[level-membership-for-basic-science-category]
Microbial contamination, spoilage and preservation of medicines
Norman A. Hodges
Chapter contents
The need to protect medicines against microbial spoilage
Products and materials vulnerable to spoilage
Sources and control of microbial contamination
Sources and types of contaminating organisms
Factors influencing the growth of spoilage organisms
Control of contamination and spoilage during manufacture
Selection and use of preservatives
Preservative interactions with formulation components and containers
Key points
The need to protect medicines against microbial spoilage
The need to protect foods against microbial spoilage is well appreciated since microbial growth results in obvious signs of deterioration. However, there is a much lower level of awareness among the general public of the need to similarly protect cosmetics, toiletries and medicines. Although most medicines present a less favourable environment for microbial growth than foods, a wide variety of potentially hazardous organisms are nevertheless capable of growing to high concentrations in unprotected products. The subject of preservation is therefore an important aspect of medicine formulation, simply because patients taking medicines are, by definition, unhealthy and so quite possibly more vulnerable to infection.
It is important to distinguish between the terms contamination and spoilage because they are sometimes used synonymously, which is incorrect.
Contamination, in this context, means the introduction of microorganisms into a product, i.e. it describes microbial ingress. Contaminating organisms can arise from many sources (considered later in this chapter) during the course of both product manufacture and subsequent use of the product. The procedures of good manufacturing practice (GMP, also considered later) are used to limit the first of these (see Rules and Guidance for Pharmaceutical Manufacturers and Distributors, 2007), but contamination arising from the patient is largely out of the control of the manufacturer except in the context of container design and labelling. There has been a trend in recent years to adopt containers that minimize contact between the patient’s body and the product, e.g. collapsible tubes are used for creams and ointments rather than open-mouthed tubs or jars into which fingers can be inserted. Similarly, single-dose eye drops may be preferred to bottles where the dropper can come into contact with an infected eye and then be replaced in the eye drop solution. Despite this, contamination by the patient is still a problem to be considered in container design and product preservation.
Spoilage follows contamination and describes the process and consequences of microbial growth in the product. Considering the potential for product spoilage and taking appropriate steps to minimize the risk of it occurring are very much the responsibility of the formulation scientist and the manufacturer.
There are three principal reasons why microorganisms should be excluded totally from medicines or their presence subjected to stringent limits set by pharmacopoeias or regulatory agencies, such as the United States Food and Drugs Administration (FDA), the European Medicines Agency (EMA) or the UK’s Medicines and Healthcare products Regulatory Agency (MHRA):
It is quite obvious that medicines should not contain pathogenic organisms that represent a source of infection. However, specifying the species and numbers of organisms representing an infection hazard is not straightforward. Certain pathogens are recognized as ‘objectionable organisms’ and must be totally excluded from particular raw materials or product types (see Chapter 14), but the risk of infection is influenced not just by the number and type of organism but by other factors too. For example, an organism may be present at a concentration that would be regarded as relatively harmless to a healthy individual but which may pose a problem for patients with impaired immunity.
In the 1960s and 1970s there were several reports in the pharmaceutical literature of infection occurring as a result of medicines containing pathogenic species, e.g. salmonellae, clostridia and Pseudomonas aeruginosa, but such reports became far less frequent towards the end of the last century with the adoption of more rigorous quality standards and regulatory control of manufacture. However, contaminated medicines are by no means a thing of the past. The FDA publishes details of product recalls on its website, and in March 2011 alone, there were three unrelated product recalls due to concerns about potential or confirmed microbial contamination. It should be emphasized that there is the possibility of an infection arising from the use of a product contaminated with a concentration of organisms that is too low to be detectable by sight or smell. This situation is potentially much more hazardous than that of a patient confronted with a medicine in which microbial growth is clearly evident.
Quite apart from representing an infection hazard, microorganisms may damage the medicine by degrading either the active ingredient or one or more excipients, thus compromising the quality and fitness for use of the product.
Degradation is usually due to either hydrolysis or oxidation, but decarboxylation, racemization and other reactions may also occur. Active ingredients known to be susceptible to microbial attack include steroids, alkaloids, analgesics and antibiotics. Much of the literature on this topic has been reviewed by Spooner (1996) and Bloomfield (2007). The numbers and variety of excipients that have been reported to be degraded are at least as great as those of active ingredients. Thus, most categories of excipients contain materials that have been shown to be susceptible to microbial enzymes, acids or other metabolic products.
Common examples of product instability or deterioration include emulsion phase separation due to surfactant degradation, loss of viscosity due to microbial effects on gums, mucilages and cellulose derivatives employed as thickening agents, and alcohol and acid accumulation following fermentation of sugars. Despite the fact that the very purpose of their use is to restrict microbial growth, even some preservatives are vulnerable to inactivation by microorganisms that, in exceptional cases, use them as a carbon and energy source. Nor should it be assumed that products whose very purpose is to kill microorganisms will necessarily be self-sterilizing: two of the three FDA product recalls mentioned above involved antiseptic wipes containing alcohol and iodine which were designed to decontaminate skin prior to injection or surgery, but the wipes were, themselves, sources of microbial contamination.
If microbial growth within the product is sufficiently extensive, it is possible for the presence of the organisms to be detectable by:
• changes in colour (pigment production)
• gas accumulation without any obvious odour (bubbles of carbon dioxide following sugar fermentation).
Clearly, any product manifesting such changes would be unlikely to be used by the patient. This may result in short-term problems for the patient of obtaining alternative supplies, and possible longer term problems for the manufacturer in terms of customer complaints, product recalls, adverse publicity and possible legal action.
Products and materials vulnerable to spoilage
Spoilage, in the sense of detectable physical or chemical change within a pharmaceutical product, nearly always follows growth and reproduction of the contaminating organisms. The pharmacopoeial and regulatory limits for the maximum permissible numbers of microorganisms in manufactured products or raw materials are typically not more than 100–1000 colony-forming units (cfu) per mL or gram. Whilst these concentrations may allow some pathogens to initiate infections, they are not normally sufficient to cause detectable changes in chemical composition, physical appearance or stability. Bacteria and fungi are just the same as all other living organisms in requiring water for growth (although not necessarily for mere survival). This means that only products containing sufficient water to permit such growth are vulnerable to spoilage. Consequently, spoilage is not normally a problem in anhydrous products like ointments and dry tablets or capsules, although hygroscopic materials like gelatin and glycerol may absorb enough water from the atmosphere to enable moulds (but not normally bacteria) to grow. Similarly, cellulosic materials, particularly paper and other packaging, may show mould growth if stored in humid atmospheres, e.g. in tropical climates.
The fact that a product contains water and is obviously a liquid does not necessarily mean that the water is available to participate in chemical reactions and enable microorganisms to grow. Some of the water present in a solution is bound to the solute due to hydrogen bonding or other mechanisms. Thus a parameter that indicates the proportion of ‘free’ or available water is a useful guide to the ease with which microorganisms might grow in the product. Such a parameter is water activity (Aw) which is the ratio of water vapour pressure of a solution to the water vapour pressure of pure water at the same temperature. Aw is expressed on a scale from zero to 1 with a value of 1.00 representing pure water. As the concentration of solute in a solution is increased, Aw falls proportionately and the range of organisms able to grow in the solution progressively diminishes. Thus, it is possible to construct a table indicating the minimum Aw values that permit the growth of different types of microorganism (Table 50.1).
Table 50.1
Water activity (Aw) minima permitting growth of various organisms
| Organism | Approximate minimum water activity |
| Many common water-borne or soil organisms and non-skin pathogens e.g. Pseudomonas aeruginosa, clostridia, E. coli. | 0.95 |
| Staphylococci and micrococci | 0.87 |
| Many yeasts, e.g. Saccharomyces and Candida spp. | 0.88–0.92 |
| Many fungi, e.g. Penicillium. Aspergillus. Mucor | 0.8–0.9 |
| Osmophilic yeasts, e.g. Zygosaccharomyces rouxii | 0.65 |
To a certain extent, the values of Aw in Table 50.1 are reflective of the natural habitats of the organisms concerned; pseudomonads and other waterborne organisms therefore tend to require high Aw values for optimum growth, whilst skin organisms like staphylococci and micrococci that exist in relatively high salt concentrations (from sweat glands) will tolerate significantly lower values. Pharmaceutical materials that have high solute concentrations, e.g. syrups, may to a large extent be self-preserving just like salted foods. Syrup BP, for example, is 67% by weight sucrose, has an Aw value of 0.86 and so is not susceptible to bacterial growth, but may contain chemical preservatives to protect it from mould spoilage.
Variations in water activity may arise within a single container of a manufactured medicine by, for example, water evaporating from the bulk liquid during storage in high temperatures and that water vapour condensing on cool glass round the neck of a bottle as the storage temperature drops, then running back to dilute the surface layer of product. It is for this reason that syrups should not be stored in fluctuating temperatures.
The possibility also exists for contaminants to grow and generate water from respiration and so produce localized increases in Aw which allow other less osmotolerant organisms to grow subsequently. Reducing the water activity of a product as a means of diminishing its susceptibility to spoilage is a formulation strategy that should not be overlooked. However, sugars and glycerol are the only common and acceptable ingredients that may be used in this way in oral products; with topical products alcohols and glycols may also be employed.
Sources and control of microbial contamination
In order to manufacture medicines of acceptable microbiological quality, it is necessary to know the common sources of microbial contaminants in the manufacturing environment and the typical organisms that might arise from each source. It is also useful to know how quickly and to what concentration those organisms might grow in pharmaceutical materials in order to put in place good manufacturing procedures that will minimize contamination and spoilage.
Sources and types of contaminating organisms
Microbial contamination of medicines arises from three principal sources:
1. The raw materials, including water, from which the product is manufactured.
2. The manufacturing environment including the atmosphere, equipment and work surfaces.
The relative contributions of these three sources vary depending upon the type of product in question. It has been noted (see Chapter 14) that raw materials of different origin may vary significantly in their extent of microbial contamination. ‘Natural’ materials originating from animals (e.g. gelatin), vegetables (starch, cellulose derivatives, alginates) or minerals (talc, kaolin, magnesium trisilicate, bentonite) usually have much higher bioburdens than synthesized chemicals in which organisms are often killed by heat, extremes of pH or organic solvents. Despite the high levels of microorganisms to be found in locations where many natural materials arise (gelatin, for example, originates in the slaughterhouse where faecal contamination of animal carcasses is not uncommon), the cleaning and purification procedures currently employed mean that contamination levels are only one or two orders of magnitude higher than those for synthesized chemicals. This is reflected in the pharmacopoeial limit of not more than 104 colony forming units (cfu) of aerobic bacteria per gram for some oral products containing materials of natural origin compared with 102 per gram otherwise.
Generally, the types of contaminating organisms are reflective of the origins of the product and this, in turn, is reflected in the objectionable organisms that must be absent. For example, salmonellae and E. coli might arise in faeces, so gelatin is subject to tests for the absence of these species. The same organisms might originate from natural fertilizers used on commercial crops and so they must be absent too from vegetable drugs, starches, mucilages, etc. Both vegetable drugs and mined minerals may contain organisms originating from the soil, e.g. Bacillus and Clostridium species, usually as spores. Vegetable drugs may be contaminated with spores of such fungal plant pathogens as Cladosporium that rarely arise in other circumstances.
Water is the most commonly used raw material for manufacturing medicines. Not only is it obviously present in the majority of liquid medicines, it may be added, then removed, during manufacture of dry products too, e.g. during tablet granulation. It is also used in the factory for cleaning equipment, work surfaces, mixing vessels and bottles or other product containers. As a consequence, the microbiological quality of both ingredient and cleaning water can have a profound effect on the final bioburden of the manufactured product. To those unfamiliar with methods of water purification, it is a paradox that pharmaceutical purified water is more likely to contain high levels of bacteria (particularly after storage) than the mains water that is used as the source material. This is simply a consequence of the fact that mains water (potable water) is chlorinated, and the chlorine which acts as a preservative is removed during purification. Despite this purification process, purified water still contains sufficient dissolved nutrients to support the growth of several species of Gram-negative bacteria to population levels well in excess of 105/mL. Such levels may be attained within days rather than weeks of room temperature storage after chlorine removal. The species commonly found are described as low-demand organisms, i.e. they are metabolically versatile and can efficiently utilize as nutrients low concentrations of a diverse range of carbon-containing compounds. Pseudomonads, i.e. the organisms resembling the pathogen Pseudomonas aeruginosa, are good examples of low-demand Gram-negative bacteria, although organisms of other genera, such as Flavobacterium and Alcaligenes also arise.
Product contaminants originating from the manufacturing environment all tend to have one characteristic in common, i.e. they survive well in dry conditions. So the Gram-negative bacteria that are prevalent in water are rarely seen in this situation. Most of these environmental contaminants are spore-formers, both bacteria and fungi, or Gram-positive bacteria like micrococci and staphylococci. All of those can persist for long periods whilst attached to dust particles suspended in the atmosphere or settled onto floors, work surfaces or equipment. Modern pharmaceutical factories are supplied with filtered air, so the level of particulate contamination in the atmosphere in a room where there is no activity (i.e. operators are absent) is usually very low. The main component of the dust in a manufacturing area that is in operation is skin scales shed by operational personnel. Humans replace skin constantly and so they are continually shedding particles with attached skin bacteria; these are typically about 20 µm in size and so cannot be seen with the naked eye. The extent to which skin scales are shed depends upon many factors like the design and coverage of protective clothing, general health, personal hygiene and, in particular, levels of activity. People standing or sitting still normally shed far fewer particles than those who are in motion. Statistics and estimates of the extent to which humans shed skin scales vary substantially, but approximately 109 per day is a commonly quoted value (Cosslett 2007).
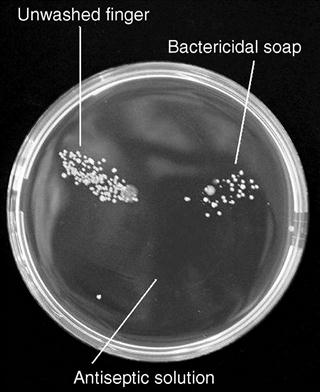
Swabbing with antiseptics or washing with bactericidal soap reduces the numbers of microorganisms on the skin, but is by no means totally effective. Figure 50.1 shows a Petri dish of nutrient medium used to take ‘finger dabs’ from fingers treated in this way. In the upper left segment bacterial colonies were cultured from an unwashed finger; the upper right shows the colonies from a finger washed with bactericidal soap and the bottom sector shows those from a finger swabbed for one minute with cotton wool soaked in antiseptic. It is clear that short periods of exposure to bactericidal soaps cannot be relied upon to eliminate contamination, but a recognized antiseptic is far more effective.
The methods and equipment used for monitoring levels of contaminants arising from water, raw materials and the environment are described in Chapter 14.
Factors influencing the growth of spoilage organisms
In addition to water activity that was considered earlier in this chapter, factors influencing the rate and extent of growth of a contaminant within a pharmaceutical raw material or manufactured medicine include:
Microorganisms differ enormously in their metabolic capabilities. Some, like E. coli, Pseudomonas aeruginosa and several Bacillus species, can synthesize all the amino acids and vitamins they need from a variety of simple carbon and nitrogen sources. The minerals that they require are often present in sufficient concentration as impurities in the ingredients of the medicine. Thus, in the absence of antimicrobial chemicals, organisms of this type may grow to concentrations of 104 per mL or gram, or even higher in products like syrups, linctuses and creams. Products containing glycerol, sugars, amino acids or proteins would clearly represent such ideal media for microbial growth that their preservation is sometimes difficult to achieve even with added preservatives. Even in the absence of these nutritionally rich materials, many bacteria and fungi are still able to utilize other components of the formulation as food sources. Several of these have already been mentioned, but in addition to surfactants and various viscosity-raising agents, the volatile and fixed oils used as flavourings or emulsion components are particularly suitable as nutrients for microorganisms.
The rate of spoilage progression will vary with temperature, although the period of time for which a manufactured medicine is usually stored before use is normally so long that the difference in bacterial growth rate between, say, 15 °C and 20 °C may become insignificant in the context of a 2-year shelf-life. However, there is the possibility of organisms growing during the course of manufacture, and so it is important for production scientists to be aware just how rapidly the population of contaminants may rise. Figure 14.1 shows that the concentration of Pseudomonas aeruginosa rose 10,000-fold in 44 hours at ambient room temperature in a multidose veterinary injection that was supposedly preserved with benzethonium chloride. Clearly, the potential for a rapid increase in numbers is even greater where there is no antimicrobial agent present at all.
Most bacteria have an optimum pH for growth that is near neutrality, whilst most fungi favour slightly acidic conditions and grow best at pH values of 5–6. Although product pH may markedly influence growth rate itself, it also has a bearing on the activity and stability of any antimicrobial chemicals present, so the magnitudes of these various effects may have to be considered at the product formulation stage, and a compromise value selected for the product pH. This is considered further in the next section of this chapter.
Redox potential (oxidation-reduction potential; Eh) is a term that indicates whether oxidizing or reducing conditions exist in a liquid. It is expressed as a positive or negative value on a scale in millivolts. Oxidizing conditions (favouring the growth of aerobic organisms) prevail in culture media or liquids with positive Eh values, and reducing conditions (favouring anaerobes) apply at negative values. Facultative anaerobic organisms, such as E. coli and many similar intestinal pathogens will grow under both conditions from +150 mV to −600 mV (Dempsey 1996). Most pharmaceutical products possess positive redox potentials and so anaerobic growth is not common. There is the potential for aerobic primary contaminants to utilize the available dissolved oxygen and so render the product vulnerable to secondary spoilage by anaerobes.
Antimicrobial chemicals are usually specifically added as preservatives in multidose sterile medicines and in non-sterile medicines. Their properties and the factors influencing their selection are considered later in this chapter. However, it is not uncommon for other ingredients of the product to possess antimicrobial activity or enhance the activity of recognized preservatives. Alcohols used as co-solvents are good examples: ethanol, isopropanol, propylene glycol and glycerol all possess useful antimicrobial activity, although, with the exception of glycerol, their use tends to be limited to topical products. Ethylenediamine tetraacetic acid (EDTA) has a dual role in many pharmaceutical products. It is a chelating agent used to remove metal ions that may catalyse the oxidation of certain active ingredients and, although it possesses little or no antimicrobial activity in its own right, it may markedly potentiate the action of many established preservatives (see below). Indeed, EDTA is present in 19 of the 23 proprietary multidose anti-inflammatory eye drops currently available in the UK. In every case, one of its functions is to potentiate the activity of benzalkonium chloride used as the preservative.
Control of contamination and spoilage during manufacture
Whilst product contamination during use is largely outside the manufacturer’s control, there are many steps that can be taken to minimize contamination whilst the product is made, both by restricting the entry of new organisms into it and limiting the opportunities for growth of organisms that are unavoidably present at the start: those from water or raw materials, for example. Many of these procedures and precautions are described in the Rules and Guidance for Pharmaceutical Manufacturers and Distributors (2007; the ‘Orange Guide’). That publication and other regulatory and pharmacopoeial requirements make it clear that employing good manufacturing practices to limit microbial contamination in the first place is the preferred strategy, rather than, for example, permitting contaminants to enter and proliferate in the product and then attempting to kill them by physical or chemical means.
The factors impacting on the hygienic manufacture of medicines are shown in Figure 50.2. Much of this is self-explanatory, although certain aspects merit more detail. Standards for atmospheric cleanliness in the manufacturing area apply not just for the production of sterile products, but also for non-sterile medicines. Thus, the air supply is invariably HEPA (high efficiency particulate air) filtered and since air-conditioning plants often act as a breeding ground for microorganisms, filters need to be installed downstream of such equipment. The factors to be considered in the design of premises and equipment are detailed in the Orange Guide, as are the procedures for cleaning and disinfection. There is evidence that persistent use of a single disinfectant might predispose to the development of bacterial resistance to it, although this evidence is far less strong than that concerning antibiotics. Nevertheless, there is a requirement that disinfectants are used on a rotational basis to minimize this risk, and in aseptic areas where sterile products are manufactured the disinfectant solutions are filter-sterilized because they may, themselves, be a source of contamination with resistant organisms.
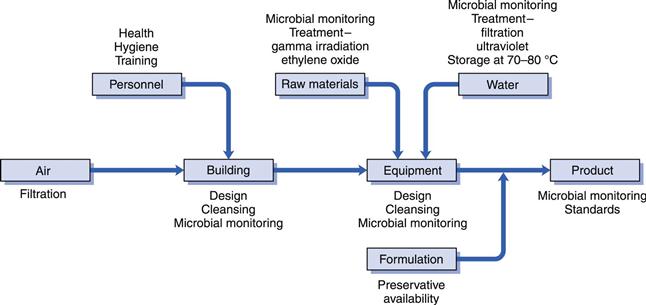
Fig. 50.2 Factors in hygienic manufacture.
Since human beings are frequently the principal source of microbial contamination in the manufacturing environment, their health, hygiene, clothing and training may all have an impact on product contamination. The design of clothing for use in different areas is described in detail in the Orange Guide. Whilst it is an unacceptable practice to use heat, radiation or antimicrobial chemicals to ‘clean up’ a product which has been allowed to acquire a high level of contaminants that could have been avoided, it is acceptable to use these methods to reduce high bioburdens that are unavoidable, e.g. in raw materials of natural origin. Thus, raw materials may be exposed to radiation or ethylene oxide for this purpose, and filtration units or ultraviolet (UV) light sources employed to reduce the bioburden in water. If the water is to be used as an ingredient of an injectable product, filtration is preferable to UV light because it physically removes the contaminating Gram-negative bacteria that may act as a source of endotoxins, which could cause fever on injection. Although UV light kills the bacteria, the endotoxins remain because the lipopolysaccharide component of the bacterial outer membrane from which they are derived is not destroyed. Because of the ability of Gram-negative bacteria to grow readily in stored purified water, it is also common for the water to be maintained at a high temperature, typically 80 °C, whenever possible during the manufacturing process in order to prevent such growth.
The final factor shown in Figure 50.2 to impact on hygienic manufacture is that of preservative availability. Whilst the inclusion of a preservative to protect the product from spoilage sounds simple in principle, there are several ways in which the preservative activity may be diminished or virtually abolished as a result of interaction with other components of the formulation or the container. These are considered below.
Selection and use of preservatives
The antimicrobial chemicals commonly used as preservatives in medicines are described in Chapter 15. The properties that are normally required in such a preservative include the following:
• low toxicity for humans, enabling it to be used in topical, oral and parenteral products
• good solubility in water; low oil solubility
• stable and effective over a wide pH range and compatible with common excipients
Not surprisingly, no single preservative satisfies all these criteria; if there were such an agent it would be universally used to the exclusion of all others. Thus, the selection of a preservative (or combination of preservatives) for a newly developed product is inevitably a compromise determined by the formulation characteristics and intended use of the product. Unfortunately, the list of available preservatives is diminishing rather than expanding. This is due both to the high cost of safety testing that would be a prerequisite for the introduction of an entirely new preservative, and to toxicity concerns resulting in the use of some agents being largely discontinued, e.g. phenyl mercury salts and chloroform, which were formerly employed in ophthalmic/parenteral products and in oral medicines, respectively.
Since the function of a preservative is to kill, or at least prevent the growth of, microorganisms, it might be expected that the first item on the above list would be a major determinant in preservative selection, but, in fact, the intended use and route of administration of a product are usually the major factors limiting the choice. The range of potential preservatives is greatest for topical products, and becomes much more restricted when the toxicity considerations applying to oral and parenteral products are applied. Thus, there are several preservatives whose use is restricted largely to topical medicines, e.g. bronopol, isothiazolones and imidazolodinyl ureas.
It has been estimated that fewer than eight preservatives are in common use in the UK (Hiom 2004), with parabens being by far the most frequently selected for topical and oral products, although sodium benzoate is also regularly chosen for the latter products. Multidose injections are now rarely used in human medicine but are still found in veterinary practice. Again parabens are used as preservatives with benzyl alcohol, phenol or chlorbutanol being common alternatives. Eye drops are usually, but not invariably, multidose products which, although initially sterile, may require protection against patient-derived contaminants during use. Benzalkonium chloride, often with EDTA, is more common in eye drops intended for the UK market than all other preservatives put together. Despite this popularity, there is increasing concern about the potential for benzalkonium chloride to cause corneal irritation. Single dose or unpreserved eye drops are also used.
Due to the limited and diminishing range of acceptable preservatives, there has been increasing attention paid in recent years to the possible benefits of using preservatives in combination. Not only is there scope for reducing the concentrations of the agents, which should confer the benefits of reduced toxicity or irritancy, but employing two or more preservatives together might also result in a broader spectrum of antimicrobial cover, a lower risk of resistance development and enhanced activity due to synergy. There are combinations in which each component fills a gap in the antimicrobial spectrum of the other, e.g. parabens and imidazolidinyl ureas which, individually, have weak activity against Pseudomonas aeruginosa and fungi respectively. The practically useful examples of synergistic combinations have been listed by Hiom (2004). Generally, synergy is most likely to be exhibited when the two agents have dissimilar modes of action. If two agents from the same chemical class or with the same target site in the microbial cell are combined together, the result is commonly found to be additive. Care is required, however, in the investigation and reporting of synergy for two reasons:
• Synergy must be divorced from the effects of concentration exponents (see Chapter 15). It is tempting to assume that doubling the concentration of a preservative results in a doubling of its antimicrobial activity, and so two agents together producing more than twice the effect of either one alone must be synergy. This logic is incorrect, however, because some agents that have high concentration exponents, e.g. phenols, exhibit a large change in activity for a small change in concentration. Combining two such agents can result in a dramatic increase in effect that might be erroneously interpreted as synergy whereas, in fact, the increase is only that to be expected from doubling the concentration of either component alone.
Preservative interactions with formulation components and containers
The adequacy with which a formulated medicine is protected from spoilage by the use of a preservative cannot easily be predicted from a study of the activity of the preservative in simple aqueous solutions. A person with limited knowledge of pharmaceutical microbiology might, for example, expect that it would be possible to confirm that a medicine was adequately preserved simply by conducting an assay for the preservative and showing it to be present at a concentration higher than that required to inhibit the growth of common contaminants (i.e. higher than minimum inhibitory concentrations (MIC values) quoted in reference books and explained in Chapter 14). Not infrequently, however, this logic does not apply because the preservative activity is reduced as a result of it interacting with other components of the formulation or the container, or by a change in the environmental conditions within the product. As a consequence, it is necessary to measure preservative efficacy not by a chemical assay but by a pharmacopoeial preservative efficacy test where the manufactured medicine is inoculated with a range of test organisms whose death rate is measured over a 28-day period (see Chapter 14).
Contaminating microorganisms do not invariably grow uniformly throughout a medicine packed in its final market container; they may be concentrated near the surface as a result of the higher oxygen availability there or, in the case of an emulsion, they grow in the water phase rather than the oil. The concentration of preservative available at the site where the organisms are growing is therefore the principal determinant of how effectively the medicine is protected from spoilage, and the ‘free’ preservative concentration (that which is actually available to kill microorganisms) may be significantly lower than the calculated value. Figure 50.3 shows the major factors that influence preservative activity.
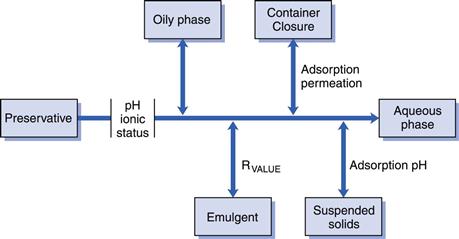
Fig. 50.3 Preservative availability.
Several groups of common preservatives are affected by pH. This may be a consequence of:
The weak organic acids, e.g. benzoic and sorbic acids, are the most commonly cited examples of preservatives whose ionization and activity are pH-dependent, but there are several others. These acids are effective in formulations that are naturally acidic or can be buffered to a low pH. This is because in these conditions they exist as the undissociated molecules which are more lipid soluble and more effective than the ionized forms that predominate when the ambient pH exceeds the molecule’s pKa value. Phenolic preservatives exhibit similar but less marked pH dependence. Parabens are also slightly affected in the same way.
This situation contrasts with that seen with quaternary ammonium compounds which are most effective in neutral or slightly alkaline conditions. Bacterial cells are usually negatively charged, and a rise in pH increases the number of such charges and so promotes the binding of positively charged molecules like quaternary ammonium compounds.
Even though many liquid medicines contain a buffer to restrict pH change, it is not uncommon for the product specification to quote a permissible pH range that is sufficiently large to have a significant impact on preservative activity and for the product pH to drift within that range during its shelf-life, which may be 2 years or more. Slow precipitation during storage (e.g. parabens precipitating in falling pH) is a further problem that is not necessarily detected by chemical assays because the assay procedure may re-dissolve the precipitate.
The oil/water partition coefficient is another molecular property that can have a marked influence on preservatives when they are used in emulsions. Since microorganisms grow in the aqueous phase, a preservative that partitions into the oil is essentially inactive, although again, this will not necessarily be apparent because a chemical assay is likely to show that the correct amount of preservative is present in a given weight or volume of sample. This is a factor that makes parabens less useful choices as cream preservatives because they are usually a lot more soluble in vegetable oil than they are in water, although their partitioning might be reduced by substitution of mineral oil for vegetable oil. Again phenolics and some other preservatives are similarly affected, so when selecting preservatives for multiphase formulations, it is useful to consult publications like the Handbook of Pharmaceutical Excipients (Rowe et al 2012) where lists of solubilities and partition coefficients may indicate the likely extent of the problem.
Entrapment of preservatives within micelles of surfactants or emulsifying agents is a related phenomenon where again, the preservative is present but unavailable to inhibit microbial spoilage. Surfactants like lecithin, Tween (polysorbate) 80 and Lubrol W are so effective in removing preservative in this way that they are commonly used as inactivators (neutralizers) to prevent preservative carry-over and erroneous results in bioburden and preservative efficacy determinations (see Chapter 14). Dissolution and dialysis techniques together with mathematical models may provide some indication of the extent of preservative loss in such complex formulations. Complexation between anionic surfactants (e.g. sodium lauryl (docedyl) sulfate) and cationic preservatives (e.g. chlorhexidine and quaternary ammonium compounds) is also a potential problem in emulsion formulation.
Preservatives may be removed from solution by adsorption onto suspended solids like bentonite, kaolin, magnesium trisilicate and talc. Indeed, in the case of bentonite the potential to adsorb cationic drugs has been investigated as a means of retarding drug release to achieve a long-acting formulation. Antacid products, in particular, may prove difficult to protect due to preservative adsorption and there have been several reports of aluminium hydroxide-, magnesium trisilicate- and kaolin-based products being vulnerable to preservative inactivation. This is reflected in the United States Pharmacopeia (2010) that specifies less stringent preservative performance criteria for antacids than for other oral products. The problems posed by adsorption are compounded by the fact that the phenomenon may also be pH-dependent, so the most favourable pH for preservative activity itself may be one that promotes adsorption. Other hydrocolloids employed as viscosity-raising agents in oral and topical products, e.g. alginates, tragacanth, cellulose derivatives and polyvinyl pyrrolidone, may also reduce preservative activity. In some cases this is simply a charge effect where an anionic polymer, e.g. alginate, complexes a cation, e.g. a quaternary ammonium compound.
Another major mechanism by which preservative activity may be compromised is interaction with the container or, in the case of volatile agents like chlorbutanol, permeation through the container and loss by evaporation. Preservatives may adsorb onto the internal surface or penetrate into the material of the container itself. This problem has become more significant as plastic has tended to replace glass as a packaging material. Rubber stoppers in vials may also cause preservative loss. Most plastics, but particularly those commonly employed for container manufacture such as polypropylene and polyethylene, have the potential to remove significant amounts of parabens and other common preservatives. The surface-to-volume ratio of the product in its container is likely to have a bearing on the magnitude of the problem; small containers have a relatively larger surface and may exhibit proportionately greater loss.
References
1. Bloomfield SF. Microbial contamination: spoilage and hazard. In: Denyer S, Baird R, eds. Guide to Microbiological Control in Pharmaceuticals. 2nd edn London: CRC Press; 2007;23–50.
2. Cosslett AG. The design of controlled environments. In: Denyer S, Baird R, eds. Guide to Microbiological Control in Pharmaceuticals. 2nd edn London: CRC Press; 2007;69–88.
3. Dempsey G. The effect of container materials and multiple-phase formulation components on the activity of antimicrobial agents. In: Baird RM, Bloomfield SF, eds. Microbial Quality Assurance in Cosmetics, Toiletries and Non-sterile Pharmaceuticals. London: Taylor and Francis; 1996;87–98.
4. Hiom S. Preservation of medicines and cosmetics. In: Fraise AP, Lambert PA, Maillard J-M, eds. Principles and Practice of Disinfection, Preservation and Sterilization. Oxford: Blackwell; 2004;484–513.
5. Rowe RC, Sheskey PJ, Cook WG, Fenton ME, eds. Handbook of Pharmaceutical Excipients. 7th edn London: Pharmaceutical Press; 2012.
2007. Rules and Guidance for Pharmaceutical Manufacturers and Distributors. London: Pharmaceutical Press; 2007.
7. Spooner DF. Hazards associated with the microbiological contamination of cosmetics, toiletries and non-sterile pharmaceuticals. In: Baird RM, Bloomfield SF, eds. Microbial Quality Assurance in Cosmetics, Toiletries and Non-sterile Pharmaceuticals. London: Taylor and Francis; 1996;9–30.
8. United States Pharmacopeia (2010) 51 Antimicrobial Effectiveness Testing. US Pharmacopeial Commission, Rockville, Maryland.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Microbial contamination, spoilage and preservation of medicines
Norman A. Hodges
Chapter contents
The need to protect medicines against microbial spoilage
Products and materials vulnerable to spoilage
Sources and control of microbial contamination
Sources and types of contaminating organisms
Factors influencing the growth of spoilage organisms
Control of contamination and spoilage during manufacture
Selection and use of preservatives
Preservative interactions with formulation components and containers
Key points
The need to protect medicines against microbial spoilage
The need to protect foods against microbial spoilage is well appreciated since microbial growth results in obvious signs of deterioration. However, there is a much lower level of awareness among the general public of the need to similarly protect cosmetics, toiletries and medicines. Although most medicines present a less favourable environment for microbial growth than foods, a wide variety of potentially hazardous organisms are nevertheless capable of growing to high concentrations in unprotected products. The subject of preservation is therefore an important aspect of medicine formulation, simply because patients taking medicines are, by definition, unhealthy and so quite possibly more vulnerable to infection.
It is important to distinguish between the terms contamination and spoilage because they are sometimes used synonymously, which is incorrect.
Contamination, in this context, means the introduction of microorganisms into a product, i.e. it describes microbial ingress. Contaminating organisms can arise from many sources (considered later in this chapter) during the course of both product manufacture and subsequent use of the product. The procedures of good manufacturing practice (GMP, also considered later) are used to limit the first of these (see Rules and Guidance for Pharmaceutical Manufacturers and Distributors, 2007), but contamination arising from the patient is largely out of the control of the manufacturer except in the context of container design and labelling. There has been a trend in recent years to adopt containers that minimize contact between the patient’s body and the product, e.g. collapsible tubes are used for creams and ointments rather than open-mouthed tubs or jars into which fingers can be inserted. Similarly, single-dose eye drops may be preferred to bottles where the dropper can come into contact with an infected eye and then be replaced in the eye drop solution. Despite this, contamination by the patient is still a problem to be considered in container design and product preservation.
Spoilage follows contamination and describes the process and consequences of microbial growth in the product. Considering the potential for product spoilage and taking appropriate steps to minimize the risk of it occurring are very much the responsibility of the formulation scientist and the manufacturer.
There are three principal reasons why microorganisms should be excluded totally from medicines or their presence subjected to stringent limits set by pharmacopoeias or regulatory agencies, such as the United States Food and Drugs Administration (FDA), the European Medicines Agency (EMA) or the UK’s Medicines and Healthcare products Regulatory Agency (MHRA):
It is quite obvious that medicines should not contain pathogenic organisms that represent a source of infection. However, specifying the species and numbers of organisms representing an infection hazard is not straightforward. Certain pathogens are recognized as ‘objectionable organisms’ and must be totally excluded from particular raw materials or product types (see Chapter 14), but the risk of infection is influenced not just by the number and type of organism but by other factors too. For example, an organism may be present at a concentration that would be regarded as relatively harmless to a healthy individual but which may pose a problem for patients with impaired immunity.
In the 1960s and 1970s there were several reports in the pharmaceutical literature of infection occurring as a result of medicines containing pathogenic species, e.g. salmonellae, clostridia and Pseudomonas aeruginosa, but such reports became far less frequent towards the end of the last century with the adoption of more rigorous quality standards and regulatory control of manufacture. However, contaminated medicines are by no means a thing of the past. The FDA publishes details of product recalls on its website, and in March 2011 alone, there were three unrelated product recalls due to concerns about potential or confirmed microbial contamination. It should be emphasized that there is the possibility of an infection arising from the use of a product contaminated with a concentration of organisms that is too low to be detectable by sight or smell. This situation is potentially much more hazardous than that of a patient confronted with a medicine in which microbial growth is clearly evident.
Quite apart from representing an infection hazard, microorganisms may damage the medicine by degrading either the active ingredient or one or more excipients, thus compromising the quality and fitness for use of the product.
Degradation is usually due to either hydrolysis or oxidation, but decarboxylation, racemization and other reactions may also occur. Active ingredients known to be susceptible to microbial attack include steroids, alkaloids, analgesics and antibiotics. Much of the literature on this topic has been reviewed by Spooner (1996) and Bloomfield (2007). The numbers and variety of excipients that have been reported to be degraded are at least as great as those of active ingredients. Thus, most categories of excipients contain materials that have been shown to be susceptible to microbial enzymes, acids or other metabolic products.
Common examples of product instability or deterioration include emulsion phase separation due to surfactant degradation, loss of viscosity due to microbial effects on gums, mucilages and cellulose derivatives employed as thickening agents, and alcohol and acid accumulation following fermentation of sugars. Despite the fact that the very purpose of their use is to restrict microbial growth, even some preservatives are vulnerable to inactivation by microorganisms that, in exceptional cases, use them as a carbon and energy source. Nor should it be assumed that products whose very purpose is to kill microorganisms will necessarily be self-sterilizing: two of the three FDA product recalls mentioned above involved antiseptic wipes containing alcohol and iodine which were designed to decontaminate skin prior to injection or surgery, but the wipes were, themselves, sources of microbial contamination.
If microbial growth within the product is sufficiently extensive, it is possible for the presence of the organisms to be detectable by:
• changes in colour (pigment production)
• gas accumulation without any obvious odour (bubbles of carbon dioxide following sugar fermentation).
Clearly, any product manifesting such changes would be unlikely to be used by the patient. This may result in short-term problems for the patient of obtaining alternative supplies, and possible longer term problems for the manufacturer in terms of customer complaints, product recalls, adverse publicity and possible legal action.
Products and materials vulnerable to spoilage
Spoilage, in the sense of detectable physical or chemical change within a pharmaceutical product, nearly always follows growth and reproduction of the contaminating organisms. The pharmacopoeial and regulatory limits for the maximum permissible numbers of microorganisms in manufactured products or raw materials are typically not more than 100–1000 colony-forming units (cfu) per mL or gram. Whilst these concentrations may allow some pathogens to initiate infections, they are not normally sufficient to cause detectable changes in chemical composition, physical appearance or stability. Bacteria and fungi are just the same as all other living organisms in requiring water for growth (although not necessarily for mere survival). This means that only products containing sufficient water to permit such growth are vulnerable to spoilage. Consequently, spoilage is not normally a problem in anhydrous products like ointments and dry tablets or capsules, although hygroscopic materials like gelatin and glycerol may absorb enough water from the atmosphere to enable moulds (but not normally bacteria) to grow. Similarly, cellulosic materials, particularly paper and other packaging, may show mould growth if stored in humid atmospheres, e.g. in tropical climates.
The fact that a product contains water and is obviously a liquid does not necessarily mean that the water is available to participate in chemical reactions and enable microorganisms to grow. Some of the water present in a solution is bound to the solute due to hydrogen bonding or other mechanisms. Thus a parameter that indicates the proportion of ‘free’ or available water is a useful guide to the ease with which microorganisms might grow in the product. Such a parameter is water activity (Aw) which is the ratio of water vapour pressure of a solution to the water vapour pressure of pure water at the same temperature. Aw is expressed on a scale from zero to 1 with a value of 1.00 representing pure water. As the concentration of solute in a solution is increased, Aw falls proportionately and the range of organisms able to grow in the solution progressively diminishes. Thus, it is possible to construct a table indicating the minimum Aw values that permit the growth of different types of microorganism (Table 50.1).
Table 50.1
Water activity (Aw) minima permitting growth of various organisms
| Organism | Approximate minimum water activity |
| Many common water-borne or soil organisms and non-skin pathogens e.g. Pseudomonas aeruginosa, clostridia, E. coli. | 0.95 |
| Staphylococci and micrococci | 0.87 |
| Many yeasts, e.g. Saccharomyces and Candida spp. | 0.88–0.92 |
| Many fungi, e.g. Penicillium. Aspergillus. Mucor | 0.8–0.9 |
| Osmophilic yeasts, e.g. Zygosaccharomyces rouxii | 0.65 |
To a certain extent, the values of Aw in Table 50.1 are reflective of the natural habitats of the organisms concerned; pseudomonads and other waterborne organisms therefore tend to require high Aw values for optimum growth, whilst skin organisms like staphylococci and micrococci that exist in relatively high salt concentrations (from sweat glands) will tolerate significantly lower values. Pharmaceutical materials that have high solute concentrations, e.g. syrups, may to a large extent be self-preserving just like salted foods. Syrup BP, for example, is 67% by weight sucrose, has an Aw value of 0.86 and so is not susceptible to bacterial growth, but may contain chemical preservatives to protect it from mould spoilage.
Variations in water activity may arise within a single container of a manufactured medicine by, for example, water evaporating from the bulk liquid during storage in high temperatures and that water vapour condensing on cool glass round the neck of a bottle as the storage temperature drops, then running back to dilute the surface layer of product. It is for this reason that syrups should not be stored in fluctuating temperatures.
The possibility also exists for contaminants to grow and generate water from respiration and so produce localized increases in Aw
[/not-level-membership-for-basic-science-category]
