CHAPTER 88 METABOLIC MYOPATHIES (INCLUDING MITOCHONDRIAL DISEASES)
Muscle contraction is dependent on the high-energy molecule adenosine triphosphate (ATP), and deficiency of ATP synthesis leads to premature muscle fatigue and weakness. Carbohydrate metabolism, fatty acid oxidation, and the oxidative phosphorylation are all important in the generation of ATP from metabolic fuels, and defects in all three pathways result in metabolic myopathies. Although this chapter is primarily concerned with the muscle symptoms of these metabolic disorders, it is important to remember that many of them also affect other tissues and organs, particularly when the final common pathway of energy metabolism is involved—mitochondrial oxidative phosphorylation.
GLYCOGEN STORAGE DISORDERS
The glycogen storage disorders generally manifest clinically in one of two ways: either with exercise intolerance, muscle cramps, and intermittent rhabdomyolysis or with slowly progressive proximal weakness (Table 88-1). Unusual manifestations include insidious neuromuscular ventilatory failure observed in some adults with α-glucosidase deficiency. Although the biochemical and genetic bases are well established for most of these disorders, the reasons for the phenotypical variability are unknown, and both environmental and epistatic genetic factors play roles (such as the interaction between phosphofructokinase deficiency and adenosine monophosphate deaminase/myoadenylate kinase).
The Glycolytic Pathway
Glycolysis provides energy for high-intensity muscle activity when oxygen availability limits aerobic respiration (Fig. 88-1). Muscle phosphorylase (also called myophosphorylase) initiates the liberation of glucose from muscle glycogen stores. Phosphofructokinase catalyzes the rate-limiting step in glycolysis. Under anaerobic conditions, glycolysis ultimately results in the conversion of pyruvate to lactate. This generates only a fraction of the ATP that would be produced if the glucose were fully oxidized to carbon dioxide and water by aerobic metabolism. The accumulation of lactate and of the major components of ATP hydrolysis (inorganic phosphate, adenosine diphosphate, and adenosine monophosphate) play an important role in causing muscle fatigue.
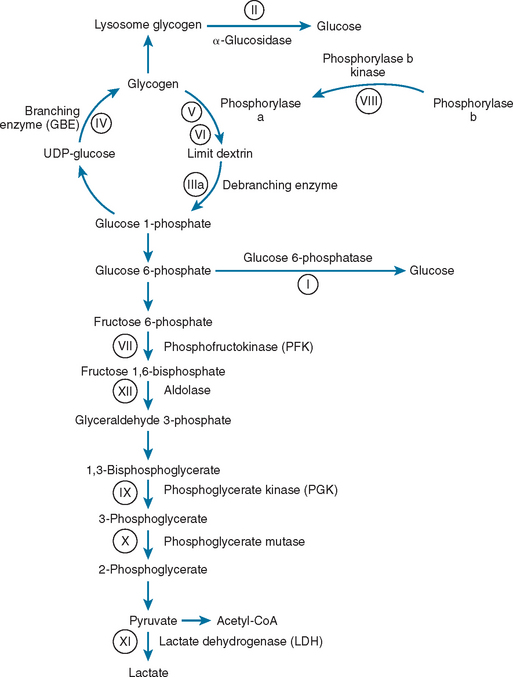
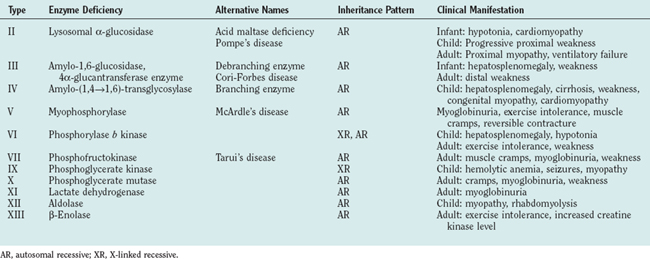
Figure 88-1 Glycogen and glucose metabolism. the principal enzyme defects causing glycogen storage disorders are shown (see Table 88-1). CoA, coenzyme A; UDP, uridine diphosphate.
For a full description of the metabolic pathways, see Matthews and van Holder.1
Clinical Features, Diagnosis, and Management
α-Glucosidase Deficiency (Acid Maltase Deficiency, Pompe’s Disease, Type II Glycogenosis)
α-Glucosidase deficiency can manifest in three ways. In the neonatal period, it typically causes hypotonia, cardiomyopathy and, less frequently, hepatomegaly, and enlargement of the tongue. Cardiac and respiratory difficulties usually lead to death by 2 years of age.2 The childhood manifestations are less dramatic, with progressive proximal weakness and no cardiac involvement. Adult patients usually present with a slowly progressive proximal myopathy, but up to one third of them may have respiratory failure.3
The serum creatine kinase level is typically raised, and electromyography reveals myopathic features, often in association with myotonic discharges. Muscle biopsy reveals a vacuolar myopathy: Vacuoles contain glycogen and show increased acid phosphatase activity. These features are nonspecific, and the diagnosis must be confirmed by enzyme assay either in muscle, fibroblasts, or lymphocytes. Genetic analysis of the α-glucosidase (GAA) gene often reveals the underlying mutation, and there is often a good relationship among type of mutation, biochemical defect, and clinical phenotype.4
Treatment is largely supportive, often involving nocturnal respiratory support after presentation in childhood or adulthood. Clinical trials of enzyme replacement therapy show promise, providing some hope for the future.5,6
Debranching Enzyme Deficiency (Type III Glycogenosis)
Debranching enzyme deficiency can manifest in childhood or adulthood. Symptoms are more prominent in childhood and can include episodes of hypoglycemia. Manifestation in adulthood is usually with a slowly progressive distal myopathy without hepatosplenomegaly. The serum creatine kinase level is usually elevated, and electromyography demonstrates myopathy with occasional myotonic discharges.l Muscle biopsy reveals a vacuolar myopathy, and the diagnosis is confirmed by biochemical assay in red or white blood cells or in muscle. Different clinical subtypes of debranching enzyme deficiency appear to be associated with different mutations in the AGL gene.7
Branching Enzyme Deficiency (Type IV Glycogenosis)
Branching enzyme deficiency can manifest in infants with muscle hypotonia and hepatosplenomegaly, leading to fatal liver cirrhosis. An infantile neuromuscular manifestation, often with cardiac or central nervous system involvement and sometimes simulating spinal muscular atrophy, is probably more common than previously suspected and should be considered in the differential diagnosis of the floppy infant syndrome.8,9 The muscle biopsy reveals periodic acid–Schiff stain–positive, diastase-fast deposits. The diagnosis is confirmed by enzyme assay, and mutations may be found in the GBE gene.9 Treatment is supportive, including liver transplantation in early childhood.
Myophosphorylase Deficiency (McArdle’s Disease, Type V Glycogenosis)
The serum creatine kinase level is usually elevated between acute attacks but rises dramatically immediately after an episode of muscle pain. Electromyography may reveal nonspecific myopathic features, but the appearance is often normal. Forearm exercise testing characteristically reveals a high or even supranormal ammonia response, with a minimal or no increase in venous blood lactate levels during or immediately after exercise. This test is not without risk, inasmuch as it may provoke rhabdomyolysis or a forearm compartment syndrome. Skeletal muscle histochemistry study demonstrates glycogen storage and the absence of phosphorylase activity. Muscle phosphorylase deficiency can be confirmed biochemically (levels usually <5% of normal), but this is rarely necessary. In the majority of patients of European descent, molecular genetic analysis reveals the R49X mutation (>70%) in the PYGM gene,10 which is either homozygous or compound heterozygous, with another mutation in the coding region of the gene. Different common mutations are found in other populations.11
Most patients adapt their lifestyles to avoid precipitating factors. There is evidence that an oral sucrose load before activity can increase exercise tolerance, but this may not be practical in every circumstance.12 Myoglobinuria and rhabdomyolysis should be treated aggressively by increasing the fluid intake. Careful monitoring of the serum creatine kinase and phosphate levels is essential. Intravenous fluids, furosemide, and hemodialysis are sometimes required.
Phosphorylase b Kinase (Type VIII Glycogenosis)
The serum creatine kinase level is often elevated. Glycogen is deposited within muscle fibers (typically type II), but with normal muscle phosphorylase activity. The diagnosis is confirmed by enzyme assay. Genetic analysis is complex because of the many subunits, and even with exhaustive sequencing of known genes, the results are often negative.13 Management is essentially supportive.
Phosphofructokinase Deficiency (Tarui’s Disease, Type VII Glycogenosis)
Approximately 90 cases of phosphofructokinase deficiency have been described in the literature.13a The disorder has a clinical manifestation similar to that of myophosphorylase deficiency (McArdle’s disease, type V glycogenosis). In both disorders, the forearm ischemic exercise test produces a flat lactate response and a marked rise in ammonia levels. In myophosphorylase deficiency, the rise in venous ammonia can be abolished by infusing 5% dextrose, but in phosphofructokinase deficiency, this causes an even more dramatic rise in the ammonia level. Both disorders show a similar pattern of subsarcolemmal glycogen storage, but pockets of polyglucosan deposits are present in phosphofructokinase deficiency. The diagnosis is confirmed by biochemical assay of the muscle specific isoform of phosphofructokinase, followed by PFK-M gene analysis.14 Management is largely supportive. A high-protein diet may help, and rhabdomyolysis must be treated vigorously.
Phosphoglycerate Kinase Deficiency (Type IX Glycogenosis)
Phosphoglycerate kinase deficiency is an X-linked disorder that can manifest with either prominent myopathic or hemolytic features. A mixed phenotype has been observed, and central nervous system features may include mental retardation and epilepsy. A range of different mutations have been found in the PGK gene.15
Lactate Dehydrogenase Deficiency (Type XI Glycogenosis)
Lactate dehydrogenase deficiency typically manifests with exercise intolerance, myalgia, and, sometimes, a nonitchy erythematous rash on the extensor surfaces of the ankles and feet. The disorder is diagnosed by a biochemical assay of muscle lactate dehydrogenase activity and by mutation analysis of the LDH gene.16
Aldolase Deficiency (Type XII Glycogenosis)
Aldolase deficiency 2004 was described for the first time in one child. It is characterized by rhabdomyolysis, often linked to fever and associated with viral infection, leading to fixed weakness. The diagnosis is confirmed by measuring aldolase activity in skeletal muscle and by mutation analysis of the ALDOA gene.17
β-Enolase Deficiency (Type XIII Glycogenosis)
This enzyme defect has also been described in a single patient, a man with adult-onset exercise intolerance and chronically increased serum creatine kinase.17a The patient was a compound heterozygote for mutations in the ENO3 gene, which encodes β-enolase, the isoform expressed predominantly in skeletal muscle.
FATTY ACID OXIDATION DISORDERS
Since the first description of carnitine palmitoyltransferase (CPT) deficiency in 1973,18 there has been a steady increase in both the number of different fatty acid oxidation disorders recognized and the number of affected patients identified. Defects involving many of the different enzymes and transport proteins involved in fatty acid oxidation have been described.19
Fatty Acid Oxidation
Mitochondrial fatty acid β-oxidation is especially important in conditions of fasting and exercise.20 The switch from predominantly carbohydrate metabolism in early exercise to fatty acid oxidation depends on several factors, including the intensity of exercise and the relative fitness of the individual.
At the outer mitochondrial membrane, fatty acids are converted into their acyl-coenzyme A (CoA) esters by the ATP-dependent acyl-CoA synthetases (Fig. 88-2). These acyl-CoA esters are then converted into acylcarnitine and free CoA by CPT-I at the outer mitochondrial membrane. The resulting acylcarnitine is then transported across the inner mitochondrial membrane by the carnitine:acylcarnitine translocase in exchange for free carnitine. Once inside the mitochondrial matrix, the acyl-CoA ester is reformed by CPT-II, and carnitine is released for further exchange by the carnitine:acylcarnitine translocase.
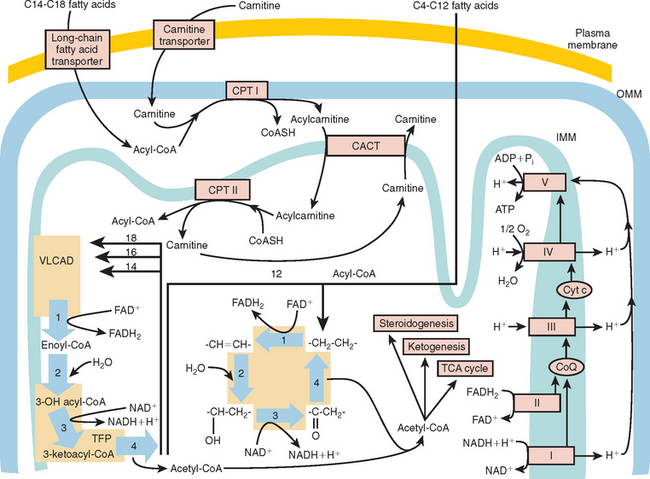
The β-oxidation of fatty acids involves the concerted action of a series of four chain length–specific reactions, which remove a molecule of acetyl-CoA (C2) per cycle from the original fatty acid molecule. The fatty acid therefore is eventually completely broken down to acetyl-CoA, which is the main substrate of the citric acid cycle. The first reaction is catalyzed by the acyl-CoA dehydrogenases, a family of flavin adenine dinucleotide–requiring oxidoreductases. The electrons generated by this process are transferred via electron-transfer flavoprotein (ETF) and ETF ubiquinone oxidoreductase to the respiratory chain. Very-long-chain acyl-CoA dehydrogenase (VLCAD) is responsible for reducing acyl-CoA esters of chain length C12 to C18. Medium-chain acyl-CoA dehydrogenase (MCAD) is responsible for the acyl-CoA esters of chain length C6 to C10, and short-chain acyl-CoA dehydrogenase for C4 to C6 substrates. Long-chain acyl-CoA dehydrogenase has substrate specificity intermediate between VLCAD and MCAD; this is the least characterized of the enzymes, and its deficiencies have not yet been described.
Clinical Features of Mitochondrial Fatty Acid Oxidation Disorders
Carnitine Palmitoyltransferase I Defect
There are three tissue-specific isoforms of CPT-I: the so-called liver (CPT-IA), muscle (CPT-IB), and brain (CPT-IC) isoforms. At present, only patients with deficiency of the liver enzyme have been well documented; they have severe hepatic encephalopathy.21
Carnitine: Acylcarnitine Translocase Defect
These patients usually have severe liver and cardiac problems early in life, but a milder form in which muscle weakness is common has been described.22
Carnitine Palmitoyltransferase II Defect
The myopathic form of this deficiency usually manifests with exercise-induced muscle pain and no involvement of other tissues. A neonatal form is severe and often associated with neonatal death. The difference between the two forms of the disease is related to the degree of residual enzyme activity.21
Very-Long-Chain Acyl-Coenzyme A Dehydrogenase Defect
Severe VLCAD manifests in newborns, but milder defects of this enzyme mimic CPT-II deficiency, with exercise-induced muscle pain.23,24
Trifunctional Protein Deficiency and Isolated Long-Chain L-3-Hydroxyacyl-Coenzyme A Dehydrogenase Deficiency
Although LCHAD is part of the TFP complex, isolated deficiencies of LCHAD and deficiencies of the whole TFP have been described. LCHAD deficiency tends to manifest with profound liver disease and death in infancy. Clinical features may also include cardiomyopathy, myopathy, pigmentary retinopathy, peripheral neuropathy, and sudden death.25 TFP deficiency manifests in a similar manner26 but seems to be less common than isolated LCHAD deficiency, because of a common point mutation in the α subunit (1528G>C) of LCHAD.27
Medium-Chain Acyl-CoA Dehydrogenase Deficiency
MCAD deficiency is the most frequent inborn error of fatty acid oxidation, and the overall frequency of the disease is estimated between 1 per 6500 and 1 per 17,000. Most affected patients present with acute metabolic crises in childhood,28 and muscle symptoms are rarely encountered.
Short-Chain 3-Hydroxyacyl–Coenzyme A Dehydrogenase Deficiency
Few patients affected with this condition have been described, but muscle involvement with rhabdomyolysis and cardiomyopathy is one phenotype.29
Medium-Chain Thiolase Deficiency
This is another rare condition, often associated with death in infancy from rhabdomyolysis.
Glutaric Aciduria Type 2
This condition is caused by deficiency of either ETF or ETF ubiquinone oxidoreductase. There are different phenotypes:30 one with severe congenital abnormalities, one manifesting in the neonatal period with severe metabolic abnormalities and cardiomyopathy, and one manifesting with muscle weakness later in life. It is particularly important to recognize the third phenotype: Affected patients may have relapsing-remitting weakness, often involving the neck muscles, and many of them respond to treatment with riboflavin at high doses.31
Investigation of Fatty Acid Oxidation Disorders
Biochemical Evaluation
The introduction of sophisticated biochemical screening methods, particularly tandem mass spectrometry of free carnitine and acylcarnitines, has revolutionized the investigation of fatty acid oxidation disorders.32–36 In the authors’ view, the ease of these investigations mandates that clinicians always explore the possibility of fatty acid oxidation disorders in patients with unexplained weakness or muscle pain. Quantitative profiles of carnitine, acylcarnitines, and fatty acids in plasma and of organic acids and acylglycines in urine are major diagnostic tools. In children, the diagnostic possibilities are more varied and the manifestations often more acute, necessitating consultation with metabolic pediatricians. In adults, the authors measure fasting acylcarnitine levels in blood first thing in the morning. If the diagnosis is in doubt or if a fatty acid oxidation defect is still highly suspected despite normal acylcarnitine levels, then in vitro measures of fatty acid oxidation or quantitative metabolic profiles in cultured skin fibroblasts may be appropriate.
Genetic Studies
Defects of mitochondrial fatty acid oxidation are autosomal recessive, and genetic defects have now been defined in several disorders. For some defects, there are common mutations that make genetic screening possible, although the advantage of genetic studies over biochemical assays is questionable. Patients with CPT-II deficiency often have a common point mutation (439C>T, S113L),37 which has been reported in several different series and is present in about 50% of mutant alleles. The common point mutation for LCHAD deficiency is 1538G>C,27 and the common mutation for MCAD (985A>G, K304E) is present in homozygous form in 80% of all affected patients. These point mutations have proved useful in assessing the frequency of fatty acid oxidation defects within populations; for example, the carrier frequency of the K304E mutation is approximately 1:40 in people of Northern European descent.
DEFECTS OF MITOCHONDRIAL OXIDATIVE PHOSPHORYLATION
Mitochondrial Oxidative Phosphorylation and Mitochondrial Genetics
The major function of the respiratory chain is the coupling of reducing equivalents generated by the oxidation of fatty acids and carbohydrates to generate readily usable energy in the form of ATP (Fig. 88-3). This process involves the transfer of electrons along the respiratory chain to molecular oxygen. The respiratory chain involves four multi-subunit complexes (I, II, III, and IV) and two mobile electron carriers, ubiquinone and cytochrome c, which transfer the electrons between the complexes. The electrons are transferred to molecular oxygen at complex IV (also called cytochrome c oxidase), and at complexes I, III, and IV, an electrochemical gradient is developed. ATP is generated at complex V (also called ATP synthetase) by the discharge of this gradient and the conversion of adenosine diphosphate to ATP. The overall process is called oxidative phosphorylation.
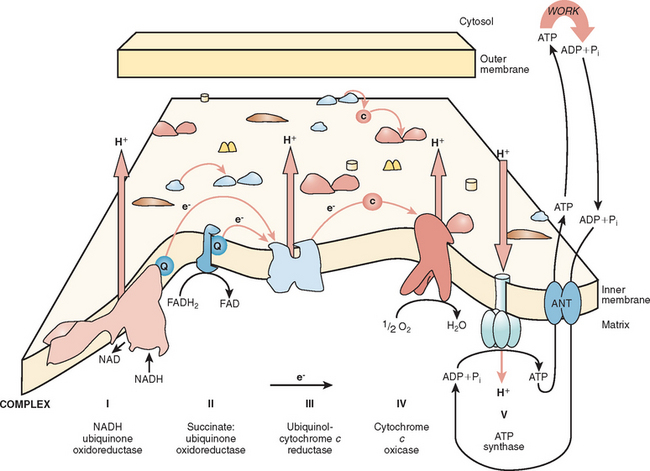
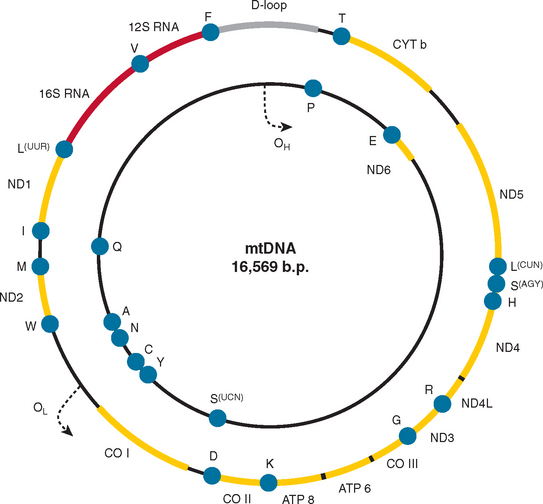
Mitochondria are under the dual genetic control of both nuclear DNA and the mitochondrial genome. The mitochondrial genome consists of a circular double-stranded DNA molecule (16.6 kilobases in humans) that encodes 13 essential polypeptides of the oxidative phosphorylation system and the necessary RNA machinery (2 ribosomal RNAs and 22 transfer RNAs) for their translation within the organelle (Fig. 88-4). The remaining protein subunits that make up the respiratory chain complexes, together with those required for mtDNA maintenance, are nuclear DNA–encoded; they are synthesized on cytoplasmic ribosomes and specifically targeted and sorted to their correct location within the organelle. As a result, mitochondrial disorders can result from mutations in mtDNA or nuclear DNA. This has important implications for the recurrence risks within families. Mitochondrial disorders can be sporadic, maternally inherited, X-linked, or transmitted as autosomal dominant and recessive traits.
The genetics of mtDNA is very different from mendelian genetics. From a clinical perspective, two features stand out. The polyploid nature of the mitochondrial genome, consisting of up to several thousand copies per cell, gives rise to two important features of mitochondrial genetics: homoplasmy (when all copies of the mitochondrial genome are identical) and heteroplasmy (the mixture of two or more mitochondrial genotypes). The relevance of these terms is apparent in consideration of mtDNA mutations that lead to disease. Some mutations appear to affect all copies of the mitochondrial genome (homoplasmic mutations), whereas others are present only in some copies of the mitochondrial genome (heteroplasmic mutations). In the presence of heteroplasmy, a threshold level of mutation is needed both for clinical expression of the disease and for the development of biochemical defects.38
The standard paradigm of mtDNA inheritance is that it is strictly maternal.39 This model has been challenged by several findings: (1) Low levels of paternal transmission of mtDNA have been seen in interspecies crosses; (2) recombination may have affected the distribution of mtDNA polymorphisms within the human population; and (3) paternal mtDNA was documented in muscle of a patient with a single deletion of the mitochondrial genome.40–42 At present, however, such observations are rare, and the maternal pattern of inheritance is the model for genetic counseling.43
Clinical Features of Mitochondrial Disease
Mitochondria are vital components of all nucleated cells. It is therefore not surprising that mitochondrial diseases affect many different tissues and that the clinical features are so variable (Table 88-2).
TABLE 88-2 Common Features Observed in Patients with Defects of Oxidative Phosphorylation
Classic Mitochondrial Syndromes
Genetic defects of the human mitochondrial genome were first described in 198844,45 and arose from investigation of two syndromes: Kearns-Sayre syndrome and Leber’s hereditary optic neuropathy. In Kearns-Sayre syndrome, single, large-scale mtDNA deletions (usually sporadic) were detected in muscle biopsy specimens, and mitochondrial ultrastructural and cytochemical abnormalities in muscle were apparent. In Leber’s hereditary optic neuropathy, strict maternal pattern of inheritance was evident, and point mutations involving the MTND genes encoding subunits of complex I were identified. Since 1988, a large number of mutations46 of the mitochondrial genome have been identified and associated with disease (Table 88-3).
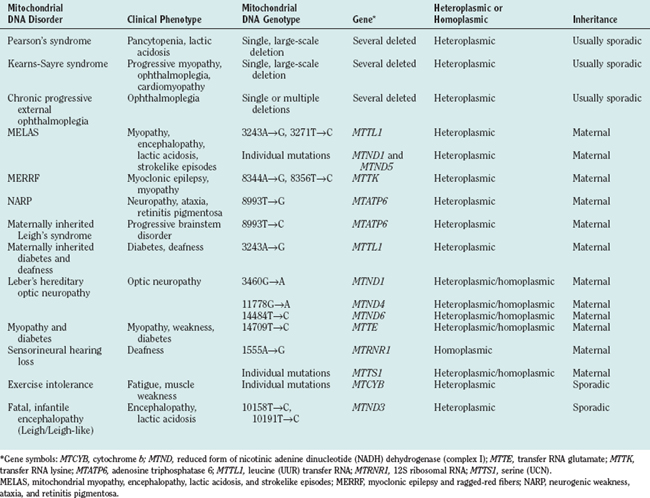
Clinical Syndromes with High Risk of Mitochondrial DNA Involvement
Since the mid-1990s, clinicians have become aware of several clinical syndromes in which an mtDNA mutation is either likely or possible. The increasing recognition of mtDNA involvement in disease is partially a result of the relative ease of sequencing the mitochondrial genome, although defining pathogenicity of specific base substitutions can be difficult.47 Examples of mtDNA-related disorders include progressive external ophthalmoplegia,48 Pearson’s syndrome,49 Leigh’s syndrome,47,50 exercise-induced muscle pain, premature fatigue and rhabdomyolysis,51 and aminoglycoside-induced hearing loss.52 For some of these conditions, such as progressive external ophthalmoplegia, mtDNA mutations are the predominant causes, whereas for others, such as Leigh’s syndrome there is a long list of potential genetic causes, only some of which involve mtDNA.
Mitochondrial DNA Involvement in Common Disease Phenotypes
The principal difficulties for clinical neurologists are that patients with mtDNA-related disease rarely present with classic phenotypes and that mtDNA enters into the differential diagnosis of many common neurological syndromes. The list of common clinical features seen in patients with mtDNA disease shows how difficult it is to decide which patients, children or adults, should undergo investigation for possible mtDNA disease. Important clinical clues in favor of mtDNA-related disease include the presence of a maternal family history, the coexistence of symptoms (such as myopathy and diabetes), or the presence of abnormal laboratory test results (see later discussion).
Diagnosis of Mitochondrial Respiratory Chain Disease
Histopathological and Histochemical Assessments of Mitochondrial Function
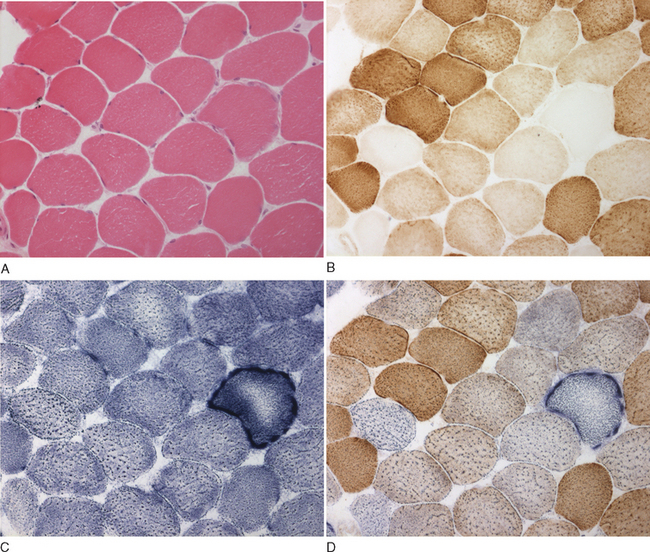
The histological and histochemical assessments of the muscle biopsy remain diagnostic tests crucial for documenting mitochondrial dysfunction. Classic changes include “ragged red” fibers visible with Gomori trichrome stain and abnormal mitochondria visible on electron microscopy study. However, these methods have been superseded by direct histochemical measurements of enzyme activity: succinate dehydrogenase and cytochrome c oxidase. The succinate dehydrogenase reaction shows subsarcolemmal accumulation of mitochondria, which is characteristic of the “ragged red fiber” (Fig. 88-5C and D). The cytochrome c oxidase reaction is particularly useful in the evaluation of mitochondrial myopathies because cytochrome c oxidase contains subunits that are encoded by both the mitochondrial and the nuclear genomes. A mosaic pattern of cytochrome c oxidase activity is highly suggestive of a heteroplasmic mtDNA disorder, and most ragged red fibers are deficient in cytochrome c oxidase (see Fig. 88-5B). In cases in which only a low percentage of cytochrome c oxidase–deficient fibers are present, the sequential cytochrome c oxidase–succinate dehydrogenase histochemistry study is especially valuable for identifying abnormal fibers, which might otherwise go undetected against a background of normal cytochrome c oxidase activity. A global decrease in the activity of cytochrome c oxidase is usually suggestive of a nuclear mutation in one of the ancillary proteins required for cytochrome c oxidase assembly and function, such as SURF1,52,53 although a similar pattern is observed in some patients with pathogenic, homoplasmic mitochondrial transfer RNA gene mutations.54
Molecular Genetic Analyses
The molecular genetic investigation of suspected mitochondrial disease can be complex. Pediatric cases are less likely to represent one of the classic mtDNA-related clinical syndromes and are more likely than adults to manifest nuclear DNA defects. A clear autosomal inheritance pattern (usually recessive) provides evidence of a nuclear DNA defect, but is not usually apparent. Patients with isolated complex IV deficiency may harbor mutations in one of five genes identified thus far that encode accessory proteins necessary for assembly of the cytochrome c oxidase holoenzyme complex: SURF1,52,53 SCO1,55 SCO2,56 COX10,57 and COX1558 or LRPPRC, the protein product of which is required for the translation of mtDNA subunits.59 Children with isolated complex I deficiency, in whom myopathy may be a feature, are more likely to harbor mutations in one of the many nuclear DNA–encoded structural subunits of this enzyme (reviewed by Triepels and coworkers60). Data from a 2004 publication indicate that pathogenic mtDNA mutations are also important in this pediatric population.61 Finally, mtDNA depletion syndrome commonly manifests in infancy. This clinically heterogeneous group of disorders is characterized by a significant reduction in mtDNA copy number. Some affected patients present with severe myopathy caused by mutations in the mitochondrial thymidine kinase (TK2) gene62 or in the SUCLA2 gene63; others present with hepatic or hepatocerebral syndromes caused by mutations in DGUOK64 or POLG1.65
Clues useful in directing the investigation in adults may also come from understanding genotype-phenotype relationships for specific mitochondrial mutations and from information concerning inheritance pattern. Patients with histochemical evidence of a mosaic distribution of cytochrome c oxidase deficiency and autosomal dominant inheritance should be screened for multiple mtDNA deletions, a disorder of intergenomic communication that results from mutations in one of several nuclear genes.66 Multiple mtDNA deletions may also be inherited in an autosomal recessive manner or may manifest with no family history at all.67 Patients with this abnormal genotype typically present with chronic PEO and proximal myopathy, but this may be complicated by cerebellar ataxia or sensory ataxia caused by peripheral neuropathy. A clear pattern of maternal transmission indicates a pathogenic mtDNA point mutation, although mtDNA heteroplasmy and the late onset of syndromes related to such mutations means that relatives may report few symptoms suggestive of mitochondrial disease and that a family history is not always clear. In addition, many point mutations, particularly those in the cyt b gene, which cause exercise intolerance, are sporadic in nature.68 This is also true of patients with chronic PEO or Kearns-Sayre syndrome resulting from single, large-scale mtDNA deletions,69 although rare cases of maternal transmission have been reported in this latter group.70,71 In chronic PEO, mtDNA deletions are reliably detected only in skeletal muscle, and investigation of this tissue is essential for confirming the diagnosis.
Some mtDNA-related disorders may be reliably diagnosed in blood, but in others the mtDNA mutations are expressed at high levels only in muscle. Patients suspected of having the T14709C (myopathy, ataxia and diabetes) or the A8344G (MERRF) mutations affecting transfer RNA(Glu) and transfer RNA(Lys), respectively, commonly exhibit high levels of heteroplasmy in blood cells. In contrast, other point mutations may be present at only very low levels or may even be undetectable in circulating lymphocytes.72 In addition, all patients in whom mitochondrial disorder is strongly suspected clinically but with unremarkable histochemical and biochemical test results should, in the authors’ opinion, undergo investigation at the molecular level.
Treatment
Although much progress has been made in defining the clinical features and in establishing the molecular diagnosis of mitochondrial respiratory chain disease, treatment is still very limited. Apart from the very rare occurrence of primary ubiquinone deficiency, there is no curative therapy. There is evidence that supportive therapy—such as correction of ptosis, provision of pacemakers for cardiac conduction defects, and provision of digital hearing aids for deafness—can all significantly improve the quality of life for patients. A Cochrane review of all published clinical trials in this area showed very few properly controlled clinical studies,72a and these did not show a definite effect for any specific treatment.
One approach that may be helpful is exercise. In control subjects, lack of exercise leads to an overall reduction in mitochondrial enzyme activity. This can be reversed by endurance training. Endurance training may therefore improve function in patients with mtDNA disease by increasing wild-type mtDNA levels. There do exist concerns, however, that mutated mtDNA might be preferentially amplified and that this increase might become clinically relevant after deconditioning.73,74 Studies are currently under way to address both the improvements and the concerns arising from these earlier reports. Resistance training or muscle necrosis stimulates the incorporation of satellite cells into existing muscle fibers.73,75 It is postulated that in patients with sporadic mutations, resistance training might lead to an overall reduction in the proportion of mutated mtDNA versus wild-type mtDNA, inasmuch as satellite cells contain low or negligible levels of mutated mtDNA.75,76
DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656-2668.
McFarland R, Taylor RW, Turnbull DM. The neurology of mitochondrial DNA disease. Lancet Neurol. 2002;1:343-351.
Moxley RT, Chinnery P, Turnbull DM. The metabolic myopathies. In: Karpati G, Hilton-Jones D, Griggs RC, editors. Disorders of Voluntary Muscle. 7th ed. Cambridge, UK: Cambridge University Press; 2001:560-579.
1 Matthews CK, van Holder KE. Biochemistry, 2nd ed., San Francisco: Benjamin Cummings; 1995:445-516.
2 van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332-340.
3 Hagemans ML, Janssens AC, Winkel LP, et al. Late-onset Pompe disease primarily affects quality of life in physical health domains. Neurology. 2004;63:1688-1692.
4 Hermans MM, van Leenen D, Kroos MA, et al. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat. 2004;23:47-56.
5 Brady RO, Schiffmann R. Enzyme-replacement therapy for metabolic storage disorders. Lancet Neurol. 2004;3:752-756.
6 Winkel LP, Van den Hout JM, Kamphoven JH, et al. Enzyme replacement therapy in late-onset Pompe’s disease: a three-year follow-up. Ann Neurol. 2004;55:495-502.
7 Lucchiari S, Donati MA, Parini R, et al. Molecular characterisation of GSD III subjects and identification of six novel mutations in AGL. Hum Mutat. 2002;20:480.
8 Bruno C, Servidei S, Shanske S, et al. Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann Neurol. 1993;33:88-93.
9 Bruno C, van Diggelen OP, Cassandrini D, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. 2004;63:1053-1058.
10 Martin MA, Rubio JC, Buchbinder J, et al. Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): a genotype-phenotype correlation study. Ann Neurol. 2001;50:574-581.
11 Tsujino S, Shanske S, Nonaka I, et al. The molecular genetic basis of myophosphorylase deficiency (McArdle’s disease). Muscle Nerve. 1995;3:S23-S27.
12 Vissing J, Haller RG. The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. N Engl J Med. 2003;349:2503-2509.
13 Burwinkel B, Hu B, Schroers A, et al. Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet. 2003;11:516-526.
13a Nakajima H, Raben N, Hamaguchi T, Yamasaki T. Phosphofructokinase deficiency: past, present and future. Curr Mol Med. 2002;2:197-212.
14 Raben N, Sherman JB. Mutations in muscle phosphofructokinase gene. Hum Mutat. 1995;6:1-6.
15 Tsujino S, Shanske S, DiMauro S. Molecular genetic heterogeneity of phosphoglycerate kinase (PGK) deficiency. Muscle Nerve. 1995;3:S45-S49.
16 Tsujino S, Shanske S, Brownell AK, et al. Molecular genetic studies of muscle lactate dehydrogenase deficiency in white patients. Ann Neurol. 1994;36:661-665.
17 Yao DC, Tolan DR, Murray MF, et al. Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase, ALDOA(Arg303X/Cys338Tyr). Blood. 2004;103:2401-2403.
17a Comi GP, Fortunato F, Lucchiari S, et al. Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann Neurol. 2001;50:202-207.
18 DiMauro S, DiMauro PM. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science. 1973;182:929-931.
19 Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477-502.
20 Eaton S, Bartlett K, Pourfarzam M. Mammalian mitochondrial beta-oxidation. Biochem J. 1996;320(Pt 2):345-357.
21 Bonnefont JP, Djouadi F, Prip-Buus C, et al. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. 2004;25:495-520.
22 Stanley CA, Hale DE, Berry GT, et al. Brief report: a deficiency of carnitine-acylcarnitine translocase in the inner mitochondrial membrane. N Engl J Med. 1992;327:19-23.
23 Yamaguchi S, Indo Y, Coates PM, et al. Identification of very-long-chain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with long-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1993;34:111-113.
24 Andresen BS, Olpin S, Poorthuis BJ, et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479-494.
25 Wanders RJ, IJlst L, van Gennip AH, et al. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 1990;13:311-314.
26 Jackson S, Kler RS, Bartlett K, et al. Combined enzyme defect of mitochondrial fatty acid oxidation. J Clin Invest. 1992;90:1219-1225.
27 IJlst L, Ruiter JP, Vreijling J, et al. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a new method to identify the G1528C mutation in genomic DNA showing its high frequency (approximately 90%) and identification of a new mutation (T2198C). J Inherit Metab Dis. 1996;19:165-168.
28 Stanley CA, Hale DE, Coates PM, et al. Medium-chain acyl-CoA dehydrogenase deficiency in children with non-ketotic hypoglycemia and low carnitine levels. Pediatr Res. 1983;17:877-884.
29 Tein I, De Vivo DC, Hale DE, et al. Short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency in muscle: a new cause for recurrent myoglobinuria and encephalopathy. Ann Neurol. 1991;30:415-419.
30 Loehr JP, Goodman SI, Frerman FE. Glutaric acidemia type II: heterogeneity of clinical and biochemical phenotypes. Pediatr Res. 1990;27:311-315.
31 Gregersen N, Rhead W, Christensen E. Riboflavin responsive glutaric aciduria type II. Prog Clin Biol Res. 1990;321:477-494.
32 Millington DS, Norwood DL, Kodo N, et al. Application of fast atom bombardment with tandem mass spectrometry and liquid chromatography/mass spectrometry to the analysis of acylcarnitines in human urine, blood, and tissue. Anal Biochem. 1989;180:331-339.
33 Rashed MS, Ozand PT, Bucknall MP, et al. Diagnosis of inborn errors of metabolism from blood spots by acylcarnitines and amino acids profiling using automated electrospray tandem mass spectrometry. Pediatr Res. 1995;38:324-331.
34 Chace DH, Hillman SL, Van Hove JL, et al. Rapid diagnosis of MCAD deficiency: quantitatively analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin Chem. 1997;43:2106-2113.
35 Vreken P, van Lint AE, Bootsma AH, et al. Quantitative plasma acylcarnitine analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidaemias and fatty acid oxidation defects. J Inherit Metab Dis. 1999;22:302-306.
36 Costa CG, Struys EA, Bootsma A, et al. Quantitative analysis of plasma acylcarnitines using gas chromatography chemical ionization mass fragmentography. J Lipid Res. 1997;38:173-182.
37 Taroni F, Verderio E, Dworzak F, et al. Identification of a common mutation in the carnitine palmitoyltransferase II gene in familial recurrent myoglobinuria patients. Nat Genet. 1993;4:314-320.
38 Sciacco M, Bonilla E, Schon EA, et al. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3:13-19.
39 Giles RE, Blanc H, Cann HM, et al. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A. 1980;77:6715-6719.
40 Gyllensten U, Wharton D, Josefsson A, et al. Paternal inheritance of mitochondrial DNA in mice. Nature. 1991;352:255-257.
41 Awadalla P, Eyre-Walker A, Smith JM. Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science. 1999;286:2524-2525.
42 Schwartz M, Vissing J. Paternal inheritance of mitochondrial DNA. N Engl J Med. 2002;347:576-580.
43 Filosto M, Mancuso M, Vives-Bauza C, et al. Lack of paternal inheritance of muscle mitochondrial DNA in sporadic mitochondrial myopathies. Ann Neurol. 2003;54:524-526.
44 Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717-719.
45 Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427-1430.
46 Brandon MC, Lott MT, Nguyen KC, et al. MITOMAP: a human mitochondrial genome database—2004 update. Nucl Acid Res. 2005;33(Database Issue):D611-D613.
47 McFarland R, Kirby DM, Fowler KJ, et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann Neurol. 2004;55:58-64.
48 Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320:1293-1299.
49 Rotig A, Cormier V, Blanche S, et al. Pearson’s marrow pancreas syndrome. A multisystem mitochondrial disorder of infancy. J Clin Invest. 1990;86:1601-1608.
50 de Vries DD, van Engelen BG, Gabreels FJ, et al. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh’s syndrome. Ann Neurol. 1993;34:410-412.
51 Andreu AL, Hanna MG, Reichmann H, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341:1037-1044.
52 Tiranti V, Hoertnagel K, Carrozzo R, et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet. 1998;63:1609-1621.
53 Zhu Z, Yao J, Johns T, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337-343.
54 McFarland R, Clark KM, Morris AA, et al. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet. 2002;30:145-146.
55 Valnot I, Osmond S, Gigarel N, et al. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am J Hum Genet. 2000;67:1104-1109.
56 Jaksch M, Ogilvie I, Yao J, et al. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9:795-801.
57 Valnot I, von Kleist-Retzow JC, Barrientos A, et al. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9:1245-1249.
58 Agostino A, Valletta L, Chinnery PF, et al. Mutations of ANT1, Twinkle, and POLG1 in sporadic progressive external ophthalmoplegia (PEO). Neurology. 2003;60:1354-1356.
59 Mootha VK, Lepage P, Miller K, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100:605-610.
60 Triepels RH, van den Heuvel L, Trijbels F, et al. Respiratory chain complex I deficiency. Am J Med Genet. 2001;106:37-45.
61 Kirby DM, McFarland R, Ohtake A, et al. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J Med Genet. 2004;41:784-789.
62 Saada A, Shaag A, Mandel H, et al. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342.
63 Elpeleg O, Miller C, Hershkovitz E, et al. Deficiency of the ADPforming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081-1086.
64 Mandel H, Szargel R, Labay V, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337.
65 Davidzon G, Mancuso M, Ferraris S, et al. POLG mutations and Alpers syndrome. Ann Neurol. 2005;57:921-923.
66 Suomalainen A, Kaukonen J. Diseases caused by nuclear genes affecting mtDNA stability. Am J Med Genet. 2001;106:53-61.
67 Antonicka H, Mattman A, Carlson CG, et al. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72:101-114.
68 Andreu AL, Hanna MG, Reichmann H, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341:1037-1044.
69 Holt IJ, Cooper JM, Morgan-Hughes JA, et al. Deletions of muscle mitochondrial DNA. Lancet. 1988;1:1462.
70 Bernes SM, Bacino C, Prezant TR, et al. Identical mitochondrial DNA deletion in mother with progressive external ophthalmoplegia and son with Pearson marrow-pancreas syndrome. J Pediatr. 1993;123:598-602.
71 Shanske S, Tang Y, Hirano M, et al. Identical mitochondrial DNA deletion in a woman with ocular myopathy and in her son with Pearson syndrome. Am J Hum Genet. 2002;71:679-683.
72 Chinnery P, Howell N, Lightowlers R, et al. Molecular pathology of MELAS and MERRF: the relationship between mutation load and clinical phenotype. Brain. 1997;120:1713-1721.
72a Chinnery P, Majamaa K, Turnbull D, Thorbura D. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2006. CD004426.
73 Taivassalo T, Fu K, Johns T, et al. Gene shifting: a novel therapy for mitochondrial myopathy. Hum Mol Genet. 1999;8:1047-1052.
74 Taivassalo T, Shoubridge EA, Chen J, et al. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol. 2001;50:133-141.
75 Clark KM, Bindoff LA, Lightowlers RN, et al. Reversal of a mitochondrial DNA defect in human skeletal muscle. Nat Genet. 1997;16:222-224.
76 Fu K, Hartlen R, Johns T, et al. A novel heteroplasmic tRNAleu(CUN) mtDNA point mutation in a sporadic patient with mitochondrial encephalomyopathy segregates rapidly in skeletal muscle and suggests an approach to therapy. Hum Mol Genet. 1996;5:1835-1840.