[level-membership-for-neurology-category]
CHAPTER 83 METABOLIC, IMMUNE-MEDIATED, AND TOXIC NEUROPATHIES
Acquired peripheral neuropathy is a highly prevalent and often overlooked condition. Most patients and many physicians are not sufficiently aware of the range of disorders associated with neuropathy outside of the most common cause—diabetes mellitus. Although many neuropathies, including the roughly 30% that have no discernible cause, have no effective treatment, symptomatic therapy is available and therapeutic options are continually expanding. In contrast, many disorders discussed in this chapter can be potentially reversed or at least slowed by treating the underlying condition, removing offending toxins or medications, or limiting immune-mediated injury. For this to happen, proper diagnosis is essential. Several immune-mediated neuropathies are included here, but other important types of neuropathy are not discussed or are considered in more general sections on specific topics. Noteworthy examples include paraneoplastic neuropathy, hereditary and acquired amyloidosis, axonal paraprotein- and myeloma-associated, and acute immune-mediated neuropathies. Topics discussed in other sections include sarcoidosis, environmental toxins, nerve injury, and collagen vascular diseases.
NEUROPATHIES ASSOCIATED WITH DIABETES MELLITUS
Diabetes mellitus is the most common cause of neuropathy in the Western world, as noted since the early 1800s. The most common presentation is distal, predominantly sensory neuropathy; however, diabetes is associated with numerous other forms of neuropathy, ranging from asymptomatic to disabling motor, autonomic, or pain syndromes (Table 83-1). This syndromic list is both clinically and experimentally useful because the differing underlying mechanisms that are likely to occur directly influence treatment choices and efficacy, clinical trial selection, and mechanistic bench research. Most published lists differ slightly in syndrome numbers and entity names, and a separate list could be based on probable pathophysiological mechanisms.1 Important clinical features include nerve modalities, symmetry, speed of onset, distribution, and related diabetic complications, especially renal failure. A consensus statement in 2005 reviewed clinical syndromes, pathogenic mechanisms, and recommended treatment.2 There is growing evidence that symptomatic peripheral neuropathy can occur in the presence of mild diabetes or glucose intolerance even in the absence of notable damage to other end organs. However, in most large series, neuropathy is associated with both retinopathy and nephropathy.3 Less common forms of diabetic neuropathy are not associated with diabetic complications, suggesting a different pathophysiology, such as inflammatory or immune-mediated injury. Conversely, the coexistence of diabetes and neuropathy in a particular patient is not in itself proof of diabetic neuropathy, as unrelated processes may be present that warrant appropriate diagnostic investigation. In general, any of the entities listed can occur with either type 1 or type 2 diabetes (Table 83-1).
TABLE 83-1 Clinical Neuropathy Patterns Associated With Diabetes Mellitus
Distal Diabetic Polyneuropathy
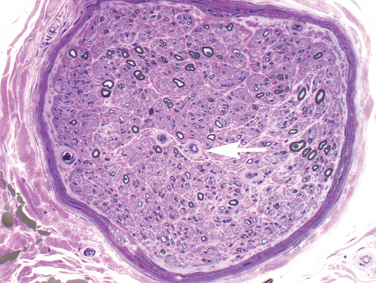
Foot numbness and paresthesia have gradual onset and ascend slowly. The process can spread to the hands, arms, trunk, and scalp in advanced cases, but more commonly hand and arm involvement is caused by superimposed processes, such as compressive mononeuropathy.3 Loss of light touch, pain, and temperature precede loss of proprioception; distal weakness and atrophy are late findings. Patients generally notice “positive” or unpleasant symptoms, especially burning pain, electrical or stabbing sensations, paresthesia, and deep pain; loss of sensation and anesthesia are less common. Symptoms are frequently worse at night when there are less sensory distractions. Approximately 20% to 50% of diabetic patients have at least one symptom of diabetic neuropathy but even more have asymptomatic disease. Lack of sensation can lead to unperceived injury, ulcers, and Charçot joints—a significant form of morbidity. Not surprisingly, objective evidence of functional loss on clinical examination and laboratory testing often exceeds symptoms. Onset may occur after several years of diabetes or with subclinical diabetes, and the neuropathy may lead to the discovery of underlying diabetes. Sensory fibers in the foot and distal leg are predominantly affected, especially small diameter sensory and autonomic fibers. Examination of the lower leg usually reveals loss of vibration, pressure, pain, and temperature perception (both small and large fiber-mediated) and absent ankle deep tendon reflexes. Signs of peripheral autonomic (sympathetic) dysfunction are frequent, including altered skin temperature (cold or warm), dry, often scaly, skin, and calluses in pressure-bearing areas. Loss of sensation for 128-Hz vibration, monofilament touch, and loss of ankle jerks are considered 87% sensitive for detecting neuropathy and predicting foot ulcer.2 The diagnosis is ultimately clinical and requires exclusion of other reasonable causes of neuropathy. Even if symptoms remain confined to the legs and feet, objective testing such as nerve or skin biopsy and clinical electrophysiological studies often show evidence of more widespread involvement. Electrodiagnostic studies are an important adjunct to assess severity and to parse out superimposed mononeuropathy, regional syndromes, superimposed radiculopathy, and demyelinating neuropathy. Nerve biopsy is not routinely necessary but in some cases it demonstrates signs of microvascular changes in addition to axonal loss (Fig. 83-1). Skin punch biopsy to assess epidermal nerve fiber density is increasingly available and minimally invasive, as discussed later.
Diabetic autonomic neuropathy affects up to one half of diabetics.3 Genitourinary and sexual dysfunction, such as impotence, are most common, but orthostatic intolerance and hypotension, gastrointestinal dysmotility (diarrhea or constipation), gastroparesis, fatigue and exercise intolerance, vasomotor disturbance, and pupil dysfunction are also common. Patients with diabetic autonomic neuropathy have shorter survival than do unaffected diabetic patients, but the relative risk is less than early reports suggested. Patients may become intolerant of autonomically active medications, such as antihypertensive or anticholinergic agents, as the neuropathy slowly worsens, producing symptoms such as orthostatic complaints, syncope, urinary retention, excessively dry eyes, and focusing difficulties. Formal autonomic testing is widely available and is especially helpful when the clinical picture is equivocal or the cause of orthostatic symptoms is unclear. Treatment and assessment are discussed in Chapters 29 through 32 Chapter 30 Chapter 31 Chapter 32. The pathophysiology is presumed to be the same as in distal diabetic polyneuropathy and is discussed later.
Ischemic mononeuropathies are presumably due to occlusion of arterioles supplying individual nerves and produce acute ischemia and aching pain, followed by loss of nerve function. The process can affect either cranial or peripheral nerves or nerve roots. Pupil-sparing oculomotor palsy is the most common cranial neuropathy, followed by sixth and seventh neuropathies, but all are relatively uncommon. Diabetics are more susceptible to compression or entrapment neuropathy, and at least 30% of diabetics have carpal tunnel syndrome, which can range from asymptomatic to very distressing.1 Ulnar neuropathy at the elbow, peroneal neuropathy at the fibular head, and lateral femoral cutaneous neuropathy (meralgia paresthetica) are also common.
Diabetic amyotrophy (diabetic radiculoplexus neuropathy) is a monophasic illness seen primarily in type 2 diabetic patients and characterized by subacute proximal leg weakness progressing stepwise over weeks to months, weight loss, atrophy, and aching proximal leg pain. The weakness affects mostly but not exclusively the femoral and obturator nerve distributions, plateaus over weeks, and slowly improves over 12 to 36 months.4 Sensory involvement is less marked and often overshadowed by the underlying distal polyneuropathy. Anatomic boundaries are not respected, and there is often evidence of both nerve root and plexus involvement, hence the descriptive label. Findings may be unilateral or bilateral but asymmetric. If involvement is extensive, a diagnosis of chronic inflammatory demyelinating polyneuropathy, which is more common in diabetic patients, should be considered. Thoracic and cervical forms also occur but are less common and must be distinguished from ischemic injury and unrelated compressive spine disease. Although various neuropathological findings have been reported, an immune-mediated process is suspected and a long-awaited treatment trial is ongoing.
Etiology
There are numerous lines of evidence and hypotheses attempting to explain diabetic polyneuropathy but most start with perturbations initiated by excessive glucose levels. Duration of diabetes, hemoglobin A1c levels, and other signs of poor glucose control correlate with neuropathy development and severity. Microvascular injury appears to be a crucial process in the development of neuropathy as well as in damage of other end organs, and interruption of this process has long been a target of therapeutic intervention. Four major pathways of glucose metabolism are implicated. Excess intracellular glucose is processed, in part, through the polyol pathway in a series of reactions catalyzed by aldose reductase. This pathway leads to sorbitol and fructose accumulation, NAD(P)H-redox imbalances, changes in signal transduction, and excessive production of reactive oxygen radicals.5 Also, nonenzymatic glycation of proteins produces advanced glycation end products in peripheral nerve, and these impair axonal transport, neurotrophic factor production, and gene expression. In addition, protein kinase C activation starts a cascade of stress responses and increases hexosamine pathway flux; both processes lead to oxygen radical generation.5 Specific inhibitors of each pathway, aimed at blocking one or more microvascular complications, have shown considerable promise in rodent models: they include nine different aldose reductase inhibitors, various neurotrophins, blood flow and angiogenesis enhancers, free radical scavengers, and others. Unfortunately, virtually all have failed or have produced equivocal results, but several trials are presently ongoing, including a gene therapy trial of vascular endothelial growth factor. A broader approach targeting several pathways at once may be needed to affect human disease. Other risk factors have gathered recent attention. A large European consortium has found the following independent risks factors for the cumulative incidence of diabetic neuropathy: low-density lipoprotein cholesterol and triglycerides, high body mass index, high von Willebrand factor levels and urinary albumin excretion rate, hypertension, and smoking.6 Several factors are potentially adjustable and some might explain why neuropathy develops in patients with early disease or with simple glucose intolerance, as we discuss later. More acute or subacute entities suggest an immune-mediated mechanism, and some may respond to immunomodulating therapies.
Treatment
Tight glycemic control is the most important and only intervention proved to prevent or limit diabetic polyneuropathy and autonomic neuropathy, but it does not reverse existing nerve injury.7,8 Treatment of other modifiable potential risk factors (lipids, blood pressure, weight) is an evolving but likely important approach. The efficacy of preventative medications currently in clinical trials is yet to be determined.
Numerous symptomatic treatments are effective in reducing—but usually not eliminating—the discomfort of diabetic and other painful polyneuropathies. Randomized double-blind, controlled trials have demonstrated symptomatic relief, primarily for painful neuropathy, with a number of agents, including several tricyclic antidepressants, gabapentin, pregabalin, tramadol hydrochloride, and the serotonin and norepinephrine reuptake inhibitor duloxetine.1,9–11 The selective serotonin reuptake inhibitor paroxetine was better than placebo but not better than imipramine, and citalopram was similar to paroxetine; fluoxetine showed no significant benefit. However, only pregabalin and duloxetine have received U.S. Food and Drug Administration indications for treatment of painful neuropathy. Numerous other medications and therapies have undergone small randomized or incompletely controlled trials and are empirically used. Proper foot care and prompt attention to injury are essential.
Glucose Intolerance and Neuropathy
There is increasing recognition of impaired glucose tolerance, sometimes designated as prediabetes, in patients with painful sensory neuropathy, as many as 34% in one study.12–14 Fasting plasma glucose of 100 to 125 mg/dL or 2-hour glucose of 140 to 199 mg/dL (impaired glucose tolerance) defines prediabetes. The 2-hour oral glucose tolerance test is the usual detection method and is recommended in patients with otherwise unexplained sensory neuropathy. Autonomic dysfunction appears to be common in these patients.15 The mechanism of nerve damage in hyperglycemia is still unknown, but early indications suggest that aggressive lifestyle modifications affect neuropathy progression and are the target of a multicenter clinical trial. Most patients with neuropathy associated with prediabetes are overweight and show metabolic manifestations of insulin resistance. Treatment of hyperglycemia, insulin resistance, neuropathic pain, and individualized diet and exercise counseling have been more effective than glucose-lowering medications in preventing progression from impaired glucose tolerance to diabetes.14 Diet and exercise also seem to reduce neuropathic pain in these patients.
Small Fiber Neuropathy
Because most conventional tests, such as nerve conduction studies, do not assess small-diameter fibers, other objective means to confirm this syndrome are desirable. Quantitative sensory testing assesses small and large sensory fibers by examining temperature and vibratory thresholds; although useful for sequential measures in clinical trials, it is not recommended for routine clinical diagnostic use.16,17 Epidermal nerve fiber density measured from skin punch biopsies is rapidly increasing in popularity and availability as a simple and minimally invasive technique. The small skin sample is stained immunohistochemically with antibodies against nerve-specific protein gene product 9.5 (PGP9.5).18–20 Most frequently, there is a reduction in nerve fiber numbers, worse distally, but axonal swellings also correlate with disease21 (Fig. 83-2). Sensitivity ranges from 74% to 87%. Distal autonomic function measures correlate better with epidermal nerve fiber density than cold perception thresholds and are significantly better than sural sensory nerve amplitude in documenting the selective loss of small diameter fiber function.17,19 Quantitative sudomotor axon reflex testing, which evaluates postganglionic sympathetic sudomotor function by measuring evoked sweat after acetylcholine iontophoresis, is the best validated technique in this condition, but other methods are also employed; current clinical and common research methods are listed in Table 83-2. Treatment is similar to that of diabetic and other painful forms of neuropathy covered previously.20,22
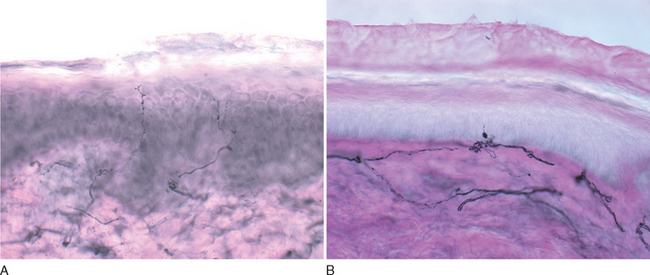
TABLE 83-2 Clinical and Research Tools for Small Fiber Neuropathy Testing
Uremic Neuropathy
Neuropathy is associated with chronic renal failure; some patients also have coincident diabetes. This is a distal symmetrical sensorimotor polyneuropathy possibly caused by uncharacterized uremic toxins. It is present in as many as 60% to 70% of patients with chronic renal failure, but patients are often asymptomatic and the neuropathy is identified only by nerve conduction studies.23 Symptoms are insidious and include painful dysesthesia, stocking-glove loss of sensation, and mild distal muscle weakness. Autonomic dysfunction is generally not as severe as with diabetes. Neuropathy typically occurs when the glomerular filtration rate falls below 20 mL/min and undialyzable toxins accumulate.24 Sensory and motor conduction velocities are mildly reduced, and evoked motor and sensory nerve response amplitudes are reduced. Pathologically, there is axonal degeneration of the most distal nerve trunks with secondary segmental demyelination.
Without dialysis or renal transplantation, prognosis is poor and the neuropathy progresses. With peritoneal or hemodialysis, the neuropathy stabilizes. Improvement after transplantation, sometimes with complete resolution of symptoms in as few as 1 to 3 months, is reported.25,26
SELECTED CHRONIC IMMUNE-MEDIATED NEUROPATHIES
Acquired Chronic Immune-Mediated Neuropathies
There is some disagreement on which entities in this category should be grouped under the unifying term chronic inflammatory demyelinating neuropathy and which variants should be separated into discrete entities. The definitive diagnosis of the classic form of chronic inflammatory demyelinating neuropathy is also debated and a number of diagnostic criteria have been proposed.27–32 The cardinal features of classic chronic inflammatory demyelinating neuropathy include progressive symptoms for more than 2 months (to differentiate from Guillain-Barré syndrome), predominant motor findings, symmetrical distal and proximal arm and leg involvement, reduced or absent deep tendon reflexes, increased cerebrospinal fluid protein, primary signs of demyelination on electrodiagnostic studies, and segmental demyelination on nerve biopsy. Not all treatment-responsive patients have all clinical or laboratory characteristics. Elevated cerebrospinal fluid protein and nerve biopsy are not required by all classifications. Magnetic resonance imaging may demonstrate gadolinium enhancement or increased T2-signal abnormalities in proximal nerves or nerve roots.
The stringent 1991 American Academy of Neurology subcommittee criteria were intended for uniformity in clinical trial enrollment, not for routine clinical application, but were used for lack of widely accepted scales31; more recent criteria deemphasize some features, sacrificing specificity to improve sensitivity.30,32 The cited prevalence is 0.5 of 100,000 children and 1 to 2 of 100,000 adults.29 The course may be relapsing or progressive but relapsing forms are more common in younger patients.33 Secondary axonal loss occurs, likely as a concomitant injury, but is critical for treatment response, long-term disability, and prognosis. Early effective treatment is the best means to limit this process.
Treatment
Randomized controlled trials have demonstrated the effectiveness of corticosteroids, plasmapheresis, and intravenous immunoglobulin and found no significant difference between these treatments, but intravenous immunoglobulin and steroids are recommended as first line treatment.34 Most studies, however, have been short in duration. Interferon β-1a was effective in open label but not in controlled trials of treatment-resistant patients. Adhering to published criteria does not seem to predict treatment responsiveness.35 Use of other medications is supported by open trials or empiric use: these include azathioprine, cyclophosphamide, mycophenolate mofetil, etanercept, and cyclosporin A. To further complicate matters, a rare treatment-responsive chronic inflammatory demyelinating neuropathy-like illness appears to be triggered by the immunomodulatory agents interferon-α or -β, tumor necrosis factor α antagonists, tacrolimus, and cyclosporin A.
Associated Conditions
A number of conditions are associated with otherwise typical CIDP, including HIV infection, hepatitis virus B and C infection, collagen vascular disease (especially systemic lupus erythematosus and Sjögren syndrome), lymphoma, melanoma, inflammatory bowel disease, and Charçot-Marie-Tooth disease. Increased incidence of demyelinating neuropathy has been noted in patients with diabetes, who appear to respond well to immunomodulatory treatment.36 The appearance of a subacute predominantly motor, symmetrical proximal and distal subacutely worsening neuropathy should raise this concern.
Chronic Inflammatory Demyelinating Neuropathy Variants
Sensory Chronic Inflammatory Demyelinating Neuropathy
From 10% to 15% of patients have a predominantly sensory syndrome but also slow motor nerve conduction velocity and other demyelinating signs; treatment response is similar to that for chronic inflammatory demyelinating neuropathy. Cerebrospinal fluid protein is elevated and residual pain is more frequent. A search for a monoclonal paraprotein and differentiation from other large fiber sensory neuropathies are necessary.37
Paraprotein-Associated Demyelinating Neuropathy
Demyelinating polyneuropathy is also associated with paraproteins, most commonly IgM and less clearly IgA or IgG. Overall, polyneuropathy occurs in 5% of patients with paraproteins, but most have only mild distal axonal sensorimotor neuropathy; some have associated amyloidosis. One third of patients with isolated monoclonal gammopathy of uncertain significance eventually develop myeloma or other hematological malignancy. Osteosclerotic myeloma is a rare form of myeloma (3%) in which one half of patients develop neuropathy, usually demyelinating and usually discovered prior to the myeloma. This entity overlaps with POEMS syndrome and associated variants that are characterized—in addition to neuropathy—by organomegaly, endocrinopathy, and skin changes, although only about 15% have the complete syndrome. Sensory neuropathy is most commonly associated with antibodies to sulfatide, but 30% of patients with antibody titers have demyelinating neuropathy with or without a paraprotein.38
Multifocal Motor Neuropathy
Multifocal motor neuropathy is an acquired immune-mediated neuropathy characterized by slowly progressive asymmetrical arm and leg weakness sparing cranial nerves, and beginning in a single peripheral nerve distribution. Typically, there is painless weakness, focal wasting, and sporadic fasciculations with preserved or absent tendon reflexes, although minor sensory complaints may also be present.39 These features can be confused with lower motor neuron disease in the early stages. However, unlike motor neuropathy, clinical weakness is generally out of proportion to atrophy, manifestations are characteristically asymmetrical and progression is slow. Men are more frequently affected with an average age of onset around 40 years.
Electrophysiological evidence of denervation is accompanied by the defining abnormality—multifocal motor conduction block, that is, the failure of a nerve impulse to conduct through an intact axon away from typical sites of nerve compression. However, lesser signs of demyelination, such as temporal dispersion, and minor sensory abnormalities are also described.40 Multifocal conduction block is the diagnostic hallmark but treatment-responsive patients with other electrophysiological abnormalities have prompted a search for additional diagnostic techniques. Ultrasonographic nerve enlargement has been noted.41 More sophisticated nerve stimulation techniques, such as magnetic stimulation and multiple stimuli, have documented proximal conduction block undetectable with conventional techniques.42 The underlying pathology is presumed to be immune mediated but predominantly restricted to motor fibers, making biopsy of a sensory nerve uninformative; a few such biopsy samples have shown little or no pathological changes. Motor nerve biopsies have shown variable features, including demyelination, rare focal inflammatory changes, frequent axonal loss, and signs of axonal regeneration.43 Some of these features are illustrated in Figure 83-3.
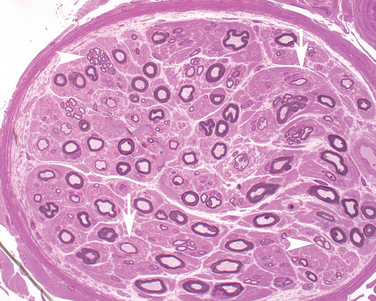
Increased titers of IgM anti-GM1 gangliosides are found in 30% to 50% or even more patients in different series; anti-GD1a antibodies are found less frequently.39,44 The pathogenicity of these antibodies remains uncertain, especially because most patients have no measurable titers. Other proposed mechanisms include antibody-mediated blockade of Na+ and K+ channels at the nodes of Ranvier. Sophisticated electrophysiological techniques suggest that focal depolarization or hyperpolarization may explain how conduction may be impaired without easily identifiable conduction block and how some patients may respond promptly to treatment.45 Cerebrospinal fluid protein is generally normal.
Multifocal Sensorimotor Demyelinating Neuropathy
Multifocal motor neuropathy is distinctively motor but may include minor sensory symptoms and findings. A separate syndrome, described in 1982 by Lewis and Sumner, is characterized by multifocal motor and sensory involvement, unlike either chronic inflammatory demyelinating neuropathy or multifocal motor neuropathy (Lewis-Sumner syndrome).46 Some prefer the cumbersome descriptive term multifocal axonal acquired demyelinating sensory and motor neuropathy. Also in contrast to multifocal motor neuropathy, male predominance is not seen, steroid-responsiveness is common, GM1 antibodies are absent, and sensory nerves demonstrate segmental demyelination and inflammatory cells. The clinical picture is closer to vasculitic neuropathy, from which it must be differentiated, but some cases are difficult to distinguish from multifocal motor neuropathy or chronic inflammatory demyelinating neuropathy. Many but not all respond to prednisone or intravenous immunoglobulin, but most respond to some form of immunotherapy.47
Vasculitic neuropathy characteristically manifests as mononeuropathy (mononeuritis) multiplex or distal symmetrical polyneuropathy. The disorder is associated with primary vasculitis, connective tissue disorders, and certain infections and malignancies. The process is most commonly systemic but isolated neuropathy can also occur. Onset is usually acute or subacute with painful severe weakness and sensory loss in the distribution of individual peripheral and cranial nerves. Progress is most commonly stepwise, but 25% to 30% of patients have distal and symmetrical neuropathy with steady progressive worsening.48,49 Rarely, patients have a chronic and indolent course.49
The etiology of vasculitic neuropathies is unknown, but the process is presumed to be immune mediated. One half of the cases occur in the setting of collagen vascular disease, and 10% are associated with malignancy, drug ingestion, or infection. Neuropathology reveals necrotizing arteritis with transmural inflammatory infiltration and fibrinoid vessel wall necrosis, lumen reduction, and signs of focal nerve ischemia, mostly in myelinated fibers. The ischemia leads to focal axonal degeneration of individual nerve fascicles.50 The diagnosis is established by the characteristic findings in biopsy samples of nerve, muscle, or both. The biopsy specimen, however, may show only axonal degeneration if the sample is distal to the infarct or if the specimen includes no affected vessels. Multiple levels may need to be sampled to increase the diagnostic yield. Nerve conduction studies may show electrical inexcitability of nerve segments distal to a site of infarct. If some nerve fascicles are selectively spared, conduction velocity is preserved but the evoked amplitude is diminished.
Immunosuppression is the primary treatment. For systemic vasculitis, prednisone (1 to 1.5 mg/kg) and cyclophosphamide (2 mg/kg) are most commonly used. In nonsystemic vasculitic neuropathy, prednisone is usually given alone. Plasmapheresis is used for cryoglobulinemia; empirical open-label benefit of intravenous immunoglobulin in treatment-resistant patients is reported.51
Prognosis has been described only in anecdotal studies. Eighty percent of nonsystemic vasculitis patients improve, but the 5-year survival rate is 37% to 48% for treated systemic vasculitis patients and only 15% for untreated patients.48,50
IATROGENIC AND TOXIC NEUROPATHY
Iatrogenic and Toxic Neuropathies
Although patients frequently suspect medications or toxins to cause otherwise unexplained peripheral neuropathies, drugs and toxins are, in fact, the cause of an estimated 2% to 4% of peripheral neuropathies.52,53 Toxic neuropathies, including medication-induced forms, generally induce axonal degeneration in a “dying back” axonal pattern. Any disturbance of axonoplasmic flow disproportionately affects the distal segments of the most vulnerable nerves, producing a uniform distal stocking-glove pattern of involvement. However, some agents cause segmental demyelination; injure Schwann cells, dorsal root, or autonomic neurons; or damage peripheral myelin. Onset is generally over weeks to months, and causation is best established by a strong dose-response relationship, consistent manifestations, close proximity of symptoms to exposure, stabilization or improvement after drug cessation, reproduction in animal models, and exclusion of other causes.54 However, some agents have idiosyncratic, delayed, or cumulative effects, their mechanisms of clearance are different in animals, and recovery is poor or delayed recovery because of axonal degeneration or neuron loss, making a definitive association problematic. In addition, some patients continue to worsen even after agent cessation, usually for several weeks, a phenomenon termed “coasting,” which clouds definitive diagnosis. A variety of neurotoxic mechanisms are known, including direct toxicity or toxicity mediated by metabolites, secondary vitamin deficiency, interference with DNA or metabolic function, mitochondrial injury, apoptosis, and immune triggers. Other factors, such as preexisting neuropathy, decreased toxin metabolism or clearance, or genetic profile may predispose to neurotoxicity. Despite these limitations, it is critical to look for the associations described above, so that an offending agent can be promptly stopped, which sometimes leads to improvement. Some agents have only a weak temporal association with neuropathy in rare patients, whereas others are clearly linked to neuropathy, which limits their use.
The blood-nerve barrier is partially protective but is less efficient than its blood-brain barrier counterpart. This and other factors make peripheral nerves more susceptible than central nerves to certain toxins. Decreased drug metabolism or clearance due to impaired hepatic or renal function can lead to the buildup of toxic agent levels, notably in the elderly. The presence of a preexisting neuropathy, whether hereditary or acquired, can predispose to neurotoxicity. Some common and more clinically relevant associations are briefly discussed; others are listed in Table 83-3.
TABLE 83-3 Medications With Probable to Well-Established Toxic Neuropathy Association
Antibiotics
Nucleoside analogs—some, but not all, nucleoside reverse transcriptase inhibitors are associated with a dose-dependent peripheral neuropathy. Examples include zalcitabine (ddC), didanosine (ddI), stavudine (ddT), and lamivudine (3TC). This distal painful sensory axonal neuropathy usually begins 6 to 8 weeks after initiating treatment. Signs and symptoms include burning dysesthesia in the legs, impaired sensation, minimal weakness, and absent ankle jerks. The process may be indistinguishable from AIDS-related neuropathy, including electrophysiological testing. The neuropathy improves after drug discontinuation, often within a month. The mechanism of neuropathy may be related to the inhibition of DNA polymerase gamma and mitochondrial DNA replication. Risk factors for nucleoside reverse transcriptase inhibitor neuropathy include a CD4 count of less than 100 cells/mm3, combination drug therapy, and underlying neuropathy.52
Linezolid, approved in 2000, is an oxazolidinone, the first new class of antibiotics approved in 40 years. It has activity against drug-resistant enterococci, Staphylococcus aureus, and Bacillus tuberculosis, making prolonged use desirable. Optic and sensory neuropathies are both well documented and growing concerns with extended use.55,56
Metronidazole is used to treat both protozoan and anaerobic bacterial infections. A predominantly sensory axonal neuropathy is a potential and underappreciated neurotoxic effect.57 Chronic treatment and high doses are risk factors, although symptomatic polyneuropathy was reported after a total of 1200 mg/day for only 9 days.58 Sural nerve biopsies show degeneration of both myelinated and unmyelinated sensory fibers. The radiosensitizing agent misonidazole is chemically related and also associated with neuropathy.
Cardiovascular Agents
Statins are notoriously associated with myopathy, but the evidence for toxic polyneuropathy was based on a series of case reports until a well-publicized large case-control series from Denmark suggested a 4- to 14-fold increased risk of developing “idiopathic” neuropathy in patients on statins.59 However, the methodology used in this study has generated some criticism.60 Identifiable causes of neuropathy were carefully excluded but the study was unable to control for undiagnosed or latent diabetes; also, recent evidence suggests that hyperlipidemia may be an independent neuropathy risk factor.6 The ascribed neuropathy is a nonspecific axonal sensorimotor neuropathy. Because these agents are so widely used, concern for causation is frequent in patients with new-onset neuropathy; a period off medication is probably the best current method to test the association in a patient. However, this entity is probably very rare.
In amiodarone users, polyneuropathy is the second most common neurological side effect, after tremor, occurring in 6% of those taking it for several months,61,62 usually in dosages greater than 400 mg/day. Sensorimotor neuropathy is most common and can be severe; motor and autonomic involvement is also seen and can also be severe. Nerve conduction studies and neuropathology show evidence of axonal loss, demyelinating features, or a combination of the two. Symptoms usually improve after drug cessation or lowering the dosage. The mechanism of toxicity is unknown but amiodarone enters lysosomes and irreversibly binds polar lipids; characteristic intralysosomal inclusions are seen in many tissues, including Schwann cells and peripheral neurons.63 Drug metabolite levels are 80-fold higher in nerve than in serum.61 Demyelinating neuropathy and lipid inclusions are also found in patients treated with another lipophilic cardiac drug, perhexiline, which has never been approved in the United States but is still used in Europe.
Chemotherapeutic Agents
Some degree of neuropathy is to be expected with certain agents, but the effects of neuropathic symptoms on the quality of life should be considered. Most agents are neurotoxic in a dose-dependent manner, either with a single high dose or with cumulative lower doses64; a lower dose may produce less toxicity but may reduce efficacy. Pretreatment with neuroprotective agents is of prime interest but not adequately investigated. Many agents have shown promise in animal models but few have performed as impressively in clinical trials. To date, amifostine is the only approved medication to blunt chemotherapeutic toxicity, primarily by reducing cisplatin toxicity on the kidney but not on peripheral nerves. Other agents have shown partial benefit in human trials, but the only drugs that appear to markedly blunt or prevent neuropathy in experimental models are neurotrophins; however, none is currently undergoing study.
Cisplatin dose-limiting neurotoxicity generally occurs at cumulative doses of 250 to 500 mg/m2, based on patient susceptibility. Patients typically have minor tingling in the distal limbs at the start of each cycle, which later resolves. Subacute numbness, paresthesia, and pain spread proximally as the cumulative dose increases; symptoms can become irreversible. Deep tendon reflexes are lost, and proprioception is disproportionately affected. Motor function is generally unaffected. Nerve conduction studies corroborate reduced sensory and normal motor responses. The drug binds to DNA in both prompts DNA repair and cell cycle entry in both dorsal root ganglia neurons and tumor cells. If the repair is unsuccessful, apoptosis may be triggered. The mechanism of toxicity is similar for carboplatin and oxaliplatin, but oxaliplatin can also cause an acute 1- to 2-day syndrome of cold-induced paresthesia, jaw and throat tightness, and cramps. Electrophysiological hallmarks of peripheral nerve hyperexcitability are seen and are thought to be caused by sodium channel dysfunction.65
Thalidomide is currently used in several neoplastic and rheumatological conditions and has antiangiogenic and anti-inflammatory properties. It causes a length-dependent sensory neuropathy, affecting both small- and large-diameter fibers. Many cases of sensory neuropathy were produced in the early 1960s but were overshadowed by the more infamous teratogenic effects. Neuropathy incidence ranges from 25% to 70%,66 and recovery is often incomplete. The mechanism is unknown but the drug inhibits the activation of necrosis factor-κB, an important factor in sensory neuron survival. Onset appears to be related to cumulative dose.
Immunosuppressants
Tacrolimus (FK-506) is a calcineurin inhibitor and macrolide antibiotic extensively used in transplant medicine. It suppresses both humoral and cellular mediated immune responses. Although central toxicity is more common, numerous cases of peripheral neuropathy are reported: this manifests as a severe multifocal demyelinating neuropathy resembling chronic inflammatory demyelinating neuropathy; patients have been treated successfully with intravenous immunoglobulin or plasmapharesis.67 Interestingly, this agent also has neuroregenerative activity, which differs from the calcineurin effects and is under investigation. Cyclosporin is rarely associated with neuropathy and sirolimus neuropathy has not been described.
Leflunomide, a novel disease-modifying rheumatoid arthritis treatment, has been associated with sometimes painful axonal sensorimotor polyneuropathy that had not been predicted in prerelease trials.68 Onset is usually after 3 to 6 months of exposure and recovery is slow but less so if therapy is stopped within 30 days of initiation. The mechanism of neurotoxicity is unknown, although medication-induced vasculitis independent from rheumatoid arthritis vasculitis is suggested in some cases.68,69
Other Agents
With chronic high dosages (serum levels greater than 20μg/mL) of phenytoin, peripheral neuropathy is known to occur, based mostly on case series. Risk factors include prolonged use (longer than 10 years), supratherapeutic levels (greater than 20μg/mL), and low folate levels.70 Neuropathy is rare at current dosages and usually produces only minimal symptoms.
Colchicine can produce a “neuromyopathy” with subacute vacuolar myopathy accompanied by mild distal pansensory axonal neuropathy. Chronic regular use and high serum levels are risk factors, which are presumed to alter axonal transport through microtubule disruption.71 Marked clinical improvement is expected after cessation.
Selected Toxins
Toxicity from heavy metals is a common patient concern but a rare cause of neuropathy.
Arsenic is the most common heavy metal to cause neuropathy: the course is slow in chronic exposure and more acute in cases of intentional poisoning. Arsenic trioxide is used to treat acute promyelocytic leukemia and can cause toxic neuropathy in a minority of treated patients. Sensory symptoms predominate initially, and pain and paresthesia precede leg weakness by days to weeks. Weakness can be severe. Stocking-glove sensory loss, especially of vibration and position sense, and distal loss of deep tendon reflexes are typical. Electrodiagnostic studies and neuropathology demonstrate signs of sensorimotor axonal loss. Chelation therapy is standard treatment.
Mercury toxicity depends on the form of mercury—elemental or organic. The organic form (methyl and ethylmercury) is most toxic to the central nervous system, although distal paresthesia and sensory ataxia are prominent (presumably from dorsal root ganglion degeneration). Ventral roots and motor function are spared. Inorganic mercury salts were used in felt hat production, and toxicity produced memory loss and tremor (hence “mad as a hatter”). Inorganic mercury may be absorbed through the gastrointestinal tract, and volatile elemental mercury may be absorbed directly through the skin or lungs. Elemental mercury exposure is a rare cause of weakness and axonal motor and sensory fiber loss. Quicksilver is used in some ethnoreligious practices. Low-level exposure is a popular concern, but no evidence supports significant toxicity from dental amalgams; exposure from dietary fish consumption and thimeresol-containing vaccines is a more controversial issue.72
Drugs of Abuse
Alcohol neuropathy is most commonly a slowly progressive predominantly sensory and sometimes painful neuropathy seen in chronic alcoholics. Symptoms and findings vary widely and are often overlooked by both patient and physician. Distal pain and dysesthesia, especially at night, are prominent; distal weakness and autonomic disturbances, although less common, are also seen. Examination reveals distal sensory loss and decreased deep tendon reflexes. Electrodiagnostic studies and nerve and muscle pathology show nonspecific, symmetrical sensory greater than motor axonal loss. Findings are common even in asymptomatic chronic alcoholics.73 Whether the cause is a nutritional deficiency or a direct toxic alcohol effect remains controversial. Although vitamin levels are often reduced in alcoholics, measures correlate poorly with incidence of neuropathy. One recent study compared alcoholic patients with normal thiamin levels to alcoholic and nonalcoholic patients with thiamin deficiencies. Patients with normal thiamin levels had typical distal sensory neuropathy and nonalcoholic thiamin-deficient patients had subacute motor-predominant neuropathy. Alcoholic thiamin-deficient patients had a mixture of findings.74 This study provided the best evidence to date of a direct toxic effect of ethanol on human peripheral nerve. Improvement after drinking cessation can occur, especially in mildly to moderately severe cases. Alcoholics are also more susceptible to nerve compression and entrapment, not limited to the classic “Saturday night palsy” affecting the radial nerve. The alcohol deterrent Dapsone can itself cause toxic neuropathy, especially at high doses, but this drug affects large-diameter sensory and motor fibers whereas alcohol disproportionately affects small fibers; however, confusion with alcoholic neuropathy sometimes occurs.75 The toxin carbon disulfide (CS2), a Dapsone metabolite, is another cause of toxic neuropathy and one possible mechanism of Dapsone toxicity.
Other drugs of abuse may lead to neuropathy, notably n-hexane and methyl-N-butyl ketone, found in widely available household solvents, fuels, and cleaning agents. Inhalation through the nose or mouth (huffing) of these materials is not rare in teens and young adults, especially if access to alcohol and hard drugs is limited. Axonal degeneration with sensory and motor impairment is seen, but focal conduction block associated with giant axonal swellings is also characteristic.76,77 Chronic industrial exposures generally lead to symmetrical axonal neuropathy. Selected other toxins are listed in Table 83-4.
Polyneuropathy Associated With Dietary States
Vitamin E deficiency contributes to neuropathy in fat malabsorption syndromes including abetalipoproteinemia, congenital biliary atresia, pancreatic dysfunction, and surgical removal of large portions of the small intestine. The clinical syndrome resembles spinocerebellar degeneration, ataxia with severe proprioception and vibration loss, and hyporeflexia. Electrodiagnostic studies show sensory but not motor changes. There is limited but preliminary evidence that vitamin E can be neuroprotective in cisplatin ototoxicity and neuropathy.78,79
Postgastrectomy Neuropathy
Bariatric surgery is rapidly increasing in popularity. Complications associated with the procedure are also increasingly noted. Peripheral neuropathy, most commonly sensorimotor axonal neuropathy, develops following surgery in a reported 5% to 16% of patients.80,81 Neuropathy risk factors include rapid and marked weight loss, poor nutritional follow-up, surgical complications, and markers of poor nutrition, such as low serum albumin. Mononeuropathies and radiculoplexus neuropathy are less common. Nerve biopsy has shown axonal degeneration and perivascular inflammation. Malnutrition, especially cobalamin, folate, thiamin, and other vitamin deficiencies, is suspected, and aggressive supplementation and nutritional counseling are recommended. Immune and inflammatory factors are also proposed but not well established.
Celiac Neuropathy
Celiac disease is a chronic inflammatory enteropathy and is increasingly recognized, with an estimated prevalence of 0.3% to 1% in Western countries. Patients with HLA-DQ2 and HLA-DQ8 alleles may show sensitivity to wheat gluten, manifested by the presence of gliadin and transglutaminase autoantibodies. In addition to ataxia, peripheral neuropathy is a common neurological association and is not attributed to nutritional deficiencies.82 Some, however, are skeptical about this association.83 The neuropathy is usually predominantly sensory and may be multifocal. Diagnosis is suspected with elevated antibody titers and confirmed by a duodenal biopsy demonstrating inflammation, crypt hyperplasia, and villous atrophy in small intestinal mucosa. Gastrointestinal—but generally not neurological—symptoms improve with a gluten-free diet. Nerve conduction studies are often normal but skin biopsy studies showed abnormalities in most patients in one small series.84 Some, but not all, recommend testing for celiac disease in idiopathic neuropathy cases, especially if irritable bowel syndrome or other gastrointestinal symptoms are present.
ACKNOWLEDGMENTS
Selected Chronic Immune-Mediated Neuropathies
Lewis RA. Chronic inflammatory demyelinating polyneuropathy and other immune-mediated demyelinating neuropathies. Semin Neurol. 2005;25:217-228.
Pratt RW, Weimer LH. Medication and toxin-induced peripheral neuropathy. Semin Neurol. 2005;25:204-216.
Said G, Lacroix C. Primary and secondary vasculitic neuropathy. J Neurol. 2005;252:633-641.
Sinnreich M, Taylor BV, Dyck PJ. Diabetic neuropathies. Classification, clinical features, and pathophysiological basis. Neurologist. 2005;11:63-79.
Van Asseldonk JT, Franssen H, Van den Berg-Vos RM, et al. Multifocal motor neuropathy. Lancet Neurol. 2005;4:309-319.
1 Sinnreich M, Taylor BV, Dyck PJ. Diabetic neuropathies. Classification, clinical features, and pathophysiological basis. Neurologist. 2005;11:63-79.
2 Boulton AJ, Vinik AI, Arezzo JC, et al. Diabetic neuropathy. A statement by the American Diabetic Association. Diabetes Care. 2005;28:956-962.
3 Dyck PJ, Kratz KM, Karnes IL, et al. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort. Neurology. 1993;43:817-824.
4 Dyck PJ, Windebank AJ. Diabetic and nondiabetic lumbosacral radiculoplexus neuropathies: new insights into pathophysiology and treatment. Muscle Nerve. 2002;25:477-491.
5 Feldman EL. Oxidative stress and diabetic neuropathy: a new understanding of an old problem. J Clin Invest. 2003;111:431-433.
6 Tesfaye S, Chaturvedi N, Simon EM, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med. 2005;352:341-350.
7 The Diabetes Control and Complications Trial Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med. 1995;122:561-568.
8 Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Eng J Med. 1993;329:977-986.
9 Mendell JR, Sahenk Z. Painful sensory neuropathy. N Engl J Med. 2003;348:1243-1255.
10 Harati Y, Gooch CL, Swenson M, et al. A double-blind, randomized trial of tramadol for treatment of the pain of diabetic neuropathy. Neurology. 1998;50:1842-1846.
11 Goldstein DJ, Lu Y, Detke MJ, et al. Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain. 2005;116:109-118.
12 Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care. 2001;24:1448-1453.
13 Sumner CJ, Sheth S, Griffin JW, et al. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology. 2003;60:108-111.
14 Singleton JR, Smith AG, Russell J, et al. Polyneuropathy with impaired glucose tolerance: implications for diagnosis and therapy. Curr Treat Options Neurol. 2005;7:33-42.
15 Rezende KF, Melo A, Pousada J, et al. Autonomic neuropathy in patients with impaired glucose intolerance. Arq Neuropsiquiatr. 1997;55:703-711.
16 Shy ME, Frohman EM, So YT, et al. Quantitative sensory testing: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2003;60:898-904.
17 Tobin K, Giuliani MJ, Lacomis D. Comparison of different modalities for detection of small-fiber neuropathy. Clin Neurophysiol. 1999;110:1909-1912.
18 Kennedy WR, Wendelschafer-Crabb G, Johnson TL, Tamura E. Use of skin biopsy and skin blister in neurologic practice. J Clin Neuromusc Dis. 2000;1:196-204.
19 Periquet MI, Novak V, Collins MP, et al. Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology. 1999;53:1641-1647.
20 Lacomis D. Small-fiber neuropathy. Muscle Nerve. 2002;26:173-188.
21 Lauria G, Morbin M, Lombardi R, et al. Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology. 2003;61:631-636.
22 Singleton JR. Evaluation and treatment of painful peripheral polyneuropathy. Semin Neurol. 2005;25:185-195.
23 Bolton CF. Peripheral neuropathies associated with chronic renal failure. Can J Neurol Sci. 1980;7:89-96.
24 Layzer RB. Neuromuscular Manifestations of Systemic Disease. Philadelphia: FA Davis, 1985;283-296.
25 Bolton CF, McKeown MJ, Chen R, et al. Subacute uremic and diabetic polyneuropathy. Muscle Nerve. 1997;20:59-64.
26 Nielsen VK. The peripheral nerve function in chronic renal failure. Recovery after renal transplantation. Clinical aspects. Acta Med Scand. 1974;195:163-170.
27 Lewis RA. Chronic inflammatory demyelinating polyneuropathy and other immune-mediated demyelinating neuropathies. Semin Neurol. 2005;25:217-228.
28 Berger AR, Bradley WG, Brannagan TH, et al. Guidelines for the diagnosis and treatment of chronic inflammatory demyelinating polyneuropathy. J Periph Nerv Sys. 2003;8:282-284.
29 Köller H, Kieseier BC, Jander S, et al. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med. 2005;352:1343-1356.
30 Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50:195-201.
31 Anonymous. Research criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). Report from an Ad Hoc Subcommittee of the American Academy of Neurology AIDS Task Force. Neurology. 1991;41:617-618.
32 Saperstein DS, Katz JS, Amato AA, et al. Clinical spectrum of chronic acquired demyelinating polyneuropathies. Muscle Nerve. 2001;24:311-324.
33 Hattori N, Misu K, Koike H, et al. Age of onset influences clinical features of chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. 2001;184:57-63.
34 Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. J Peripher Nerv Syst. 2005;10:220-228.
35 Chan YC, Allen DC, Fialho D, et al. Predicting response to treatment in chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 2006;77:114-116.
36 Haq RU, Pendlebury WW, Fries TJ, et al. Chronic inflammatory demyelinating polyradiculoneuropathy in diabetic patients. Muscle Nerve. 2003;27:465-470.
37 Gorson KC, Allam G, Ropper AH. Chronic inflammatory demyelinating polyneuropathy: clinical features and response to treatment in 67 consecutive patients with and without a monoclonal gammopathy. Neurology. 1997;48:321-328.
38 Dabby R, Weimer LH, Hays AP, et al. Antisulfatide antibodies in neuropathy: clinical and electrophysiologic correlates. Neurology. 2000;54:1448-1452.
39 Van Asseldonk JT, Franssen H, Van den Berg-Vos RM, et al. Multifocal motor neuropathy. Lancet Neurol. 2005;4:309-319.
40 Chaudhry V, Corse AM, Cornblath DR, et al. Multifocal motor neuropathy: electrodiagnostic features. Muscle Nerve. 1994;17:198-205.
41 Beekman R, van den Berg LH, Franssen H, et al. Ultrasonography shows extensive nerve enlargements in multifocal motor neuropathy. Neurology. 2005;65:305-307.
42 Attarian S, Azulay JP, Verschueren A, et al. Magnetic stimulation using a triple-stimulation technique in patients with multifocal neuropathy without conduction block. Muscle Nerve. 2005;32:710-714.
43 Taylor BV, Dyck PJ, Engelstad J, et al. Multifocal motor neuropathy: pathologic alterations at the site of conduction block. J Neuropathol Exp Neurol. 2004;63:129-137.
44 Kinsella LJ, Lange DJ, Trojaborg W, et al. Clinical and electrophysiologic correlates of elevated anti-GM1 antibody titers. Neurology. 1994;44:1278-1282.
45 Priori A, Bossi B, Ardolino G, et al. Pathophysiological heterogeneity of conduction blocks in multifocal motor neuropathy. Brain. 2005;128:1642-1648.
46 Lewis RA, Sumner AJ, Brown MJ, et al. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32:958-962.
47 Viala K, Renié L, Maisonobe T, et al. Follow-up study and response to treatment in 23 patients with Lewis-Sumner syndrome. Brain. 2004;127:2010-2017.
48 Hawke SH, Davies L, Pamphlett R, et al. Vasculitic neuropathy. A clinical and pathological study. Brain. 1991;114:2175-2190.
49 Kissel JT, Slivka AP, Warmolts JR, et al. The clinical spectrum of necrotizing angiopathy of the peripheral nervous system. Ann Neurol. 1985;18:251-257.
50 Said G, Lacroix C. Primary and secondary vasculitic neuropathy. J Neurol. 2005;252:633-641.
51 Levy Y, Uziel Y, Zandman G, et al. Response of vasculitic peripheral neuropathy to intravenous immunoglobulin. Ann NY Acad Sci. 2005;1051:779-786.
52 Pratt RW, Weimer LH. Medication and toxin-induced peripheral neuropathy. Semin Neurol. 2005;25:204-216.
53 Jain KK. Drug-induced peripheral neuropathies. In: Jain KK, editor. Drug-Induced Neurological Disorders. 2nd ed. Seattle: Hogrefe & Huber; 2001:263-294.
54 Hill AB. The environment and disease: association or causation? Proc R Soc Med. 1965;58:295-300.
55 Zivkovic SA, Lacomis D. Severe sensory neuropathy associated with long-term linezolid use. Neurology. 2005;4:926-927.
56 Frippiat F, Derue G. Causal relationship between neuropathy and prolonged linezolid use. Clin Infect Dis. 2004;39:439.
57 Kapoor K, Chandra M, Nag D, et al. Evaluation of metronidazole toxicity: a prospective study. Int J Clin Pharmacol Res. 1999;19:83-88.
58 Rustscheff S, Hulten S. An unexpected and severe neurological disorder with permanent disability acquired during short-course treatment with metronidazole. Scand J Infect Dis. 2003;35:279-280.
59 Gaist D, Jeppesen U, Andersen M, et al. Statins and risk of polyneuropathy: a case control study. Neurology. 2002;58:1333-1337.
60 Leis AA, Stokic D, Olivier J. Statins and polyneuropathy: setting the record straight. Muscle Nerve. 2005;32:428-430.
61 Fraser AG, McQueen IN, Watt AH, et al. Peripheral neuropathy during long-term high-dose amiodarone therapy. J Neurol Neurosurg Psychiatry. 1985;48:576-578.
62 Charness ME, Morady F, Scheinman MM. Frequent neurologic toxicity associated with amiodarone therapy. Neurology. 1984;34:669-671.
63 Santoro L, Barbieri F, Nucciotti R, et al. Amiodarone induced experimental acute neuropathy in rats. Muscle Nerve. 1992;15:788-795.
64 Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249:9-17.
65 Lehky TJ, Leonard GD, Wilson RH, et al. Oxaliplatin-induced neurotoxicity: acute hyperexcitability and chronic neuropathy. Muscle Nerve. 2004;29:387-392.
66 Cavaletti G, Beronio A, Reni L, et al. Thalidomide sensory neurotoxicity: a clinical and neurophysiologic study. Neurology. 2004;62:2291-2293.
67 Wilson JR, Conwit RA, Eidelman BH, et al. Sensorimotor neuropathy resembling CIDP in patients receiving FK-506. Muscle Nerve. 1994;17:528-532.
68 Bonnel RA, Grahm DJ. Peripheral neuropathy in patients treated with leflunomide. Clin Pharm Ther. 2004;75:580-585.
69 Martin K, Bentaberry F, Dumoulin C, et al. Neuropathy associated with leflunomide: a case series. Ann Rheum Dis. 2005;64:649-650.
70 Shovron SD, Reynolds EH. Anticonvulsant peripheral neuropathy: a clinical and electrophysiological study of patients on single drug treatment with phenytoin, carbamazepine or barbiturates. J Neurol Neurosurg Psychiatry. 1982;45:620-626.
71 Kuncl RW, Duncan G, Watson D, et al. Colchicine myopathy and neuropathy. N Eng J Med. 1987;316:1562-1568.
72 Clarkson TW, Magos L, Myers GJ. The toxicology of mercury: current exposures and clinical manifestations. N Engl J Med. 2003;349:1731-1737.
73 Vittadini G, Buonocore M, Colli G, et al. Alcoholic polyneuropathy: a clinical and epidemiological study. Alcohol Alcohol. 2001;36:393-400.
74 Koike H, Iijima M, Sugiura M, et al. Alcoholic neuropathy is clinicopathologically distinct from thiamine-deficiency neuropathy. Ann Neurol. 2003;54:19-29.
75 Palliyath SK, Schwartz BD, Gant L. Peripheral nerve functions in chronic alcoholic patients on disulfiram: a six month follow up. J Neurol Neurosurg Psychiatry. 1990;53:227-230.
76 Chang AP, England JD, Garcia CA, et al. Focal conduction block in n-hexane polyneuropathy. Muscle Nerve. 1998;21:964-969.
77 Pastore C, Izura V, Marhuenda D, et al. Partial conduction blocks in n-hexane neuropathy. Muscle Nerve. 2002;26:132-135.
78 Argyriou AA, Chroni E, Koutras A, et al. Vitamin E for prophylaxis against chemotherapy-induced neuropathy: a randomized controlled trial. Neurology. 2005;64:26-31.
79 Leonetti C, Biroccio A, Gabellini C, et al. Alpha-tocopherol protects against cisplatin-induced toxicity without interfering with antitumor efficacy. Int J Cancer. 2003;104:243-250.
80 Thaisetthawatkul P, Collazo-Clavell ML, Sarr MG, et al. A controlled study of peripheral neuropathy after bariatric surgery. Neurology. 2004;63:1462-1470.
81 Koffman BM, Greenfield LJ, Ali II, Pirzada NA. Neurologic complications after surgery for obesity. Muscle Nerve. 2006;33:166-176.
82 Chin RL, Sander HW, Brannagan TH. Celiac neuropathy. Neurology. 2003;60:1581-1585.
83 Rosenberg NR, Vermeulen M. Should coeliac disease be considered in the work up of patients with chronic peripheral neuropathy? J Neurol Neurosurg Psychiatry. 2005;76:1415-1419.
84 Brannagan TH, Hays AP, Chin SS, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol. 2005;62:1574-1578.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 83 METABOLIC, IMMUNE-MEDIATED, AND TOXIC NEUROPATHIES
Acquired peripheral neuropathy is a highly prevalent and often overlooked condition. Most patients and many physicians are not sufficiently aware of the range of disorders associated with neuropathy outside of the most common cause—diabetes mellitus. Although many neuropathies, including the roughly 30% that have no discernible cause, have no effective treatment, symptomatic therapy is available and therapeutic options are continually expanding. In contrast, many disorders discussed in this chapter can be potentially reversed or at least slowed by treating the underlying condition, removing offending toxins or medications, or limiting immune-mediated injury. For this to happen, proper diagnosis is essential. Several immune-mediated neuropathies are included here, but other important types of neuropathy are not discussed or are considered in more general sections on specific topics. Noteworthy examples include paraneoplastic neuropathy, hereditary and acquired amyloidosis, axonal paraprotein- and myeloma-associated, and acute immune-mediated neuropathies. Topics discussed in other sections include sarcoidosis, environmental toxins, nerve injury, and collagen vascular diseases.
NEUROPATHIES ASSOCIATED WITH DIABETES MELLITUS
Diabetes mellitus is the most common cause of neuropathy in the Western world, as noted since the early 1800s. The most common presentation is distal, predominantly sensory neuropathy; however, diabetes is associated with numerous other forms of neuropathy, ranging from asymptomatic to disabling motor, autonomic, or pain syndromes (Table 83-1). This syndromic list is both clinically and experimentally useful because the differing underlying mechanisms that are likely to occur directly influence treatment choices and efficacy, clinical trial selection, and mechanistic bench research. Most published lists differ slightly in syndrome numbers and entity names, and a separate list could be based on probable pathophysiological mechanisms.1 Important clinical features include nerve modalities, symmetry, speed of onset, distribution, and related diabetic complications, especially renal failure. A consensus statement in 2005 reviewed clinical syndromes, pathogenic mechanisms, and recommended treatment.2 There is growing evidence that symptomatic peripheral neuropathy can occur in the presence of mild diabetes or glucose intolerance even in the absence of notable damage to other end organs. However, in most large series, neuropathy is associated with both retinopathy and nephropathy.3 Less common forms of diabetic neuropathy are not associated with diabetic complications, suggesting a different pathophysiology, such as inflammatory or immune-mediated injury. Conversely, the coexistence of diabetes and neuropathy in a particular patient is not in itself proof of diabetic neuropathy, as unrelated processes may be present that warrant appropriate diagnostic investigation. In general, any of the entities listed can occur with either type 1 or type 2 diabetes (Table 83-1).
TABLE 83-1 Clinical Neuropathy Patterns Associated With Diabetes Mellitus
Distal Diabetic Polyneuropathy
Foot numbness and paresthesia have gradual onset and ascend slowly. The process can spread to the hands, arms, trunk, and scalp in advanced cases, but more commonly hand and arm involvement is caused by superimposed processes, such as compressive mononeuropathy.3 Loss of light touch, pain, and temperature precede loss of proprioception; distal weakness and atrophy are late findings. Patients generally notice “positive” or unpleasant symptoms, especially burning pain, electrical or stabbing sensations, paresthesia, and deep pain; loss of sensation and anesthesia are less common. Symptoms are frequently worse at night when there are less sensory distractions. Approximately 20% to 50% of diabetic patients have at least one symptom of diabetic neuropathy but even more have asymptomatic disease. Lack of sensation can lead to unperceived injury, ulcers, and Charçot joints—a significant form of morbidity. Not surprisingly, objective evidence of functional loss on clinical examination and laboratory testing often exceeds symptoms. Onset may occur after several years of diabetes or with subclinical diabetes, and the neuropathy may lead to the discovery of underlying diabetes. Sensory fibers in the foot and distal leg are predominantly affected, especially small diameter sensory and autonomic fibers. Examination of the lower leg usually reveals loss of vibration, pressure, pain, and temperature perception (both small and large fiber-mediated) and absent ankle deep tendon reflexes. Signs of peripheral autonomic (sympathetic) dysfunction are frequent, including altered skin temperature (cold or warm), dry, often scaly, skin, and calluses in pressure-bearing areas. Loss of sensation for 128-Hz vibration, monofilament touch, and loss of ankle jerks are considered 87% sensitive for detecting neuropathy and predicting foot ulcer.2 The diagnosis is ultimately clinical and requires exclusion of other reasonable causes of neuropathy. Even if symptoms remain confined to the legs and feet, objective testing such as nerve or skin biopsy and clinical electrophysiological studies often show evidence of more widespread involvement. Electrodiagnostic studies are an important adjunct to assess severity and to parse out superimposed mononeuropathy, regional syndromes, superimposed radiculopathy, and demyelinating neuropathy. Nerve biopsy is not routinely necessary but in some cases it demonstrates signs of microvascular changes in addition to axonal loss (Fig. 83-1). Skin punch biopsy to assess epidermal nerve fiber density is increasingly available and minimally invasive, as discussed later.
Diabetic autonomic neuropathy affects up to one half of diabetics.3 Genitourinary and sexual dysfunction, such as impotence, are most common, but orthostatic intolerance and hypotension, gastrointestinal dysmotility (diarrhea or constipation), gastroparesis, fatigue and exercise intolerance, vasomotor disturbance, and pupil dysfunction are also common. Patients with diabetic autonomic neuropathy have shorter survival than do unaffected diabetic patients, but the relative risk is less than early reports suggested. Patients may become intolerant of autonomically active medications, such as antihypertensive or anticholinergic agents, as the neuropathy slowly worsens, producing symptoms such as orthostatic complaints, syncope, urinary retention, excessively dry eyes, and focusing difficulties. Formal autonomic testing is widely available and is especially helpful when the clinical picture is equivocal or the cause of orthostatic symptoms is unclear. Treatment and assessment are discussed in Chapters 29 through 32 Chapter 30 Chapter 31 Chapter 32. The pathophysiology is presumed to be the same as in distal diabetic polyneuropathy and is discussed later.
Ischemic mononeuropathies are presumably due to occlusion of arterioles supplying individual nerves and produce acute ischemia and aching pain, followed by loss of nerve function. The process can affect either cranial or peripheral nerves or nerve roots. Pupil-sparing oculomotor palsy is the most common cranial neuropathy, followed by sixth and seventh neuropathies, but all are relatively uncommon. Diabetics are more susceptible to compression or entrapment neuropathy, and at least 30% of diabetics have carpal tunnel syndrome, which can range from asymptomatic to very distressing.1 Ulnar neuropathy at the elbow, peroneal neuropathy at the fibular head, and lateral femoral cutaneous neuropathy (meralgia paresthetica) are also common.
Diabetic amyotrophy (diabetic radiculoplexus neuropathy) is a monophasic illness seen primarily in type 2 diabetic patients and characterized by subacute proximal leg weakness progressing stepwise over weeks to months, weight loss, atrophy, and aching proximal leg pain. The weakness affects mostly but not exclusively the femoral and obturator nerve distributions, plateaus over weeks, and slowly improves over 12 to 36 months.4 Sensory involvement is less marked and often overshadowed by the underlying distal polyneuropathy. Anatomic boundaries are not respected, and there is often evidence of both nerve root and plexus involvement, hence the descriptive label. Findings may be unilateral or bilateral but asymmetric. If involvement is extensive, a diagnosis of chronic inflammatory demyelinating polyneuropathy, which is more common in diabetic patients, should be considered. Thoracic and cervical forms also occur but are less common and must be distinguished from ischemic injury and unrelated compressive spine disease. Although various neuropathological findings have been reported, an immune-mediated process is suspected and a long-awaited treatment trial is ongoing.
Etiology
There are numerous lines of evidence and hypotheses attempting to explain diabetic polyneuropathy but most start with perturbations initiated by excessive glucose levels. Duration of diabetes, hemoglobin A1c levels, and other signs of poor glucose control correlate with neuropathy development and severity. Microvascular injury appears to be a crucial process in the development of neuropathy as well as in damage of other end organs, and interruption of this process has long been a target of therapeutic intervention. Four major pathways of glucose metabolism are implicated. Excess intracellular glucose is processed, in part, through the polyol pathway in a series of reactions catalyzed by aldose reductase. This pathway leads to sorbitol and fructose accumulation, NAD(P)H-redox imbalances, changes in signal transduction, and excessive production of reactive oxygen radicals.5 Also, nonenzymatic glycation of proteins produces advanced glycation end products in peripheral nerve, and these impair axonal transport, neurotrophic factor production, and gene expression. In addition, protein kinase C activation starts a cascade of stress responses and increases hexosamine pathway flux; both processes lead to oxygen radical generation.5 Specific inhibitors of each pathway, aimed at blocking one or more microvascular complications, have shown considerable promise in rodent models: they include nine different aldose reductase inhibitors, various neurotrophins, blood flow and angiogenesis enhancers, free radical scavengers, and others. Unfortunately, virtually all have failed or have produced equivocal results, but several trials are presently ongoing, including a gene therapy trial of vascular endothelial growth factor. A broader approach targeting several pathways at once may be needed to affect human disease. Other risk factors have gathered recent attention. A large European consortium has found the following independent risks factors for the cumulative incidence of diabetic neuropathy: low-density lipoprotein cholesterol and triglycerides, high body mass index, high von Willebrand factor levels and urinary albumin excretion rate, hypertension, and smoking.6 Several factors are potentially adjustable and some might explain why neuropathy develops in patients with early disease or with simple glucose intolerance, as we discuss later. More acute or subacute entities suggest an immune-mediated mechanism, and some may respond to immunomodulating therapies.
Treatment
Tight glycemic control is the most important and only intervention proved to prevent or limit diabetic polyneuropathy and autonomic neuropathy, but it does not reverse existing nerve injury.7,8 Treatment of other modifiable potential risk factors (lipids, blood pressure, weight) is an evolving but likely important approach. The efficacy of preventative medications currently in clinical trials is yet to be determined.
Numerous symptomatic treatments are effective in reducing—but usually not eliminating—the discomfort of diabetic and other painful polyneuropathies. Randomized double-blind, controlled trials have demonstrated symptomatic relief, primarily for painful neuropathy, with a number of agents, including several tricyclic antidepressants, gabapentin, pregabalin, tramadol hydrochloride, and the serotonin and norepinephrine reuptake inhibitor duloxetine.1,9–11 The selective serotonin reuptake inhibitor paroxetine was better than placebo but not better than imipramine, and citalopram was similar to paroxetine; fluoxetine showed no significant benefit. However, only pregabalin and duloxetine have received U.S. Food and Drug Administration indications for treatment of painful neuropathy. Numerous other medications and therapies have undergone small randomized or incompletely controlled trials and are empirically used. Proper foot care and prompt attention to injury are essential.
Glucose Intolerance and Neuropathy
There is increasing recognition of impaired glucose tolerance, sometimes designated as prediabetes, in patients with painful sensory neuropathy, as many as 34% in one study.12–14 Fasting plasma glucose of 100 to 125 mg/dL or 2-hour glucose of 140 to 199 mg/dL (impaired glucose tolerance) defines prediabetes. The 2-hour oral glucose tolerance test is the usual detection method and is recommended in patients with otherwise unexplained sensory neuropathy. Autonomic dysfunction appears to be common in these patients.15 The mechanism of nerve damage in hyperglycemia is still unknown, but early indications suggest that aggressive lifestyle modifications affect neuropathy progression and are the target of a multicenter clinical trial. Most patients with neuropathy associated with prediabetes are overweight and show metabolic manifestations of insulin resistance. Treatment of hyperglycemia, insulin resistance, neuropathic pain, and individualized diet and exercise counseling have been more effective than glucose-lowering medications in preventing progression from impaired glucose tolerance to diabetes.14 Diet and exercise also seem to reduce neuropathic pain in these patients.
Small Fiber Neuropathy
Because most conventional tests, such as nerve conduction studies, do not assess small-diameter fibers, other objective means to confirm this syndrome are desirable. Quantitative sensory testing assesses small and large sensory fibers by examining temperature and vibratory thresholds; although useful for sequential measures in clinical trials, it is not recommended for routine clinical diagnostic use.16,17 Epidermal nerve fiber density measured from skin punch biopsies is rapidly increasing in popularity and availability as a simple and minimally invasive technique. The small skin sample is stained immunohistochemically with antibodies against nerve-specific protein gene product 9.5 (PGP9.5).18–20 Most frequently, there is a reduction in nerve fiber numbers, worse distally, but axonal swellings also correlate with disease21 (Fig. 83-2). Sensitivity ranges from 74% to 87%. Distal autonomic function measures correlate better with epidermal nerve fiber density than cold perception thresholds and are significantly better than sural sensory nerve amplitude in documenting the selective loss of small diameter fiber function.17,19 Quantitative sudomotor axon reflex testing, which evaluates postganglionic sympathetic sudomotor function by measuring evoked sweat after acetylcholine iontophoresis, is the best validated technique in this condition, but other methods are also employed; current clinical and common research methods are listed in Table 83-2. Treatment is similar to that of diabetic and other painful forms of neuropathy covered previously.20,22
TABLE 83-2 Clinical and Research Tools for Small Fiber Neuropathy Testing
Uremic Neuropathy
Neuropathy is associated with chronic renal failure; some patients also have coincident diabetes. This is a distal symmetrical sensorimotor polyneuropathy possibly caused by uncharacterized uremic toxins. It is present in as many as 60% to 70% of patients with chronic renal failure, but patients are often asymptomatic and the neuropathy is identified only by nerve conduction studies.23 Symptoms are insidious and include painful dysesthesia, stocking-glove loss of sensation, and mild distal muscle weakness. Autonomic dysfunction is generally not as severe as with diabetes. Neuropathy typically occurs when the glomerular filtration rate falls below 20 mL/min and undialyzable toxins accumulate.24 Sensory and motor conduction velocities are mildly reduced, and evoked motor and sensory nerve response amplitudes are reduced. Pathologically, there is axonal degeneration of the most distal nerve trunks with secondary segmental demyelination.
Without dialysis or renal transplantation, prognosis is poor and the neuropathy progresses. With peritoneal or hemodialysis, the neuropathy stabilizes. Improvement after transplantation, sometimes with complete resolution of symptoms in as few as 1 to 3 months, is reported.25,26
SELECTED CHRONIC IMMUNE-MEDIATED NEUROPATHIES
Acquired Chronic Immune-Mediated Neuropathies
There is some disagreement on which entities in this category should be grouped under the unifying term chronic inflammatory demyelinating neuropathy and which variants should be separated into discrete entities. The definitive diagnosis of the classic form of chronic inflammatory demyelinating neuropathy is also debated and a number of diagnostic criteria have been proposed.27–32 The cardinal features of classic chronic inflammatory demyelinating neuropathy include progressive symptoms for more than 2 months (to differentiate from Guillain-Barré syndrome), predominant motor findings, symmetrical distal and proximal arm and leg involvement, reduced or absent deep tendon reflexes, increased cerebrospinal fluid protein, primary signs of demyelination on electrodiagnostic studies, and segmental demyelination on nerve biopsy. Not all treatment-responsive patients have all clinical or laboratory characteristics. Elevated cerebrospinal fluid protein and nerve biopsy are not required by all classifications. Magnetic resonance imaging may demonstrate gadolinium enhancement or increased T2-signal abnormalities in proximal nerves or nerve roots.
The stringent 1991 American Academy of Neurology subcommittee criteria were intended for uniformity in clinical trial enrollment, not for routine clinical application, but were used for lack of widely accepted scales31; more recent criteria deemphasize some features, sacrificing specificity to improve sensitivity.30,32 The cited prevalence is 0.5 of 100,000 children and 1 to 2 of 100,000 adults.29 The course may be relapsing or progressive but relapsing forms are more common in younger patients.33 Secondary axonal loss occurs, likely as a concomitant injury, but is critical for treatment response, long-term disability, and prognosis. Early effective treatment is the best means to limit this process.
Treatment
Randomized controlled trials have demonstrated the effectiveness of corticosteroids, plasmapheresis, and intravenous immunoglobulin and found no significant difference between these treatments, but intravenous immunoglobulin and steroids are recommended as first line treatment.34 Most studies, however, have been short in duration. Interferon β-1a was effective in open label but not in controlled trials of treatment-resistant patients. Adhering to published criteria does not seem to predict treatment responsiveness.35 Use of other medications is supported by open trials or empiric use: these include azathioprine, cyclophosphamide, mycophenolate mofetil, etanercept, and cyclosporin A. To further complicate matters, a rare treatment-responsive chronic inflammatory demyelinating neuropathy-like illness appears to be triggered by the immunomodulatory agents interferon-α or -β, tumor necrosis factor α antagonists, tacrolimus, and cyclosporin A.
Associated Conditions
A number of conditions are associated with otherwise typical CIDP, including HIV infection, hepatitis virus B and C infection, collagen vascular disease (especially systemic lupus erythematosus and Sjögren syndrome), lymphoma, melanoma, inflammatory bowel disease, and Charçot-Marie-Tooth disease. Increased incidence of demyelinating neuropathy has been noted in patients with diabetes, who appear to respond well to immunomodulatory treatment.36 The appearance of a subacute predominantly motor, symmetrical proximal and distal subacutely worsening neuropathy should raise this concern.
Chronic Inflammatory Demyelinating Neuropathy Variants
Sensory Chronic Inflammatory Demyelinating Neuropathy
From 10% to 15% of patients have a predominantly sensory syndrome but also slow motor nerve conduction velocity and other demyelinating signs; treatment response is similar to that for chronic inflammatory demyelinating neuropathy. Cerebrospinal fluid protein is elevated and residual pain is more frequent. A search for a monoclonal paraprotein and differentiation from other large fiber sensory neuropathies are necessary.37
Paraprotein-Associated Demyelinating Neuropathy
Demyelinating polyneuropathy is also associated with paraproteins, most commonly IgM and less clearly IgA or IgG. Overall, polyneuropathy occurs in 5% of patients with paraproteins, but most have only mild distal axonal sensorimotor neuropathy; some have associated amyloidosis. One third of patients with isolated monoclonal gammopathy of uncertain significance eventually develop myeloma or other hematological malignancy. Osteosclerotic myeloma is a rare form of myeloma (3%) in which one half of patients develop neuropathy, usually demyelinating and usually discovered prior to the myeloma. This entity overlaps with POEMS syndrome and associated variants that are characterized—in addition to neuropathy—by organomegaly, endocrinopathy, and skin changes, although only about 15% have the complete syndrome. Sensory neuropathy is most commonly associated with antibodies to sulfatide, but 30% of patients with antibody titers have demyelinating neuropathy with or without a paraprotein.38
[/not-level-membership-for-neurology-category]
