affect both activation gating and slow inactivation, while various widely scattered disease mutations identified in paramyotonia congenita and other skeletal myopathies suppress slow inactivation of the Na channel.
Four different β-subunits (NaVβ1 to 4, SCN1B-SCN4B) have been isolated, and all appear to be single membrane–spanning domain (type I topology) proteins with a large extracellular V-shaped immunoglobulin (Ig) fold often found in cell adhesion molecules and small carboxyl terminal cytoplasmic domain.10 The effects of the particular β-subunit on the kinetics and voltage dependence of the α-subunit vary depending on the particular β-subunit and the cell expression system. Despite the expression of β1 mRNA, using subtype-specific antisera, the functional role of β-subunits in cardiac Na currents is still being debated. Several pieces of evidence are consistent with a role for β-subunit(s) in heart cells. First, β-subunits are found in cardiac myocytes, although no NaVβ1 is found in association with the α-subunit protein from the rat heart. Second, NaVβ1 variably modulates the function of NaV1.5 in heterologous expression systems, including the sensitivity of the channel to blockade by antiarrhythmic drugs and free fatty acids. Finally, mutations in β-subunits have been directly or indirectly implicated in heritable cardiac arrhythmias.
In contrast to PKA, protein kinase C (PKC) alters the function of all of the mammalian Na channel isoforms. The PKC effect is largely attributable to phosphorylation of a highly conserved serine in the III-IV linker (see Figure 3-5). Conventional PKC isoforms reduce the maximal conductance of the channels and alter gating in an isoform-specific fashion. The macroscopic current decay of neuronal channels is uniformly slowed by PKC, which suggests destabilization of the inactivated state. Cardiac channels exhibit a hyperpolarizing shift in the steady-state availability curve consistent with an enhancement of inactivation from closed states.
Alteration of ion channel function is an important pathophysiologic mechanism of various familial diseases of muscle and brain and of inherited arrhythmias. The Na channelopathies were among the first molecularly characterized human ion channel diseases.11 Rare allelic variants in SCN5A have been linked to inherited ventricular arrhythmias,12 conduction system disease, and sudden infant death syndrome. Complex electrophysiological phenotypes have been associated with mutations in both α- and β-subunits of Na channels. Rare variants or mutations have been associated with sudden death in women (in a population-based study) and with atrial fibrillation. More common acquired forms of long QT syndrome (LQTS), generally associated with drug ingestion, have been linked to common variants of SCN5A. Finally, disease-causing mutations in the Na channel have been associated with alterations in drug blockade. The highly variant phenotypes of these arrhythmic syndromes are, in part, explained by the variable effects of the mutations on channel subunit function or expression.
Calcium Channels: L-type
The pore-forming subunit (α1) of the calcium (Ca) channel is built on the same structural framework as the Na channel.13 As is the case with the Na channel, a number of genes encode surface membrane Ca channel α1-subunits. The predominant sarcolemmal Ca channels in the heart are the L-type (large and long-lasting) and T-type (tiny and transient) Ca channels (Table 3-2). The cardiac L-type Ca channel (CaV1.2) is a multi-subunit transmembrane protein composed of α1C (α11.2) (165 kDa), β (55 kDa), and α2 (130 kDa) to δ (32 kDa) subunits. Three genes are known to encode L-type Ca channel α1-subunits (CaVα11.x to α13.x), and the CaVα11.2 is the gene expressed in the heart (see Table 3-1). Distinct splice variants of the CaVα11.2 gene have been described, and these contribute to the diversity of the cardiac L-type Ca channel function. Similar to the α-subunit of the Na channel, the S5-S6 linkers (P segments) of the α1-subunit of the Ca channel form the ion-selective pore (Figure 3-6). However, unlike Na channels, each P segment contributes a glutamic acid to a cluster that serve to bind Ca2+ in the channel pore. The β-subunit is completely cytoplasmic and noncovalently binds to the α1C subunit, modifying its function and contributing to appropriate membrane trafficking of the channel complex. Although β2a has been proposed to be the major L-type Ca channel β-subunit, splice variants of all β-subunits β1 to β4 are expressed in the mammalian heart in a complex spatial and temporal pattern. As many as five genes have been suggested to encode the α2-δ subunit: These gene products undergo post-translational processing to produce the mature extracellular α2-subunit linked by a disulfide bond to the transmembrane δ-subunit. In heterologous expression systems, α2– to δ-subunits enhance expression of Ca channels and hasten current activation and deactivation in the presence of α1– and β-subunits.
| L-TYPE | T-TYPE | |
|---|---|---|
| Pore-forming α-subunit | α1C | α1H |
| Auxiliary subunits | β, α2-δ | ? |
| Permeability | Ba2+ > Ca2+ | Ba2+ ≅ Ca2+ |
| Activation threshold | >–30 mV | >–60 mV |
| Inactivation threshold | >–40 mV | >–90 mV |
| Inactivation | ||
| Rate | Slow | Fast |
| Calcium-dependent | Yes | No |
| Voltage-sensitive | Yes | Yes |
| Recovery | Fast | Slow |
| Localization in heart | All | Nodal > Purkinje > atria |
| Blocker sensitivity | ||
| Dihydropyridines | ++++ | + |
| Phenylalkylamines | ++++ | + |
| Benzothiazepines | ++++ | + |
| Tetralols | ++ | +++ |
| Ni2+ | + | +++ |
| Cd2+ | +++ | + |
Ba, Barium; Cd, cadmium; Ni, nickel.
Ca channels inactivate by both Ca2+-dependent inactivation (CDI) and voltage-dependent inactivation (VDI) processes. CDI and VDI are regulated by channel phosphorylation and β-subunits, which suggests shared structural mechanisms that perhaps involve the I-II domain linker. The C-terminus contains peptide sequences that bind calmodulin (CaM) and Ca2+; these are an IQ motif named for the signature amino acids (isoleucine and glutamine) in the sequence and a helix-loop-helix structural domain, referred to as an EF hand, that mediate CDI. CaM is permanently tethered to the channel complex and serves as a Ca2+ sensor for the L-type channel. The Ca2+-CaM complex facilitates the interaction of CaM with the IQ motif resulting in the occlusion of the inner mouth of the Ca channel pore and terminating the inward Ca2+ flux despite continued depolarization. As cytoplasmic Ca2+ concentration falls, calmodulin unbinds Ca2+ and the IQ motif, thus relieving Ca2+-dependent inactivation. Spatial discrimination of Ca2+ concentration by CaM may involve regions in the N-terminus of the channel. A Ca2+-binding EF hand motif in the carboxyl terminus of α1 appears to be necessary to confer Ca2+-dependent inactivation to the CaVα11.2-subunit, although not through a direct binding of Ca2+ (see Figure 3-6, bottom
[/not-level-membership-for-cardiovascular-category]
