[level-membership-for-neurology-category]
Chapter 63 Mitochondrial Disorders
The bacterial hypothesis of the origin of mitochondria suggests that approximately 1 to 2 billion years ago, alpha-purple bacteria were incorporated into evolving eukaryotic cells. During evolution, these bacteria transferred many of their essential genes to the nuclear chromosomes. Mitochondria still have many remnants of their bacterial origin, such as the use of N-formylmethionyl-tRNA (transfer ribonucleic acid) as the initiator of protein synthesis. Some unicellular eukaryote “protists” lack mitochondria, supporting the notion that these protists were offshoots of the primitive eukaryotic lineage that diverged before the mitochondrial symbiosis developed. This hypothesis has recently been challenged by the discovery of the genetic remnants of mitochondria in the nucleus and cytoplasm of mitochondria-lacking protists. These mitochondrial remnants and other cytosolic structures such as hydrogenosomes and mitosomes are joining a growing list of cryptic mitochondrial relics in eukaryotes that are capable of producing energy in even hostile and anoxic habitats and can perform functions other than respiration (for a review, see Tielens et al., 2002). There is therefore still much to be understood about the evolution of mitochondria beyond the hypothesis of bacterial symbiosis.
In 1988, a major breakthrough in our understanding of mitochondrial disorders occurred with the report of the association of sporadic human encephalomyopathies with large deletions of the mtDNA (Holt et al., 1988) and of Leber hereditary optic neuropathy (LHON) with a point mutation at nucleotide pair (np) 11778 in the mtDNA. More than 170 pathogenic point mutations of the mtDNA, large-scale mtDNA deletions, and rearrangements have been subsequently reported (Schon and DiMauro, 2007). More than 85% of mitochondrial proteins are encoded by the nuclear DNA (nDNA), and therefore many unknown mitochondrial disorders due to nDNA defects may exist. The list of nDNA defects affecting mitochondrial function, including mtDNA replication and integrity, has been growing in recent years (DiMauro and Hirano, 2005). The delineation of human mtDNA variation and genetics has also provided startling new insights into the evolution and migration of human populations (Wallace, 1995). Mitochondria may also play an important role in neurodegenerative diseases and the aging process (Schapira, 2006, 2008; Turner and Schapira, 2010).
The diseases included under the term mitochondrial disorders are so diverse and involve so many parts of the nervous system and other organs that the whole spectrum cannot be easily addressed in any one chapter. Therefore the disorders related to intermediate metabolism and mitochondrial Krebs cycle are discussed with inborn errors of metabolism in Chapter 62. The syndrome that combines epilepsy and ragged-red muscle fibers (myoclonic epilepsy and ragged-red fibers, MERRF) is discussed in Chapters 40 and 67. The syndrome of progressive external ophthalmoplegia (PEO) is discussed with other abnormalities of eye movement (Chapter 16), and LHON with other causes of vision loss (Chapter 14). This chapter overviews the principles of mitochondrial genetics and mitochondrial pathophysiology and provides a summary of the clinical features and management of patients with mitochondrial disorders.
Genetics of Mitochondrial Disorders
MtDNA is a 16,569-np double-stranded, closed, circular molecule located within the matrix of the double-membrane mitochondrion. Each human cell contains a dynamic network of mitochondria and hundreds of mtDNA molecules. Human mtDNA encodes 13 of the 89 subunits of the mitochondrial respiratory chain, as well as the small (12S) and large (16S) ribosomal RNAs (rRNAs) and 22 tRNAs necessary for intramitochondrial protein synthesis (Fig. 63.1, A).
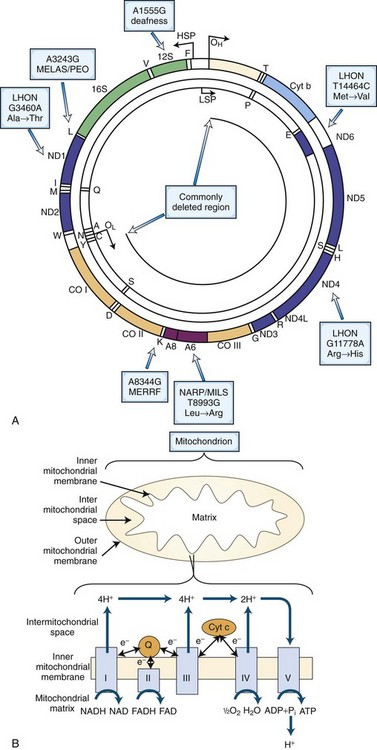
The mtDNA is replicated and transcribed by using an origin and promoter for each of the two DNA strands, the G-rich heavy (H) strand and the C-rich light (L) strand. The H- and L-strand replication origins (OH and OL; see Fig. 63.1, A) are relatively distant within the molecule, but the H- and L-strand promoters (PH and PL) are closely spaced and located adjacent to OH in the approximately 1000-np noncoding control region. This region also encompasses the D-loop, which is formed by replication initiation events from the OH. Research involving evolution of mammalian species and origin and migration of humans on earth has focused on the variation in a small hypervariable noncoding region within the D-loop. The replication mechanism of mtDNA may be more complex than originally suggested, in that the mtDNA can also replicate by the extension of both leading and lagging strands, a process resembling nDNA replication (Holt et al., 2000).
The respiratory chain (see Fig. 63.1, B) is located within the mitochondrial inner membrane and is composed of five multimeric enzyme complexes whose genes are dispersed between the mtDNA and nDNA. Complex I (NADH–ubiquinone oxidoreductase) accepts electrons from the reduced form of nicotinamide adenine dinucleotide (NADH), whereas complex II (succinate–ubiquinone oxidoreductase) collects electrons from succinate. Both NADH and succinate are the products of the Krebs cycle. Enzyme complexes I and II transfer electrons to coenzyme Q10 (CoQ10). From CoQ10, the electrons flow through complex III (ubiquinone–cytochrome c oxidoreductase) to cytochrome c, then to complex IV (cytochrome c oxidase), and finally to oxygen to yield water. The electron transfer is coupled to proton (H+) pumping by complexes I, III, and IV from the matrix to the intermembrane space, creating an electrochemical gradient across the inner membrane. This electrochemical gradient is utilized by complex V (adenosine triphosphate [ATP] synthase) as a source of energy to condense adenosine diphosphate (ADP) and inorganic phosphate (Pi) to synthesize ATP. ATP and ADP are then exchanged across the mitochondrial membrane by the adenine nucleotide translocator (ANT).
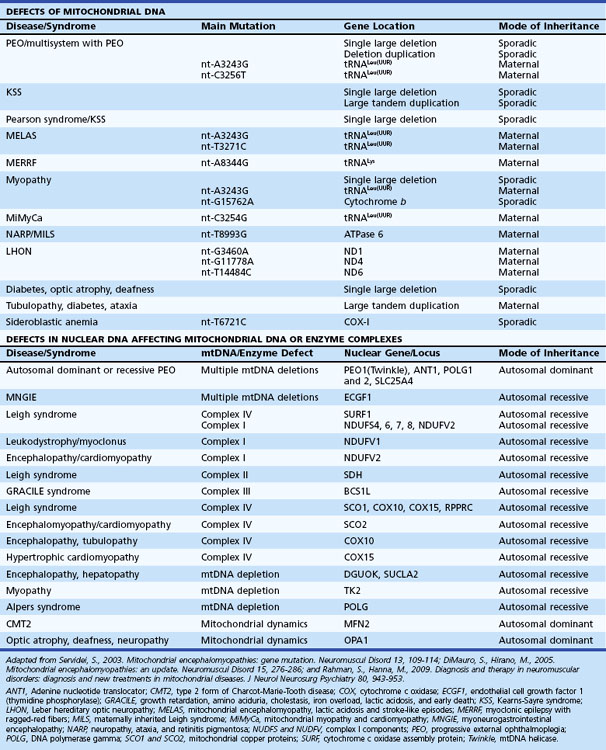
Mitochondrial diseases therefore can arise from defects in the mtDNA (sporadic or maternal inheritance, see following section) or nDNA (sporadic or Mendelian inheritance). Nuclear DNA-related mitochondrial disorders result from defects involving nDNA-encoded mitochondrial polypeptides including respiratory chain complexes, respiratory chain assembly, mtDNA maintenance, protein import, lipid dynamics, and biosynthesis of CoQ10. Table 63.1 summarizes a simplified clinical and genetic classification of mitochondrial diseases.
Maternal Inheritance of Mitochondrial DNA
Patients inherit mtDNA from their mothers, and therefore the mode of transmission of mtDNA, including pathogenic mtDNA mutations, follows maternal line inheritance. A single case report has demonstrated paternal transmission of a mtDNA mutation (Schwartz and Vissing, 2002). Maternal inheritance implies maternal transmission of mtDNA to all offspring but subsequent transmission only by females. Thus a disease expressed in all children of both sexes of an affected individual, without evidence of paternal inheritance, strongly suggests an mtDNA point mutation. However, exceptions to this general rule are encountered in clinical practice. First, a de novo point mutation in the mtDNA of the maternal germ cell line will not necessarily be transmitted to all children. Second, for unknown reasons, mtDNA mutations involving large-scale deletions, rearrangements, and point mutations in some protein-coding genes may occur sporadically and be due to mutations arising in the oocyte. Bottleneck expression of an mtDNA mutation may be enhanced in the fetus. Multiple mtDNA deletions and depletions are autosomally transmitted, as they are the consequence of mutations in nuclear-encoded factors involved in mtDNA metabolism or replication. Third, maternal inheritance may not always be clinically evident because of extreme variability of clinical expression among family members due to heteroplasmy and the threshold effect (described in the following section).
Pathophysiology of Mitochondrial Disorders
Mitochondria have several functions in maintaining cellular homeostasis, such as transient storage of intracellular calcium, fatty acid oxidation, the Krebs cycle, and iron metabolism (McBride et al., 2006; Schapira, 2006). Mitochondria also have a key role in the regulation of apoptosis (Wang, 2001). One of the most important roles of mitochondria is to catalyze the phosphorylation of the majority of cellular ADP to ATP. ATP is generated by oxidation of intermediates such as NADH and FADH2 via the process of oxidative phosphorylation within mitochondria by the respiratory chain enzymes. Several intermediary metabolic pathways from carbohydrates, fatty acids, and amino acids converge in mitochondria at the level of acetyl-CoA for the final conversion of the fuel into ATP. Pyruvate is the terminal product of anaerobic glycolysis and is transported across the inner mitochondrial membrane to the mitochondrial matrix where it is converted to acetyl CoA. The transportation of pyruvate is coupled with the influx of hydrogen ions down their electrochemical gradient across the inner mitochondrial membrane. Pyruvate can also be generated during the catabolism of the amino acids, alanine, serine, glycine, and cysteine. Transport of free fatty acids across the mitochondrial membrane requires two enzymes (carnitine palmitoyltransferases [CPT] I and II), a carrier molecule (l-carnitine), and a translocase (carnitine-acylcarnitine translocase). Fatty acids are also metabolized into acetyl-CoA. Acetyl-CoA enters the Krebs cycle, where three molecules of NADH and one molecule of the reduced form of flavin adenine dinucleotide (FADH2) are produced from each acetyl-CoA. Molecules of NADH and FADH2 donate electrons to the electron transport chain (NADH to complex I and succinate to FAD in complex II). The functional unit comprising the electron transport chain (complex I to IV) and complex V (ATP synthase) constitutes the oxidative phosphorylation (OXPHOS) system (see Fig. 63.1, B).
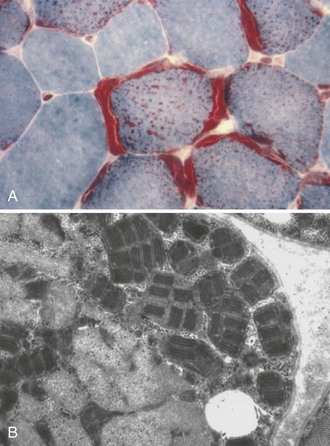
In skeletal muscle, some fibers are severely involved and others may appear normal on histological analysis. With more severe involvement, the combination of patchy myofibrillar degeneration along with mitochondrial proliferation gives rise to the so-called ragged-red appearance of fibers on modified Gomori trichrome staining (Fig. 63.2, A). Defective OXPHOS may result in compensatory mitochondrial proliferation, particularly in type I and IIA muscle fibers. Ultrastructural analysis can demonstrate abnormal mitochondrial morphology such as intramitochondrial paracrystalline inclusions (see Fig. 63.2, B). Patients with mitochondrial disorders can show a wide range of symptoms, including any combination of developmental delay, short stature, small muscle bulk, seizures, vision loss, hearing impairment, peripheral neuropathy, autonomic nervous system difficulties, gastrointestinal dysfunction, endocrine problems, hematopoietic disease, and failure to thrive (Box 63.1). The presentation of a particular mitochondrial disorder can be variable, even among affected individuals in the same family. Conversely, more than one pathogenic mutation can give rise to similar clinical phenotypes.
Approach to the Diagnosis of Mitochondrial Disorders
A detailed clinical history and examination in conjunction with experienced interpretation of a battery of complex laboratory results is often required to make an accurate diagnosis. This process is often best carried out in a specialist mitochondrial clinic and subsequently discussed at a multidisciplinary meeting involving neurologists, pediatricians, geneticists, pathologists, and biochemists. Consensus diagnostic criteria for mitochondrial disorders in infants and children (Morava et al., 2006; Wolf and Smeitink, 2002) and adults (Bernier et al., 2002) have been proposed.
Laboratory Findings
The mitochondrial metabolic test battery includes serum creatine kinase (CK), lactate and pyruvate, plasma and urine acylcarnitines, blood and urine amino acids, urine organic acids, and cerebrospinal fluid (CSF) lactate and pyruvate (if the CNS is involved). The lactate/pyruvate ratio may differentiate disorders of the OXPHOS system in comparison to more proximal metabolic defects such as pyruvate dehydrogenase deficiency. However, normal values for the lactate and lactate/pyruvate ratio in a patient do not exclude mitochondrial disease; mtDNA-related diseases are generally associated with normal or only mildly elevated resting blood lactate levels but often with a significant rise with exercise. Serum CK is usually normal or mildly elevated in patients with mitochondrial myopathies. Blood levels of free carnitine are often decreased in mtDNA-related disorders, with a relative increase in acylcarnitine levels. The interpretation of test results of free and total carnitine, blood and urine amino acids, and urine organic acids is discussed in Chapter 62.
Neuroradiology
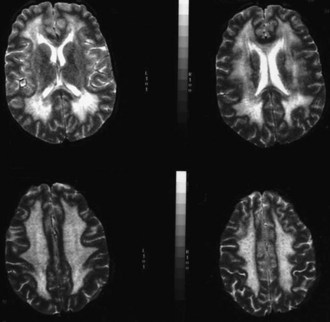
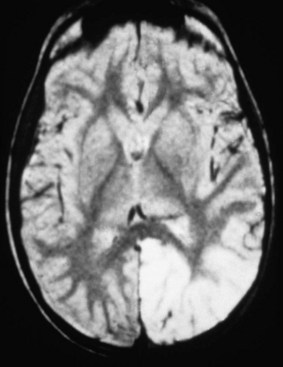
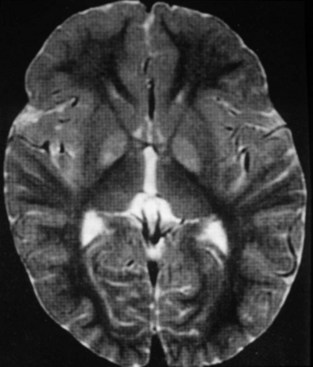
The use of neuroimaging, especially brain magnetic resonance imaging (MRI), has greatly facilitated the detection of CNS involvement in mitochondrial disorders. Brain atrophy is common in children with mitochondrial disease. Developmental delay and basal ganglia calcification are common in KSS and MELAS, and diffuse signal abnormalities of the white matter are characteristic of KSS and myoneurogastrointestinal encephalopathy (MNGIE) (Fig. 63.3). The diagnosis of MELAS can be aided by the clinical association of stroke-like episodes with radiological lesions that do not conform to the anatomical territories of blood vessels and predominantly involve cortical gray matter (Fig. 63.4). The initial or predominant lesions in MELAS are characteristically in the parietal-occipital region, and new lesions generally appear with acute illness and elevated CSF lactate levels. Leigh syndrome characteristically shows bilateral hyperintense signals on T2-weighted and fluid-attenuated inversion recovery (FLAIR) MRIs in the putamen, globus pallidus (Fig. 63.5), and thalamus. 1H-MRS often detects lactate accumulation in the CSF and in specific areas of the brain.
Muscle Biopsy
Histochemistry
The hallmark feature in mitochondrial diseases is the ragged-red fiber (RRF) (see Fig. 63.2, A). In frozen sections stained with modified Gomori trichrome, subsarcolemmal and intermyofibrillar accumulation of mitochondria appear as bright red masses on at least three sides of the fiber, against the background of the blue myofibrils. These abnormal accumulations represent a compensatory proliferation of mitochondria, some of which are ultrastructurally normal and others dystrophic (see Fig. 63.2, B). The same fibers stain intensely blue with the histochemical reaction for succinate dehydrogenase (SDH), an OXPHOS enzyme encoded entirely by the nDNA. SDH staining is more sensitive than the modified Gomori trichrome in detecting mitochondrial proliferation. NADH-tetrazolium reductase (NADH-TR) stains mitochondria-rich fibers even more intensely, but the enzyme reaction is less specific than SDH for mitochondria. RRFs are seen in most patients with mtDNA defects and in some patients with nDNA mutations, but RRFs are neither a universal feature in mitochondrial disorders, nor are they specific for primary mitochondrial diseases. RRFs can occur in other neuromuscular diseases such as inflammatory myopathies or inclusion body myositis, as well as in normal aging. They also occur in the toxic myopathy caused by the drug, zidovudine, where the underlying pathogenesis is drug-induced damage of the mtDNA. Mitochondrial proliferation in the smooth muscle of intramuscular vessels results in strongly SDH-reactive vessels in MELAS. The histochemical stain for cytochrome oxidase (COX) activity may be helpful. COX or complex IV of the mitochondrial respiratory chain has 13 subunits; three—COX I, II, and III—are encoded in mtDNA and the others are encoded in the nuclear genome. COX may be absent in myofibers from patients with defects of mtDNA, mitochondrial transcription and translation, or assembly of complex IV. RRFs which are COX-negative suggest impaired mitochondrial protein synthesis in the face of mitochondrial proliferation and are typically seen in mtDNA deletions. In the combined COX-SDH histochemical stain, COX-negative fibers with normal or high concentrations of SDH stain blue against a background of normal brown fibers that have both COX and SDH. Single-fiber polymerase chain reaction (PCR) from these fibers (RRF/SDH-rich and COX-negative) shows higher levels of mutated mtDNA molecules, suggesting that these mutations are deleterious. Discordance between RRF status and COX activity is not uncommon in muscle fibers in mitochondrial diseases. In patients with mutations in mtDNA protein-coding genes and defects in complex IV, RRF and many non-RRFs are COX negative or deficient, whereas in patients with defects of complex I or III and tRNA mtDNA point mutations, many RRFs with normal COX activity may be seen. SCO2 and SURF1 nDNA mutations, which cause complex IV deficiency, are generally associated with diffuse COX deficiency but without RRFs. The absence of either RRFs or COX-negative fibers does not rule out mitochondrial disease. For instance, patients with NARP-MILS syndrome (neuropathy, ataxia, retinitis pigmentosa and maternally inherited Leigh syndrome) or LHON do not have RRF or COX-negative fibers.
Electron Microscopy
Electron microscopy of muscle biopsy specimens from patients with mitochondrial diseases may reveal subsarcolemmal and intermyofibrillar proliferation of mitochondria and the presence of abnormal mitochondria in muscle fibers. Enlarged, elongated, irregular, and dumbbell-shaped mitochondria with hypoplastic and dystrophic cristae and paracrystalline inclusions (see Fig. 63.2, B) may be seen in a patient’s muscle biopsy with a diagnosis of mitochondrial disease. However, they are nonspecific and can be present in other neuromuscular disorders. Significant intermyofibrillar mitochondrial proliferation should be detectable by light microscopy in the modified Gomori trichrome, NADH-TR, and SDH stains. Isolated focal collections of mitochondria in the subsarcolemmal space or near the A-I junction in muscle fiber can be normal and should not be mistaken for pathological accumulation.
Blue Native Polyacrylamide Gel Electrophoresis
One of the most powerful tools to appear in the last few years for the analysis of the OXPHOS complexes is the resolution of the individual fully assembled complexes in blue native polyacrylamide gel electrophoresis (BN-PAGE). By using mild detergents, the OXPHOS complexes normally remain intact in the electrophoretic field, but they begin separating from each other if components of the OXPHOS are abnormal (Ugalde et al., 2004). The complexes and their components can be detected by direct staining of proteins, enzyme activity, or antibodies.
DNA-Based Diagnosis
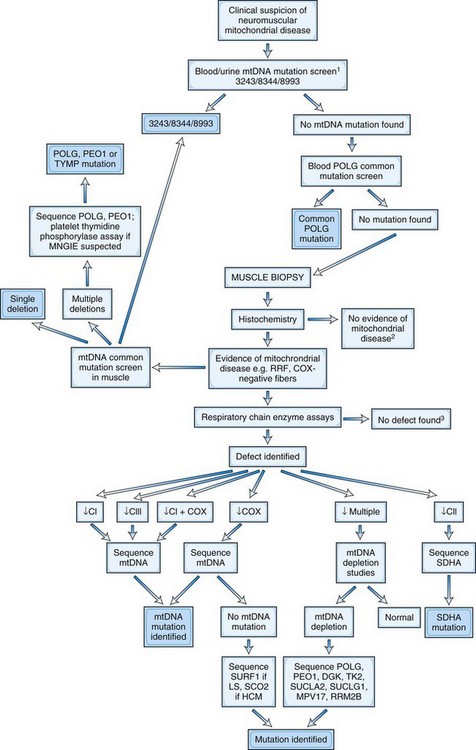
A large number of mtDNA and nDNA mutations are now known to cause mitochondrial disorders, and their number (especially nDNA gene mutations) has continued to rise in recent years (DiMauro and Hirano, 2005) Further details about mtDNA mutations can be found in the MitoMap database, http://www.mitomap.org. If a patient has a clearly defined clinical syndrome such as MELAS, MERRF, LHON, or NARP, the most common mutations associated with the syndrome can be initially screened in blood (see Table 63.1). Genetic tests for mutations at positions 3243, 8344, and 8993 are now widely available in diagnostic laboratories. The majority of LHON cases will have one of the three common point mutations at positions 11778, 14484, or 3460. The level of np-3243A>G in blood declines with age and may become undetectable over the age of 30.
Fig. 63.6 demonstrates an algorithm for the DNA diagnosis of a patient with suspected mitochondrial disease.
Mitochondrial Clinical Syndromes
Mitochondrial Myopathies without Progressive External Ophthalmoplegia
The clinical spectrum of isolated mitochondrial myopathy varies from mild nondisabling proximal limb weakness to severe infantile myopathy with lactic acidosis and death by 1 year. Exercise intolerance is common. Some of these cases present in adult life, but careful questioning usually elicits a history of lifelong exercise intolerance (weakness, fatigue, shortness of breath, and tachycardia). A sporadic form of myopathy related to somatic mutations in the cytochrome b gene of mtDNA is associated with progressive exercise intolerance and weakness and, in some cases, attacks of rhabdomyolysis (Andreu et al., 1999). Rare patterns associated with a cytochrome b mutation include weakness and exertional myoglobinuria. Less frequently, sporadic patients with exercise intolerance have been found to have mtDNA mutations in genes encoding subunits of complexes I or IV (DiMauro and Hirano, 2005). Some patients with mitochondrial myopathy without PEO will develop progressive PEO in later life, and others may have overlapping deficits with MERRF and MELAS.
Mitochondrial Peripheral Neuropathy
Patients with complex mtDNA-associated mitochondrial phenotypes involving the CNS (e.g., MERRF, MELAS) often have a mild axonal sensorimotor neuropathy that may be subclinical. A peripheral neuropathy can be the dominant clinical feature in some patients. Mutations in the mitofusin 2 (MFN2) gene, which encodes a protein that influences mitochondrial dynamics, is a cause of Charcot-Marie-Tooth disease (CMT). Some families with MFN2 mutations have additional clinical features including optic atrophy (Züchner et al., 2006). Mutations in POLG can cause a prominent large-fiber sensory neuropathy with significant proprioceptive loss in the SANDO (sensory ataxic neuropathy dysarthria and ophthalmoplegia) syndrome. Axonal sensorimotor polyneuropathy may also be a feature of dominant optic atrophy caused by mutations in the OPA1 gene encoding a mitochondrial dynamin-like protein.
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes
A typical radiological feature is that the stroke involves the cerebral cortex, spares the white matter (see Fig. 63.4), mostly affects the parietal and occipital cortices, and does not conform to vascular territories. Neuroimaging may show additional lesions that have no clinical correlates. The onset is generally in childhood or early adult life. Most patients have RRF on muscle biopsy. Roughly 80% of patients with MELAS have an A-to-G point mutation at np-3243 (tRNALeu[UUR] gene, MT-TL1). Another point mutation at np-3271 of the tRNALeu(UUR) gene accounts for 10% of cases of MELAS.
Myoclonic Epilepsy with Ragged-Red Fiber Myopathy
MERRF is a maternally inherited encephalomyopathy characterized by myoclonus, epilepsy, cerebellar ataxia, and myopathy with RRF. Onset is usually in childhood or early adulthood. The syndrome begins with stimulus-sensitive myoclonic epilepsy in childhood, which may be photosensitive. Worsening ataxia and mental retardation is seen in later childhood. Patients may also develop cardiomyopathy, short stature, deafness, optic atrophy, PEO, cutaneous lipomas, and neuropathy. Overlapping clinical features of MERRF and MELAS can occur in the same patient or among different members of the same family (Verma et al., 1996). The clinical course in MERRF is variable, but it is typically progressive.
Mitochondrial Neurogastrointestinal Encephalomyopathy
MNGIE is an autosomal recessive disease with secondary alterations of mtDNA. There is typically a combination of ptosis, PEO, severe gastrointestinal dysmotility leading to episodes of pseudoobstruction and cachexia, peripheral neuropathy, leukoencephalopathy on brain MRI (see Fig. 63.3), and evidence of mitochondrial dysfunction (e.g., lactic acidosis or RRF in muscle biopsy) (Hirano et al., 2004). Onset is usually in the late teens, and most patients die before age 40. MNGIE is caused by mutations in the gene encoding thymidine phosphorylase. The disease can be diagnosed by blood tests demonstrating loss of thymidine phosphorylase activity or elevation of plasma thymidine and deoxyuridine.
Subacute Necrotizing Encephalomyelopathy (Leigh Syndrome)
Leigh syndrome is a familial or sporadic mitochondrial disorder characterized by psychomotor regression and lesions in the basal ganglia and brainstem (see Fig. 63.5). Some cases display a maternal inheritance, such as mtDNA np-8993 and np-8344 mutations (MILS). Others follow an autosomal (pyruvate carboxylase, SURF1 gene mutations with COX deficiency, complex I deficiencies) or sex-linked (pyruvate dehydrogenase E1 gene mutations) pattern of inheritance. Over 50% of cases present in the first year of life, usually before 6 months of age. Late-onset varieties with a greater degree of clinical heterogeneity are also reported. The precise clinical boundaries of Leigh syndrome have not been defined; there is clinical heterogeneity even among members of the same family. Leigh syndrome and congenital lactic acidosis are described further in Chapter 62.
Management of Mitochondrial Diseases
Treatment of Associated Complications
Treatment of mitochondrial disease is mainly symptomatic, empirical, and often palliative (DiMauro and Mancuso, 2007). Patients and families with confirmed mitochondrial disease require management and support in a multidisciplinary clinical team setting. This is often coordinated by a neurologist with close links to a range of different disciplines such as rehabilitation medicine, physiotherapy, occupational therapy, cardiology, endocrinology, ophthalmology, audiology, and speech therapy. There is usually no specific treatment for most mitochondrial disorders, and therefore monitoring and treatment of complications arising from the disease is vital for improving quality of life and reducing morbidity.
Pharmacological Approaches
Other Pharmacological Approaches
A number of other pharmacological agents have been tried in mitochondrial disease but with limited benefit. A recent Cochrane systematic review concluded that there was insufficient evidence to recommend any standard treatment. There are anecdotal reports of benefit of various agents (e.g., riboflavin, succinate, l-carnitine, α-lipoic acid creatine (Tarnopolsky et al., 1997), and vitamins C, E, and K), but the clinical heterogeneity and unpredictable natural history of mitochondrial disease, with a frequently relapsing-remitting course, makes interpretation of the effectiveness of any given agent in a single individual difficult. The few randomized double-blind clinical trials that have been performed yielded inconclusive or conflicting results. Recently some novel pharmacological approaches have emerged, aimed at stimulating mitochondrial biogenesis via the transcriptional coactivator, PGC-1. Drugs that may stimulate this pathway include bezafibrate and resveratrol.
Removal or Neutralization of Toxic Metabolites
The pathophysiological mechanism of MNGIE syndrome (thymidine phosphorylase deficiency) is considered to result from an imbalance of intramitochondrial nucleosides, leading to stalling of the mtDNA replication apparatus. A rationale for treatment is thus to restore intramitochondrial nucleoside balance by removing accumulated nucleosides. In one study, renal dialysis was used to remove accumulated plasma thymidine and deoxyuridine in patients with MNGIE, but these metabolites reaccumulated within 24 hours of dialysis. Diuretics have also been used to increase renal excretion of thymidine and deoxyuridine, but without success. Bicarbonate may be used to correct acute or chronic lactic acidosis. Dichloroacetate (DCA) is an inhibitor of pyruvate dehydrogenase (PDH) and thus maintains PDH in its active (phosphorylated) state, resulting in reduced lactate production. DCA can be effective in lowering lactate levels in acute acidotic states. Recently, a double-blind placebo-controlled trial aimed to investigate the efficacy of DCA in the MELAS syndrome but had to be terminated prematurely because of reversible peripheral nerve toxicity (Kaufmann et al., 2006).
Enzyme and Metabolite Replacement
Gene Therapy
Resistance Exercise Training to Shift mtDNA Genotype
The proportion of mutant mtDNA in muscle correlates with the degree of reduction in oxidative capacity. Recently there has been increasing interest in the role of exercise therapy to improve muscle respiratory chain oxidative capacity by potentially reducing mutant mtDNA load—a process known as gene shifting. Certain mtDNA mutations such as deletions and some tRNA point mutations are present in high levels in mature skeletal muscle, but for reasons that remain unclear, they are absent from the muscle satellite cell population, which harbors only wild-type mtDNA. Previous experimental work demonstrated that activation of satellite cells has the potential to introduce wild-type mtDNA into mature skeletal muscle, thereby lowering the proportion of mutant mtDNA and reversing the respiratory chain defect. Certain types of exercise protocols have the potential to induce satellite cell activation and promote entry of wild-type mtDNA into mature muscle. Initial studies employed endurance training and demonstrated improved aerobic capacity (Taivassalo et al., 2006). More recent studies using 12-week progressive overload leg resistance exercise training protocols demonstrated increased muscle strength and improved muscle oxidative capacity, although there was no measurable reduction in deleted mtDNA.
Andreu A.L., Hanna M.G., Reichmann H., et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341:1037-1044.
Bernier F.P., Boneh A., Dennett X., et al. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406-1411.
DiMauro S., Hirano M. Mitochondrial encephalomyopathies: an update. Neuromuscul Disord. 2005;15:276-286.
DiMauro S, Mancuso M. Mitochondrial diseases: therapeutic approaches. Biosci Rep. 2007;27:125-137.
Hirano M., Nishigaki Y., Marti R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a disease of two genomes. Neurologist. 2004;10:8-17.
Holt I.J., Harding A.E., Morgan-Hughes J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331(6158):717-719.
Holt I.J., Lorimer H.E., Jacobs H.T. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515-524.
Kaufmann P., Engelstad K., Wei Y., et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324-330.
McBride H.M., Neuspeil M., Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16(14):R551-R560.
Morava E., van den Heuvel L., Hol F., et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823-1826.
Rahman S., Hanna M.G. Diagnosis and therapy in neuromuscular disorders: diagnosis and new treatments in mitochondrial diseases. J Neurol Neurosurg. Psychiatry. 2009;80:943-953.
Schon E.A., DiMauro S. Mitochondrial mutations: genotype to phenotype. Novartis Found Symp. 2007;287:214-225.
Schwartz M., Vissing J. Paternal inheritance of mitochondrial DNA. N Engl J Med. 2002;347:576-580.
Schapira A.H.V. Mitochondrial disease. Lancet. 2006;368(9529):70-82.
Schapira A.H.V. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008;7(1):97-109.
Taivassalo T., Gardner J.L., Taylor R.W., et al. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. 2006;129:3391-3401.
Tarnopolsky M.A., Roy B.D., MacDonald J.R. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20:1502-1509.
Tielens A.G., Rotte C., van Hellemond J.J., et al. Mitochondria as we don’t know them. Trends Biochem Sci. 2002;27:564-572.
Turner C., Schapira A.H. Mitochondrial matters of the brain: the role in Huntington’s disease. J Bioenerg Biomembr. 2010;42(3):193-198.
Ugalde C., Janssen R.J., van den Heuvel L.P., et al. Differences in assembly or stability of complex I and other mitochondrial OXPHOS complexes in inherited complex I deficiency. Hum Mol Genet. 2004;13:659-667.
Verma A., Moraes C.T., Shebert R.T., et al. MERRF/PEO overlap syndrome associated with the mitochondrial DNA 3243 mutation. Neurology. 1996;46:1334-1336.
Wallace D.C. 1994. William Allan Award Address. Mitochondrial DNA variation in human evolution, degenerative disease, and aging. Am J Hum Genet. 1995;57:201-223.
Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15(22):2922-2933.
Wolf N.I., Smeitink J.A.M. Mitochondrial disorders. A proposal for consensus diagnostic criteria in infants and children. Neurology. 2002;59:1402-1405.
Züchner S., De Jonghe P., Jordanova A., et al. Axonal neuropathy with optic atrophy (HMSN VI) is caused by mutations in mitofusin 2. Ann Neurol. 2006;59:276-281.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 63 Mitochondrial Disorders
The bacterial hypothesis of the origin of mitochondria suggests that approximately 1 to 2 billion years ago, alpha-purple bacteria were incorporated into evolving eukaryotic cells. During evolution, these bacteria transferred many of their essential genes to the nuclear chromosomes. Mitochondria still have many remnants of their bacterial origin, such as the use of N-formylmethionyl-tRNA (transfer ribonucleic acid) as the initiator of protein synthesis. Some unicellular eukaryote “protists” lack mitochondria, supporting the notion that these protists were offshoots of the primitive eukaryotic lineage that diverged before the mitochondrial symbiosis developed. This hypothesis has recently been challenged by the discovery of the genetic remnants of mitochondria in the nucleus and cytoplasm of mitochondria-lacking protists. These mitochondrial remnants and other cytosolic structures such as hydrogenosomes and mitosomes are joining a growing list of cryptic mitochondrial relics in eukaryotes that are capable of producing energy in even hostile and anoxic habitats and can perform functions other than respiration (for a review, see Tielens et al., 2002). There is therefore still much to be understood about the evolution of mitochondria beyond the hypothesis of bacterial symbiosis.
In 1988, a major breakthrough in our understanding of mitochondrial disorders occurred with the report of the association of sporadic human encephalomyopathies with large deletions of the mtDNA (Holt et al., 1988) and of Leber hereditary optic neuropathy (LHON) with a point mutation at nucleotide pair (np) 11778 in the mtDNA. More than 170 pathogenic point mutations of the mtDNA, large-scale mtDNA deletions, and rearrangements have been subsequently reported (Schon and DiMauro, 2007). More than 85% of mitochondrial proteins are encoded by the nuclear DNA (nDNA), and therefore many unknown mitochondrial disorders due to nDNA defects may exist. The list of nDNA defects affecting mitochondrial function, including mtDNA replication and integrity, has been growing in recent years (DiMauro and Hirano, 2005). The delineation of human mtDNA variation and genetics has also provided startling new insights into the evolution and migration of human populations (Wallace, 1995). Mitochondria may also play an important role in neurodegenerative diseases and the aging process (Schapira, 2006, 2008; Turner and Schapira, 2010).
The diseases included under the term mitochondrial disorders are so diverse and involve so many parts of the nervous system and other organs that the whole spectrum cannot be easily addressed in any one chapter. Therefore the disorders related to intermediate metabolism and mitochondrial Krebs cycle are discussed with inborn errors of metabolism in Chapter 62. The syndrome that combines epilepsy and ragged-red muscle fibers (myoclonic epilepsy and ragged-red fibers, MERRF) is discussed in Chapters 40 and 67. The syndrome of progressive external ophthalmoplegia (PEO) is discussed with other abnormalities of eye movement (Chapter 16), and LHON with other causes of vision loss (Chapter 14). This chapter overviews the principles of mitochondrial genetics and mitochondrial pathophysiology and provides a summary of the clinical features and management of patients with mitochondrial disorders.
Genetics of Mitochondrial Disorders
MtDNA is a 16,569-np double-stranded, closed, circular molecule located within the matrix of the double-membrane mitochondrion. Each human cell contains a dynamic network of mitochondria and hundreds of mtDNA molecules. Human mtDNA encodes 13 of the 89 subunits of the mitochondrial respiratory chain, as well as the small (12S) and large (16S) ribosomal RNAs (rRNAs) and 22 tRNAs necessary for intramitochondrial protein synthesis (Fig. 63.1, A).
The mtDNA is replicated and transcribed by using an origin and promoter for each of the two DNA strands, the G-rich heavy (H) strand and the C-rich light (L) strand. The H- and L-strand replication origins (OH and OL; see Fig. 63.1, A) are relatively distant within the molecule, but the H- and L-strand promoters (PH and PL) are closely spaced and located adjacent to OH in the approximately 1000-np noncoding control region. This region also encompasses the D-loop, which is formed by replication initiation events from the OH. Research involving evolution of mammalian species and origin and migration of humans on earth has focused on the variation in a small hypervariable noncoding region within the D-loop. The replication mechanism of mtDNA may be more complex than originally suggested, in that the mtDNA can also replicate by the extension of both leading and lagging strands, a process resembling nDNA replication (Holt et al., 2000).
The respiratory chain (see Fig. 63.1, B) is located within the mitochondrial inner membrane and is composed of five multimeric enzyme complexes whose genes are dispersed between the mtDNA and nDNA. Complex I (NADH–ubiquinone oxidoreductase) accepts electrons from the reduced form of nicotinamide adenine dinucleotide (NADH), whereas complex II (succinate–ubiquinone oxidoreductase) collects electrons from succinate. Both NADH and succinate are the products of the Krebs cycle. Enzyme complexes I and II transfer electrons to coenzyme Q10 (CoQ10). From CoQ10, the electrons flow through complex III (ubiquinone–cytochrome c oxidoreductase) to cytochrome c, then to complex IV (cytochrome c oxidase), and finally to oxygen to yield water. The electron transfer is coupled to proton (H+) pumping by complexes I, III, and IV from the matrix to the intermembrane space, creating an electrochemical gradient across the inner membrane. This electrochemical gradient is utilized by complex V (adenosine triphosphate [ATP] synthase) as a source of energy to condense adenosine diphosphate (ADP) and inorganic phosphate (Pi) to synthesize ATP. ATP and ADP are then exchanged across the mitochondrial membrane by the adenine nucleotide translocator (ANT).
Mitochondrial diseases therefore can arise from defects in the mtDNA (sporadic or maternal inheritance, see following section) or nDNA (sporadic or Mendelian inheritance). Nuclear DNA-related mitochondrial disorders result from defects involving nDNA-encoded mitochondrial polypeptides including respiratory chain complexes, respiratory chain assembly, mtDNA maintenance, protein import, lipid dynamics, and biosynthesis of CoQ10. Table 63.1 summarizes a simplified clinical and genetic classification of mitochondrial diseases.
Maternal Inheritance of Mitochondrial DNA
Patients inherit mtDNA from their mothers, and therefore the mode of transmission of mtDNA, including pathogenic mtDNA mutations, follows maternal line inheritance. A single case report has demonstrated paternal transmission of a mtDNA mutation (Schwartz and Vissing, 2002). Maternal inheritance implies maternal transmission of mtDNA to all offspring but subsequent transmission only by females. Thus a disease expressed in all children of both sexes of an affected individual, without evidence of paternal inheritance, strongly suggests an mtDNA point mutation. However, exceptions to this general rule are encountered in clinical practice. First, a de novo point mutation in the mtDNA of the maternal germ cell line will not necessarily be transmitted to all children. Second, for unknown reasons, mtDNA mutations involving large-scale deletions, rearrangements, and point mutations in some protein-coding genes may occur sporadically and be due to mutations arising in the oocyte. Bottleneck expression of an mtDNA mutation may be enhanced in the fetus. Multiple mtDNA deletions and depletions are autosomally transmitted, as they are the consequence of mutations in nuclear-encoded factors involved in mtDNA metabolism or replication. Third, maternal inheritance may not always be clinically evident because of extreme variability of clinical expression among family members due to heteroplasmy and the threshold effect (described in the following section).
Pathophysiology of Mitochondrial Disorders
Mitochondria have several functions in maintaining cellular homeostasis, such as transient storage of intracellular calcium, fatty acid oxidation, the Krebs cycle, and iron metabolism (McBride et al., 2006; Schapira, 2006). Mitochondria also have a key role in the regulation of apoptosis (Wang, 2001). One of the most important roles of mitochondria is to catalyze the phosphorylation of the majority of cellular ADP to ATP. ATP is generated by oxidation of intermediates such as NADH and FADH2 via the process of oxidative phosphorylation within mitochondria by the respiratory chain enzymes. Several intermediary metabolic pathways from carbohydrates, fatty acids, and amino acids converge in mitochondria at the level of acetyl-CoA for the final conversion of the fuel into ATP. Pyruvate is the terminal product of anaerobic glycolysis and is transported across the inner mitochondrial membrane to the mitochondrial matrix where it is converted to acetyl CoA. The transportation of pyruvate is coupled with the influx of hydrogen ions down their electrochemical gradient across the inner mitochondrial membrane. Pyruvate can also be generated during the catabolism of the amino acids, alanine, serine, glycine, and cysteine. Transport of free fatty acids across the mitochondrial membrane requires two enzymes (carnitine palmitoyltransferases [CPT] I and II), a carrier molecule (l-carnitine), and a translocase (carnitine-acylcarnitine translocase). Fatty acids are also metabolized into acetyl-CoA. Acetyl-CoA enters the Krebs cycle, where three molecules of NADH and one molecule of the reduced form of flavin adenine dinucleotide (FADH2) are produced from each acetyl-CoA. Molecules of NADH and FADH2 donate electrons to the electron transport chain (NADH to complex I and succinate to FAD in complex II). The functional unit comprising the electron transport chain (complex I to IV) and complex V (ATP synthase) constitutes the oxidative phosphorylation (OXPHOS) system (see Fig. 63.1, B).
In skeletal muscle, some fibers are severely involved and others may appear normal on histological analysis. With more severe involvement, the combination of patchy myofibrillar degeneration along with mitochondrial proliferation gives rise to the so-called ragged-red appearance of fibers on modified Gomori trichrome staining (Fig. 63.2, A). Defective OXPHOS may result in compensatory mitochondrial proliferation, particularly in type I and IIA muscle fibers. Ultrastructural analysis can demonstrate abnormal mitochondrial morphology such as intramitochondrial paracrystalline inclusions (see Fig. 63.2, B). Patients with mitochondrial disorders can show a wide range of symptoms, including any combination of developmental delay, short stature, small muscle bulk, seizures, vision loss, hearing impairment, peripheral neuropathy, autonomic nervous system difficulties, gastrointestinal dysfunction, endocrine problems, hematopoietic disease, and failure to thrive (Box 63.1). The presentation of a particular mitochondrial disorder can be variable, even among affected individuals in the same family. Conversely, more than one pathogenic mutation can give rise to similar clinical phenotypes.
