[level-membership-for-opthalmology-category]
Chapter 32 Mitochondrial Genetics of Retinal Disease
Mitochondrial origins
It is now accepted that the origin of mitochondria within eukaryotes is the result of an endosymbiotic relationship and that mitochondrial (mt) DNAs can be traced to an α-proteobacterial genome.1,2 This theory has been supported by phytogenetic patterns of gene arrangements, small subunit ribosomal RNAs (rRNA) and protein data.3,4 DNA sequencing studies have shown that tremendous variations are still present within mtDNA, with the genome of the protozoan Reclinomonas americana having the most bacteria-like mitochondrial genome and Rickettsia prowazekii the most mitochondria-like eubacterial genome.1 The mtDNA from eukaryote species show remarkable differences in size, ranging from 6 to 60 kb. They also vary in shape, with some eukaryotes having linear mtDNA while others have circular mtDNA.
Mitochondrial structure
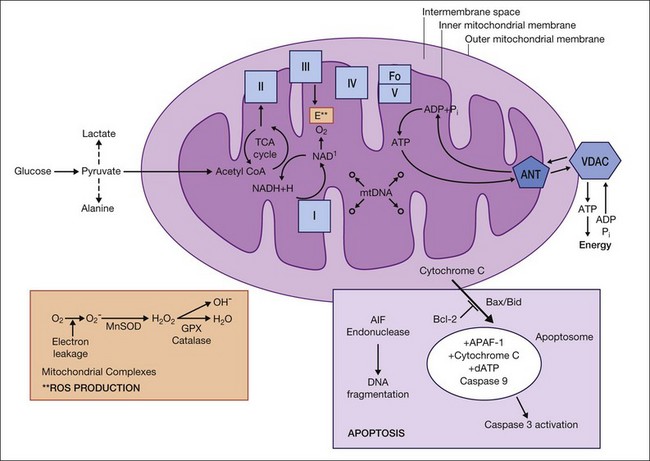
Depending on the energy requirement of each cell, the number of mitochondria varies from one to several thousand. Each mitochondrion is divided into compartments that are contained within the outer membrane, and include the intermembrane space, the inner membrane, cristae, and the matrix (Fig. 32.1). The outer membrane is permeable to molecules smaller than 5 kDa, which pass through the lipid bilayer channels called porins (voltage-dependent anion channel) into the intermembrane space. It also contains a translocase of the outer membrane complex involved in the import of resident mitochondrial proteins that are encoded by the nuclear genome and produced in the cytosol.5 The inner membrane has a high content of cardiolipin and its selective permeability allows only specific molecules into the matrix. The surface area is greatly expanded as a result of the numerous invaginations of the inner membrane, known as cristae. Embedded within the inner membrane are many of the enzyme complexes required for adenosine triphosphate (ATP) production and the translocase of the inner membrane complex that is responsible for the import of nuclear-encoded proteins into the matrix. The matrix contains numerous proteins, ribosomes, tRNA, and the mtDNA.
Mitochondrial DNA
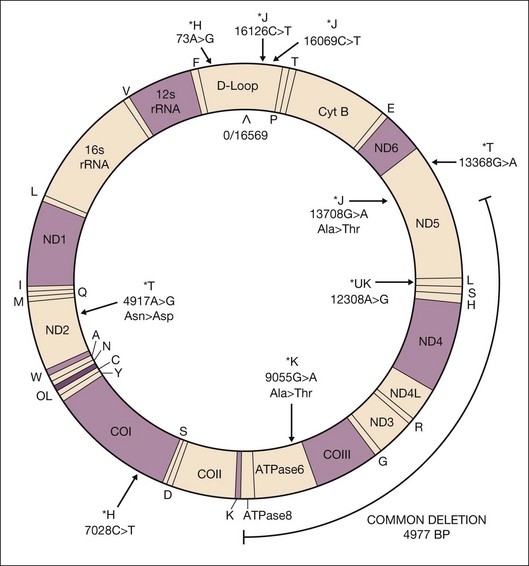
Mitochondria are unique in that they have their own DNA that is inherited through the maternal lineage. The human mtDNA forms a closed circle of double-stranded DNA, with 16 569 nucleotide pairs, comprised of two strands that are differentiated by their nucleotide content. The heavy strand is guanine-rich and encodes for 28 genes while the light strand is cytosine-rich and encodes for nine genes. Unlike the nuclear genome, mtDNA contains a unique noncoding control region but no introns. The noncoding mtDNA Dloop has within it the 1121 nucleotide control region that is important for replication and transcription. The coding region of mtDNA codes for 37 genes, including 13 protein subunits essential for oxidative phosphorylation (OXPHOS), two ribosomal RNAs, and 22 transfer RNAs (Fig. 32.2).6–8 The vast majority of mitochondrial proteins (~1500–2000) that contribute to energy biogenesis4,9 are encoded by nuclear DNA and imported into the mitochondria.
Within a cell there is a single DNA copy of the nuclear genome (nDNA) but multiple copies of mtDNA because there can be thousands of mitochondria per cell, and within each mitochondrion, 1–10 copies of mtDNA. With aging and exposure to oxidative stress, mtDNA molecules can be damaged, which results in a mixture of nonmutated (wild-type) and mutant mtDNA within the same cell. This mixture of damaged and undamaged mtDNA is termed “heteroplasmy.” When cells with heteroplasmic mitochondria divide, the two types of mtDNA are randomly or, in some instances, nonrandomly distributed into the daughter cells.10–14 Alternatively, cells may have either a pure mutant mtDNA or pure nonmutant (wild-type) mtDNA population, in which case it is referred to as homoplasmic mtDNA.15 Homoplasmy within a cell indicates that all mtDNA copies are identical. Cells can function only with relatively low levels of heteroplasmy but once this threshold is breached, abnormal function and disease can occur. Although low levels of heteroplasmic mtDNA defects may have an effect on function, the mtDNA changes are not always obvious and special technical approaches are required to ensure their detection. Correlating a phenotype with mtDNA defects can be difficult because the complexity of the phenotype can be influenced by when (time during embryogenesis/development) and where (tissue type) the mutation arises.16–18 In addition, environmental factors, such as oxidative stress, can modulate the expression of the phenotype. mtDNA is particularly susceptible to oxidative damage because it resides in the matrix and is in close proximity to sites of reactive oxygen species (ROS) formation. In addition, the mitochondria have a poor DNA repair process and a high transcription rate. Oxidative damage to mtDNA is especially prevalent in very metabolically active tissues such as the retina, brain, and muscle.
Electron leakage and ROS formation
Under normal conditions, ATP is produced via a series of oxidation–reduction reactions that involve the flow of electrons through complex I–IV of the ETC. Electron transfer generates the energy required to transport protons across the inner mitochondrial membrane into the inner-membrane space. The flow of the protons down the concentration gradient from the inner-membrane space into the matrix through complex V, also known as ATP synthase, provides the energy to generate ATP from adenosine diphosphate. Under optimal conditions, molecular oxygen captures four electrons released from complex IV to form water. However, partial reduction of oxygen can produce potentially harmful intermediates, such as superoxide, peroxide, and hydroxyl radicals, collectively known as ROS. Additionally, ineffective transfer of electrons between ETC complexes can permit partial reduction of oxygen and production of ROS byproducts during normal oxidative metabolism. In fact, approximately 2–5% of the oxygen we consume is only partially reduced and forms ROS.19
Localization of mitochondria within the retina and optic nerve
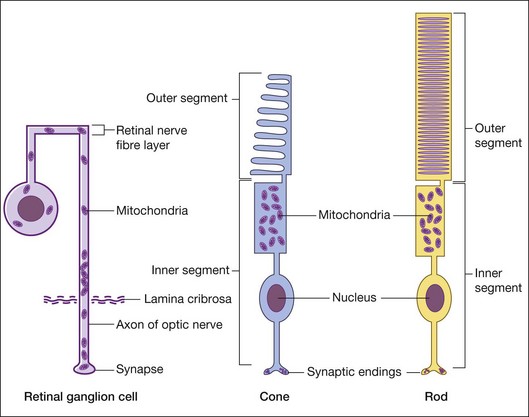
The retina has one of the highest oxygen consumption rates in the body. To meet the energy demand for vision, the photoreceptor cells have a high concentration of mitochondria within the inner segments (Fig. 32.3). The photoreceptors have high exposure to ultraviolet light and OXPHOS levels, leading to increased ROS formation, which makes these cells a target for mtDNA damage. In vitro studies show that oxidative stress leads to preferential damage of mtDNA compared to nuclear DNA.20 Animal models demonstrate increased mtDNA deletions and damage in aging and degenerating retinas.21,22 Mitochondrial abnormalities, including mtDNA damage, have been found in aging and age-related macular degeneration (AMD) retinas.20,21,23–25 The retinal ganglion cells (RGC) with their long axons are also susceptible to mitochondrial damage. In RGCs, the mitochondria are present around the nucleus and along the axons but they tend to accumulate just anterior to the lamina cribrosa (Fig. 32.2).26 The numbers of RGCs and their axons are significantly decreased in Leber’s hereditary optic neuropathy and glaucoma.27,28 This may be related to lower energy production caused by mtDNA damage and dysfunction.
Influences of mtdna ON cell function
Influences of mtDNA upon cell function can be classified into ancient adaptive polymorphic changes (haplogroups), recent mutations, and somatic mtDNA changes.29 Examples of each are presented below.
Ancient inherited mtDNA variants representing populations (haplogroups)
Definition of haplogroups
Haplogroups are mtDNA sequence polymorphism variations that have occurred over more than 150 000 years and correlate with the geographic origins of populations traced through the maternal lineages. The oldest haplogroups are from Africa and with geographic migration and climate adaptations, European, Asian, and Native American haplogroups have evolved.4 Each haplogroup has related patterns of mtDNA sequences (haplotypes) that represent that population. If the specific single polynucleotide polymorphism (SNP) variants representing the haplogroup are found in the mtDNA Dloop, it can affect the replication and transcription rates. If within the coding region, the SNP variants can be nonsynonymous (amino acid changing), which can potentially alter efficiencies of energy production, causing ROS formation, apoptosis, and cell death. This means that each haplogroup, with its different set of SNPs, can produce unique bioenergetic as well as biochemical properties. Haplogroups increasingly are being correlated with a broad spectrum of common diseases, including age-related diseases, such as Parkinson’s disease and Alzheimer’s disease.30–40
Association of haplogroups with AMD and other eye diseases
Recent studies show that AMD is associated with specific mtDNA SNPs that define northern European haplogroups.41–44 Large soft drusen and retinal pigment abnormalities, which are characteristics of AMD retinas, have been associated with J and U haplogroups.44 The haplogroup T-associated SNP A4917G located in the NADH dehydrogenase subunit 2 of complex I is an independent predictor of AMD.43 Udar and coworkers found that patients with late AMD showed strong association with noncoding mtDNA Dloop SNPs and coding region SNPs representing the J, T, and U haplogroups (Fig. 32.2).41 Two variants of the T2 haplogroup, A11812G of MT-ND4 and A14233G of MT-ND6 located in respiratory complex I, are 2.5 times more likely to be associated with advanced AMD than age-matched control subjects.42
Recent studies have reported that susceptibility to pseudoexfoliation glaucoma is decreased in patients with a U haplogroup but increased with T or L2 haplogroups.45,46 In a Saudi Arabian population, the African L haplogroups, excluding L2 haplogroup, have been associated with increased risk of primary open-angle glaucoma.47 Another study has reported that type 2 diabetic patients with the mtDNA T-haplogroup background have a higher prevalence of diabetic retinopathy.48 Haplogroups may also influence diseases via interaction with nuclear genes to increase the severity of, or protect against, disease. It has been shown that an individual harboring the milder Leber’s hereditary optic neuropathy mutations at positions 11778, 14484, and 10663 has increased probability of blindness if he or she has a J-haplogroup background.49,50 In contrast, human immunodeficiency virus (HIV)-infected patients with a J-haplogroup background were protected against progression of neuroretinal disorder.51
Some of the mtDNA SNPs that define the haplogroups may be related to partial uncoupling of OXPHOS and decreased efficiency of ATP production.39,40,52 Within the retinal cells, if the mitochondrial energy production levels fall below a bioenergetic threshold, the mitochondrial permeability transition pore (mtPTP) can be activated and photoreceptors or RGCs destroyed through apoptosis. This scenario of mitochondrial dysfunction playing a role in retinal or neuronal diseases may offer an array of new therapeutic approaches targeted toward increasing mitochondrial energy production, decreasing mitochondrial ROS, and stabilizing the mtPTP.
Recent maternally transmitted mtDNA mutations associated with retinal diseases
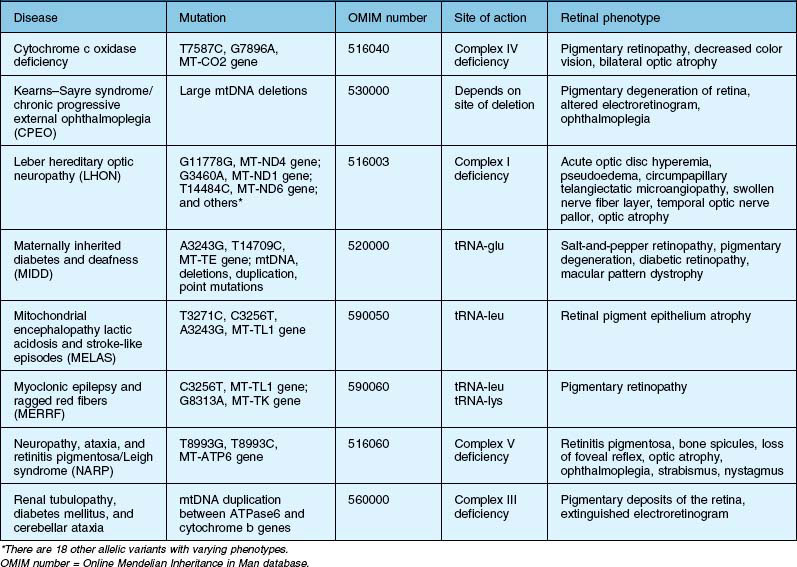
This category refers to true pathology-creating mtDNA mutations that are inherited over recent generations rather than mtDNA adaptation (haplogroups) that occur over tens of thousands of years in response to environmental challenges. Defects or mutations in the mtDNA genes can decrease OXPHOS and ATP production, resulting in energy-deficiency diseases. Tissues that are very metabolically active, such as brain, heart, muscle, retina, kidney, and the endocrine organs, can be significantly affected by these changes. Therefore, many of these patients have not only retinal abnormalities but also neurological, cardiac, metabolic, and skeletal muscle dysfunctions related to damage in these high-energy-requiring organ systems. It is now apparent that patients with mtDNA mutations that cause mitochondrial dysfunction can have a wide range of clinically abnormal ocular and systemic phenotypes.53 Table 32.1 lists mtDNA defects that result in diseases involving the retina and optic nerve. While there are many diseases caused by nuclear gene defects encoding mitochondrial proteins, they are not addressed here but are covered in an excellent review by Yu-Wai-Man and coworkers.54
Somatic mtDNA variations associated with retinal diseases
Mechanisms of mtDNA damage
The hydroxyl radical is most reactive with mtDNA and can produce multiple DNA modifications, such as strand breaks, cleavage of bases from the deoxyribose backbone to form abasic sites, and addition of oxygen to DNA, forming many different mutagenic molecules (e.g., 8-hydroxydeoxyguanosine (8-OHdG) and thymine glycol). Compared with nuclear DNA, mtDNA is particularly susceptible to oxidative damage due in part to its close proximity to the ETC, the major cellular source of ROS. Additionally, mtDNA lacks protective histones and has a limited capacity for repair, which includes only a mechanism for base excision repair.55 Enzymes involved in base excision repair that are present in the mitochondria include glycosylases, endonucleases, and GTPases. Of great importance for ocular tissue, proteins involved in nucleotide excision repair that repair ultraviolet damage are abundant in the nucleus but are absent from the mitochondria.55
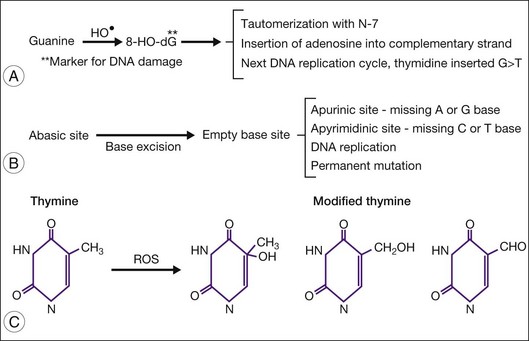
The most frequent oxidative damage to mtDNA is 8-hydroxylation/oxidation of a guanine base to 8-OHdG56,57 (Fig. 32.4). This oxidative modification is mutagenic because it inhibits methylation and can pair with adenosine (rather than cytosine) during DNA replication leading to a GC to AT conversion, which is the most frequent type of spontaneous mutation. In postmitotic tissue, such as skeletal muscle, neurons, and retina, 8-OHdG accumulates with age and disease.58–60 Abasic sites, which have neither purines (adenine and guanine) nor pyrimidines (cytosine and thymine), are also produced due to DNA damage and can lead to somatic mutations. Thymine is also susceptible to oxidative modification and produces a variety of modified thymines that are mutagenic.
In addition to DNA mutations, oxidative damage to mtDNA can also lead to mtDNA deletions by causing double-strand breaks in the DNA.61,62 Damage from ultraviolet light can also cause double-strand breaks.63 In general, mtDNA deletions occur more frequently than somatic mutations in aged postmitotic tissue.64,65 There is a frequently encountered 4977-bp deletion known as the “common deletion.” It is located in the major arc between the two origins of replication (OH and OL) and is flanked by short direct repeats (Fig. 32.2). This region of the mtDNA genome is eliminated with increasing frequency in aged postmitotic cells, such as neurons, muscle, cochlear tissue, retina, and retinal pigment epithelium (RPE), and thus is considered a hallmark of aging.58,64,66–69 The common deletion encompasses the coding region for subunits of complex I (ND3, ND4, ND4L, ND5), complex IV (cytochrome oxidase III), and complex V (ATPase 6, ATPase 8). Therefore, high levels of common deletion should have a functional impact on the cell. However, each cell contains multiple mtDNA copy numbers which may be a mixture of damaged and undamaged mtDNA (heteroplasmic). As the damaged mtDNA continues to accumulate, a “critical threshold” is reached, whereby extensive mitochondrial dysfunction causes detrimental consequences for the cell. Importantly, the “critical threshold” is tissue-specific. For example, the abundance of mtDNA deletions must reach >85% in muscles of Kearns–Sayre syndrome patients for disease phenotype to manifest.70 In aged substantia nigra neurons, the critical threshold for functional defects is between 48% and 67%.60 In RPE from elderly donors, deletion levels of ~40% were tolerated since the donors had no clinically obvious signs of retinal degeneration.69 This may explain why clinical signs of diseases (such as drusen and RPE cell loss) may take decades to present in AMD patients. Of note, the content of RPE mtDNA deletions was not significantly increased in AMD compared with age-matched controls, suggesting that the presence of the common deletion does not contribute to AMD pathology.69
Two opposing models involving either replication or repair have been proposed as the mechanism for deletion formation.71 While both models are supported by strong evidence, the presence of high levels of deletions in postmitotic cells compared with actively replicating mitotic cells favors defects in the process of repair as the mechanism responsible for generating deletions. In brief, double-stranded breaks that occur as a consequence of oxidative or ultraviolet damage stimulates activation of a 3’-5’ exonuclease that removes basepairs that are directly adjacent to the double-stranded break.71 This action exposes homologous regions of 10–15 bp and allows them to anneal with direct repeats on the opposite strand. The unbound single strands are degraded and double strands are ligated. This results in production of mtDNA containing a partially deleted sequence. Thus, the age-dependent accumulation of mtDNA deletions is likely a byproduct of a lifetime of ongoing repair.
mtDNA damage and AMD
Two recent studies using a long-extension polymerase chain reaction technique testing for mtDNA damage in the RPE from human donor eyes provided evidence that mtDNA damage is increased with AMD. Karunadharma and colleagues showed marked differences in damage accumulation with aging and disease.69 Age-related damage in the RPE appeared to be limited to the common deletion, whereas AMD was associated with global damage, which includes deletions and oxidative damage to mtDNA, throughout the mitochondrial genome. Corroborating data were also provided by Lin and colleagues, who showed that the increased DNA damage in RPE from donors with AMD was accompanied by a fivefold increase in somatic mutations compared with age-matched controls.72 Sequence analyses of the mtDNA Dloop showed a significantly greater number of SNPs per person in the AMD population compared to either older or younger normal groups.23 Finally, immunohistochemical studies using 8-OHdG antibodies showed oxidatively damaged DNA in the AMD retinas.41 Using an animal model, it has been shown that increased accumulation of mtDNA deletions and mutations impaired the cellular function and left the cells more susceptible to external stresses.73 These results support the hypothesis that defects in the mitochondria are hallmark and most probably pathologic events in AMD and other retinal diseases.
1 Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481.
2 Burger G, Gray MW, Lang BF. Mitochondrial genomes: anything goes. Trends Genet. 2003;19:709–716.
3 Gray MW, Lang BF, Cedergren R, et al. Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res. 1998;26:865–878.
4 Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407.
5 Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–667.
6 Wallace DC. Diseases of the mitochondrial DNA. Annu Rev Biochem. 1992;61:1175–1212.
7 Wallace DC. Mitochondrial DNA mutations in diseases of energy metabolism. J Bioenerg Biomembr. 1994;26:241–250.
8 McFarland R, Turnbull DM. Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med. 2009;265:210–228.
9 Yates JR, 3rd., Gilchrist A, Howell KE, et al. Proteomics of organelles and large cellular structures. Nat Rev Mol Cell Biol. 2005;6:702–714.
10 Shoubridge EA, Karpati G, Hastings KE. Deletion mutants are functionally dominant over wild-type mitochondrial genomes in skeletal muscle fiber segments in mitochondrial disease. Cell. 1990;62:43–49.
11 Bua E, Johnson J, Herbst A, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480.
12 Durham SE, Samuels DC, Cree LM, et al. Normal levels of wild-type mitochondrial DNA maintain cytochrome C oxidase activity for two pathogenic mitochondrial DNA mutations but not for m.3243A→G. Am J Hum Genet. 2007;81:189–195.
13 Chinnery PF, Zwijnenburg PJ, Walker M, et al. Nonrandom tissue distribution of mutant mtDNA. Am J Med Genet. 1999;85:498–501.
14 Fan W, Waymire KG, Narula N, et al. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008;319:958–962.
15 Wallace DC, Lott MT, Procaccio V. Mitochondrial genes in degenerative diseases, cancer and aging. Philadelphia, PA: Churchill Livingstone Elsevier; 2007.
16 Corral-Debrinski M, Horton T, Lott MT, et al. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–329.
17 Wang Y, Michikawa Y, Mallidis C, et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc Natl Acad Sci U S A. 2001;98:4022–4027.
18 Nekhaeva E, Bodyak ND, Kraytsberg Y, et al. Clonally expanded mtDNA point mutations are abundant in individual cells of human tissues. Proc Natl Acad Sci U S A. 2002;99:5521–5526.
19 Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605.
20 Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403.
21 Bravo-Nuevo A, Williams N, Geller S, et al. Mitochondrial deletions in normal and degenerating rat retina. Adv Exp Med Biol. 2003;533:241–248.
22 Wang AL, Lukas TJ, Yuan M, et al. Age-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retina. Neurobiol Aging. 2010;31:2002–2010.
23 Kenney MC, Atilano SR, Boyer D, et al. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest Ophthalmol Vis Sci. 2010;51:4289–4297.
24 Nag TC, Wadhwa S, Chaudhury S. The occurrence of cone inclusions in the ageing human retina and their possible effect upon vision: an electron microscope study. Brain Res Bull. 2006;71:224–232.
25 Nordgaard CL, Karunadharma PP, Feng X, et al. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2848–2855.
26 Sadun AA, Carelli V, Salomao SR, et al. A very large Brazilian pedigree with 11778 Leber’s hereditary optic neuropathy. Trans Am Ophthalmol Soc. 2002;100:169–178. discussion 78–9
27 Carelli V. Leber’s hereditary optic neuropathy. Boston, MA: Butterworth-Heinemann; 2002.
28 Kong GY, Van Bergen NJ, Trounce IA, et al. Mitochondrial dysfunction and glaucoma. J Glaucoma. 2009;18:93–100.
29 Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10:12–31.
30 van der Walt JM, Nicodemus KK, Martin ER, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet. 2003;72:804–811.
31 van der Walt JM, Dementieva YA, Martin ER, et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365:28–32.
32 Fesahat F, Houshmand M, Panahi MS, et al. Do haplogroups H and U act to increase the penetrance of Alzheimer’s disease? Cell Mol Neurobiol. 2007;27:329–334.
33 Hassani-Kumleh H, Houshmand M, Panahi MS, et al. Mitochondrial D-loop variation in Persian multiple sclerosis patients: K and A haplogroups as a risk factor!. Cell Mol Neurobiol. 2006;26:119–125.
34 Dunaief JL, Dentchev T, Ying GS, et al. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442.
35 Huerta C, Castro MG, Coto E, et al. Mitochondrial DNA polymorphisms and risk of Parkinson’s disease in a Spanish population. J Neurol Sci. 2005;236:49–54.
36 Castro MG, Huerta C, Reguero JR, et al. Mitochondrial DNA haplogroups in Spanish patients with hypertrophic cardiomyopathy. Int J Cardiol. 2006;112:202–206.
37 Torroni A, Huoponen K, Francalacci P, et al. Classification of European mtDNAs from an analysis of three European populations. Genetics. 1996;144:1835–1850.
38 Torroni A, Schurr TG, Cabell MF, et al. Asian affinities and continental radiation of the four founding Native American mtDNAs. Am J Hum Genet. 1993;53:563–590.
39 Ruiz-Pesini E, Mishmar D, Brandon M, et al. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226.
40 Mishmar D, Ruiz-Pesini E, Golik P, et al. Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci U S A. 2003;100:171–176.
41 Udar N, Atilano SR, Memarzadeh M, et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2966–2974.
42 SanGiovanni JP, Arking DE, Iyengar SK, et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE. 2009;4:e5508.
43 Canter JA, Olson LM, Spencer K, et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE. 2008;3:e2091.
44 Jones MM, Manwaring N, Wang JJ, et al. Mitochondrial DNA haplogroups and age-related maculopathy. Arch Ophthalmol. 2007;125:1235–1240.
45 Wolf C, Gramer E, Müller-Myhsok B, et al. Mitochondrial haplogroup U is associated with a reduced risk to develop exfoliation glaucoma in the German population. BMC Genet. 2010;11:8.
46 Abu-Amero KK, Cabrera VM, Larruga JM, et al. Eurasian and Sub-Saharan African mitochondrial DNA haplogroup influences pseudoexfoliation glaucoma development in Saudi patients. Mol Vis. 2011;17:543–547.
47 Abu-Amero KK, Gonzalez AM, Osman EA, et al. Mitochondrial DNA lineages of African origin confer susceptibility to primary open-angle glaucoma in Saudi patients. Mol Vis. 2011;17:1468–1472.
48 Kofler B, Mueller EE, Eder W, et al. Mitochondrial DNA haplogroup T is associated with coronary artery disease and diabetic retinopathy: a case control study. BMC Med Genet. 2009;10:35.
49 Hofmann S, Bezold R, Jaksch M, et al. Wolfram (DIDMOAD) syndrome and Leber hereditary optic neuropathy (LHON) are associated with distinct mitochondrial DNA haplotypes. Genomics. 1997;39:8–18.
50 Brown MD, Sun F, Wallace DC. Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet. 1997;60:381–387.
51 Hendrickson SL, Jabs DA, Van Natta M, et al. Mitochondrial haplogroups are associated with risk of neuroretinal disorder in HIV-positive patients. J Acquir Immune Defic Syndr. 2010;53:451–455.
52 Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101:10726–10731.
53 Chinnery PF. Mitochondrial disorders overview. NCBI Bookshelf. Seattle, WA: National Library of Medicine, National Institutes of Health; 2010.
54 Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies – disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114.
55 LeDoux SP, Druzhyna NM, Hollensworth SB, et al. Mitochondrial DNA repair: a critical player in the response of cells of the CNS to genotoxic insults. Neuroscience. 2007;145:1249–1259.
56 Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–318.
57 Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat Res. 1992;275:331–342.
58 Wang AL, Lukas TJ, Yuan M, et al. Increased mitochondrial DNA damage and down-regulation of DNA repair enzymes in aged rodent retinal pigment epithelium and choroid. Mol Vis. 2008;14:644–651.
59 Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623.
60 Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517.
61 Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood). 2002;227:671–682.
62 Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Radic Biol Med. 1995;18:1033–1077.
63 Berneburg M, Plettenberg H, Medve-Konig K, et al. Induction of the photoaging-associated mitochondrial common deletion in vivo in normal human skin. J Invest Dermatol. 2004;122:1277–1283.
64 Meissner C, Bruse P, Mohamed SA, et al. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Exp Gerontol. 2008;43:645–652.
65 Kraytsberg Y, Kudryavtseva E, McKee AC, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520.
66 Markaryan A, Nelson EG, Hinojosa R. Quantification of the mitochondrial DNA common deletion in presbycusis. Laryngoscope. 2009;119:1184–1189.
67 Barreau E, Brossas JY, Courtois Y, et al. Accumulation of mitochondrial DNA deletions in human retina during aging. Invest Ophthalmol Vis Sci. 1996;37:384–391.
68 Barron MJ, Johnson MA, Andrews RM, et al. Mitochondrial abnormalities in ageing macular photoreceptors. Invest Ophthalmol Vis Sci. 2001;42:3016–3022.
69 Karunadharma PP, Nordgaard CL, Olsen TW, et al. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51:5470–5479.
70 Sciacco M, Bonilla E, Schon EA, et al. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3:13–19.
71 Krishnan KJ, Reeve AK, Samuels DC, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279.
72 Lin H, Xu H, Liang FQ, et al. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:3521–3529.
73 Kong YX, Van Bergen N, Trounce IA, et al. Increase in mitochondrial DNA mutations impairs retinal function and renders the retina vulnerable to injury. Aging Cell. 2011;10:572–583.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 32 Mitochondrial Genetics of Retinal Disease
Mitochondrial origins
It is now accepted that the origin of mitochondria within eukaryotes is the result of an endosymbiotic relationship and that mitochondrial (mt) DNAs can be traced to an α-proteobacterial genome.1,2 This theory has been supported by phytogenetic patterns of gene arrangements, small subunit ribosomal RNAs (rRNA) and protein data.3,4 DNA sequencing studies have shown that tremendous variations are still present within mtDNA, with the genome of the protozoan Reclinomonas americana having the most bacteria-like mitochondrial genome and Rickettsia prowazekii the most mitochondria-like eubacterial genome.1 The mtDNA from eukaryote species show remarkable differences in size, ranging from 6 to 60 kb. They also vary in shape, with some eukaryotes having linear mtDNA while others have circular mtDNA.
Mitochondrial structure
Depending on the energy requirement of each cell, the number of mitochondria varies from one to several thousand. Each mitochondrion is divided into compartments that are contained within the outer membrane, and include the intermembrane space, the inner membrane, cristae, and the matrix (Fig. 32.1). The outer membrane is permeable to molecules smaller than 5 kDa, which pass through the lipid bilayer channels called porins (voltage-dependent anion channel) into the intermembrane space. It also contains a translocase of the outer membrane complex involved in the import of resident mitochondrial proteins that are encoded by the nuclear genome and produced in the cytosol.5 The inner membrane has a high content of cardiolipin and its selective permeability allows only specific molecules into the matrix. The surface area is greatly expanded as a result of the numerous invaginations of the inner membrane, known as cristae. Embedded within the inner membrane are many of the enzyme complexes required for adenosine triphosphate (ATP) production and the translocase of the inner membrane complex that is responsible for the import of nuclear-encoded proteins into the matrix. The matrix contains numerous proteins, ribosomes, tRNA, and the mtDNA.
Mitochondrial DNA
Mitochondria are unique in that they have their own DNA that is inherited through the maternal lineage. The human mtDNA forms a closed circle of double-stranded DNA, with 16 569 nucleotide pairs, comprised of two strands that are differentiated by their nucleotide content. The heavy strand is guanine-rich and encodes for 28 genes while the light strand is cytosine-rich and encodes for nine genes. Unlike the nuclear genome, mtDNA contains a unique noncoding control region but no introns. The noncoding mtDNA Dloop has within it the 1121 nucleotide control region that is important for replication and transcription. The coding region of mtDNA codes for 37 genes, including 13 protein subunits essential for oxidative phosphorylation (OXPHOS), two ribosomal RNAs, and 22 transfer RNAs (Fig. 32.2).6–8 The vast majority of mitochondrial proteins (~1500–2000) that contribute to energy biogenesis4,9 are encoded by nuclear DNA and imported into the mitochondria.
Within a cell there is a single DNA copy of the nuclear genome (nDNA) but multiple copies of mtDNA because there can be thousands of mitochondria per cell, and within each mitochondrion, 1–10 copies of mtDNA. With aging and exposure to oxidative stress, mtDNA molecules can be damaged, which results in a mixture of nonmutated (wild-type) and mutant mtDNA within the same cell. This mixture of damaged and undamaged mtDNA is termed “heteroplasmy.” When cells with heteroplasmic mitochondria divide, the two types of mtDNA are randomly or, in some instances, nonrandomly distributed into the daughter cells.10–14 Alternatively, cells may have either a pure mutant mtDNA or pure nonmutant (wild-type) mtDNA population, in which case it is referred to as homoplasmic mtDNA.15 Homoplasmy within a cell indicates that all mtDNA copies are identical. Cells can function only with relatively low levels of heteroplasmy but once this threshold is breached, abnormal function and disease can occur. Although low levels of heteroplasmic mtDNA defects may have an effect on function, the mtDNA changes are not always obvious and special technical approaches are required to ensure their detection. Correlating a phenotype with mtDNA defects can be difficult because the complexity of the phenotype can be influenced by when (time during embryogenesis/development) and where (tissue type) the mutation arises.16–18 In addition, environmental factors, such as oxidative stress, can modulate the expression of the phenotype. mtDNA is particularly susceptible to oxidative damage because it resides in the matrix and is in close proximity to sites of reactive oxygen species (ROS) formation. In addition, the mitochondria have a poor DNA repair process and a high transcription rate. Oxidative damage to mtDNA is especially prevalent in very metabolically active tissues such as the retina, brain, and muscle.
Electron leakage and ROS formation
Under normal conditions, ATP is produced via a series of oxidation–reduction reactions that involve the flow of electrons through complex I–IV of the ETC. Electron transfer generates the energy required to transport protons across the inner mitochondrial membrane into the inner-membrane space. The flow of the protons down the concentration gradient from the inner-membrane space into the matrix through complex V, also known as ATP synthase, provides the energy to generate ATP from adenosine diphosphate. Under optimal conditions, molecular oxygen captures four electrons released from complex IV to form water. However, partial reduction of oxygen can produce potentially harmful intermediates, such as superoxide, peroxide, and hydroxyl radicals, collectively known as ROS. Additionally, ineffective transfer of electrons between ETC complexes can permit partial reduction of oxygen and production of ROS byproducts during normal oxidative metabolism. In fact, approximately 2–5% of the oxygen we consume is only partially reduced and forms ROS.19
Localization of mitochondria within the retina and optic nerve
The retina has one of the highest oxygen consumption rates in the body. To meet the energy demand for vision, the photoreceptor cells have a high concentration of mitochondria within the inner segments (Fig. 32.3). The photoreceptors have high exposure to ultraviolet light and OXPHOS levels, leading to increased ROS formation, which makes these cells a target for mtDNA damage. In vitro studies show that oxidative stress leads to preferential damage of mtDNA compared to nuclear DNA.20 Animal models demonstrate increased mtDNA deletions and damage in aging and degenerating retinas.21,22 Mitochondrial abnormalities, including mtDNA damage, have been found in aging and age-related macular degeneration (AMD) retinas.20,21,23–25 The retinal ganglion cells (RGC) with their long axons are also susceptible to mitochondrial damage. In RGCs, the mitochondria are present around the nucleus and along the axons but they tend to accumulate just anterior to the lamina cribrosa (Fig. 32.2).26 The numbers of RGCs and their axons are significantly decreased in Leber’s hereditary optic neuropathy and glaucoma.27,28 This may be related to lower energy production caused by mtDNA damage and dysfunction.
Influences of mtdna ON cell function
Influences of mtDNA upon cell function can be classified into ancient adaptive polymorphic changes (haplogroups), recent mutations, and somatic mtDNA changes.29 Examples of each are presented below.
Ancient inherited mtDNA variants representing populations (haplogroups)
Definition of haplogroups
Haplogroups are mtDNA sequence polymorphism variations that have occurred over more than 150 000 years and correlate with the geographic origins of populations traced through the maternal lineages. The oldest haplogroups are from Africa and with geographic migration and climate adaptations, European, Asian, and Native American haplogroups have evolved.4 Each haplogroup has related patterns of mtDNA sequences (haplotypes) that represent that population. If the specific single polynucleotide polymorphism (SNP) variants representing the haplogroup are found in the mtDNA Dloop, it can affect the replication and transcription rates. If within the coding region, the SNP variants can be nonsynonymous (amino acid changing), which can potentially alter efficiencies of energy production, causing ROS formation, apoptosis, and cell death. This means that each haplogroup, with its different set of SNPs, can produce unique bioenergetic as well as biochemical properties. Haplogroups increasingly are being correlated with a broad spectrum of common diseases, including age-related diseases, such as Parkinson’s disease and Alzheimer’s disease.30–40
