Metabolic disorders
Mucinoses
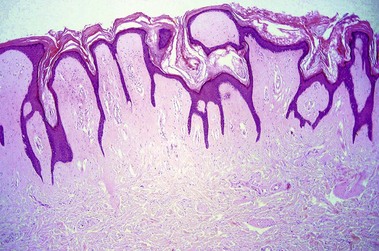
Scleredema (of Buschke)
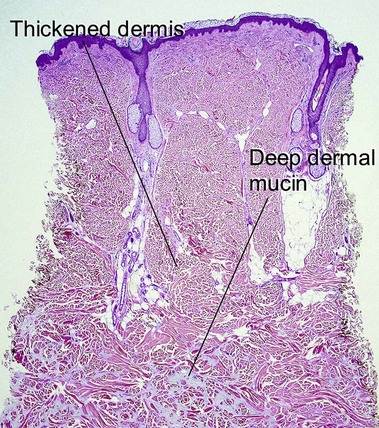
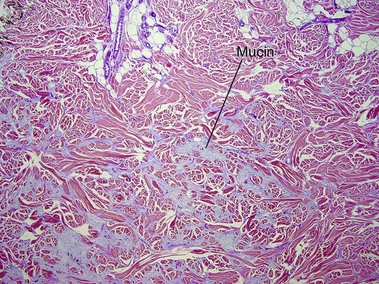
Scleredema most often occurs in the setting of insulin-dependent, adult-onset diabetes on the upper back, an area that normally displays a thick dermis. Scleredema can also occur suddenly following a streptococcal infection or in association with a monoclonal gammopathy.
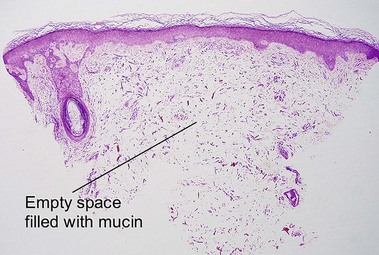
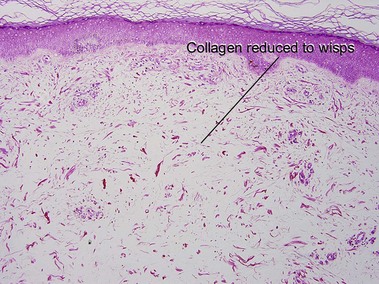
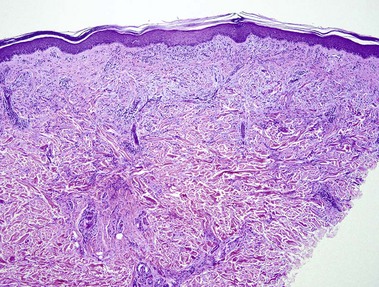
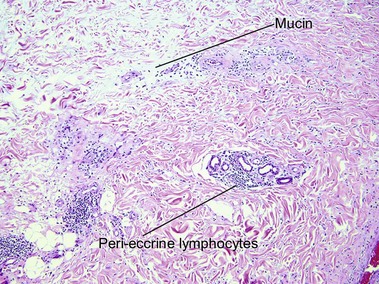
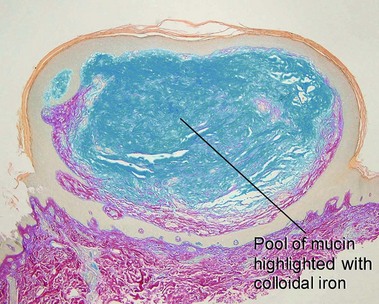
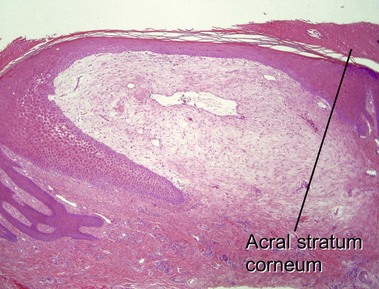
Amyloidosis
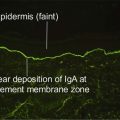
The clinical types of amyloidosis and their associated amyloid protein are listed in Table 14.1. In addition to the specific fibrillar component, amyloid P, a non-fibrillar glycoprotein, binds to all types of amyloid fibrils. Antisera to these proteins can be used immunohistochemically to identify the amyloid deposition.
Table 14-1
Clinical types of amyloidosis and their associated amyloid protein
| Clinical type | Amyloid fibril protein | Precursor substance |
| Localized cutaneous | ||
| Macular | AK | Altered keratin |
| Lichenoid | AK | Altered keratin |
| Nodular | AL (Aλ and Aκ) | Immunoglobulin light chain |
| Systemic | ||
| Primary/myeloma-associated | AL (Aλ and Aκ) | Immunoglobulin light chain |
| Secondary | AA | Serum amyloid A |
| Familial Mediterranean fever | AA | Serum amyloid A |
| Muckle–Wells’ syndrome | AA | Serum amyloid A |
| Familial amyloid polyneuropathy | Prealbumin (transthyretin) | |
| Hemodialysis-associated | β2-microglobulin |
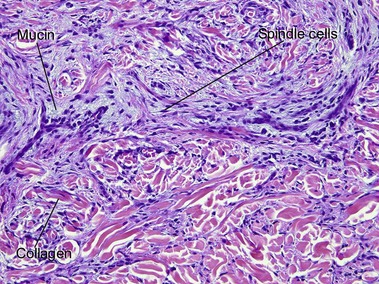
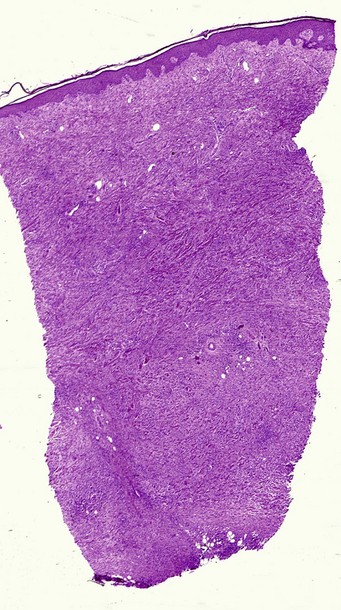
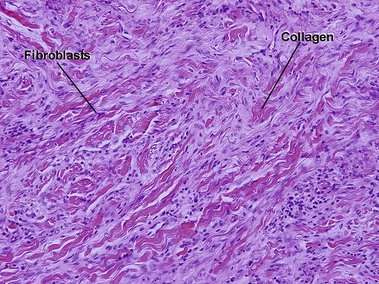
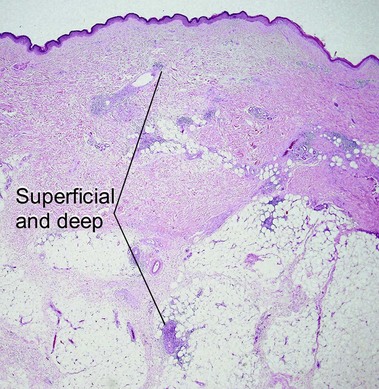
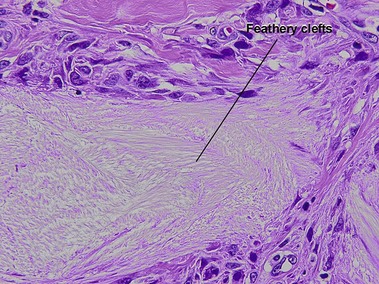
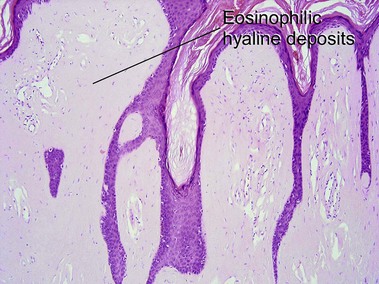
Nodular amyloidosis
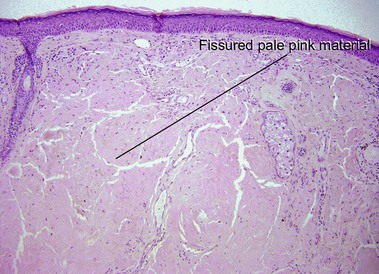
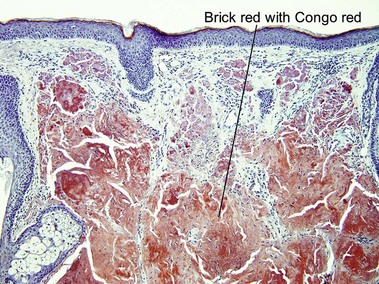
Nodular amyloid consists of light chains (AL) and may be a purely cutaneous lesion or occur in association with primary systemic amyloidosis. Lesions may be a manifestation of a localized plasmacytoma.
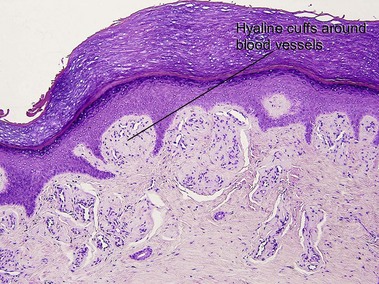
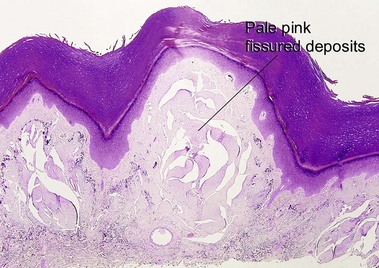
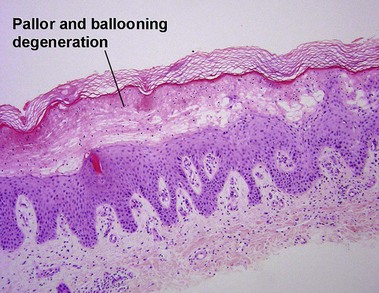
Macular amyloid
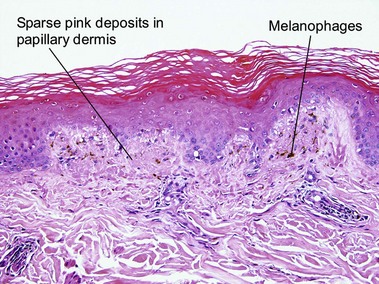
Alternatively, what initially appears to be normal skin at scan can be approached systematically, starting in the stratum corneum looking for organisms, then moving deeper to look for absence of granular layer, followed by the epidermal pattern, alteration in pigmentation, deposition in the dermal papillae, perivascular or interstitial infiltrate, and down to the eccrine glands looking for silver granules.
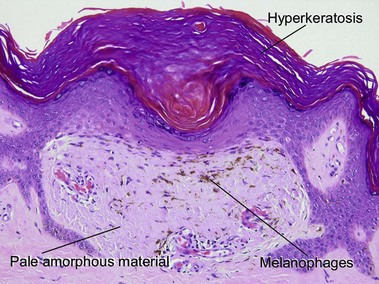
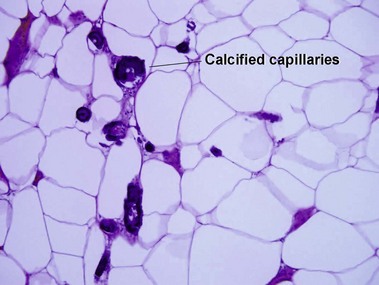
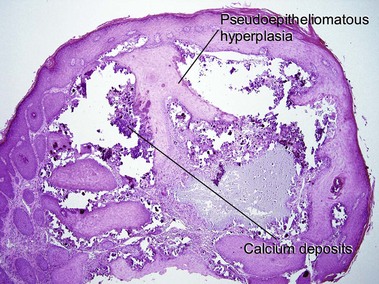
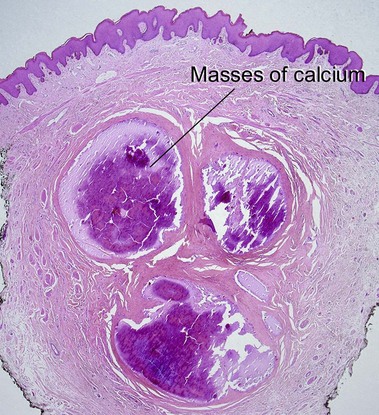
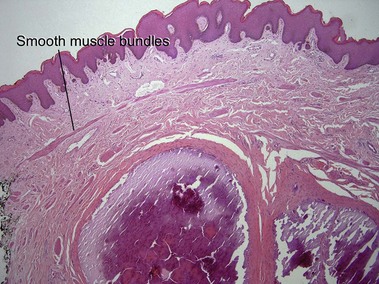
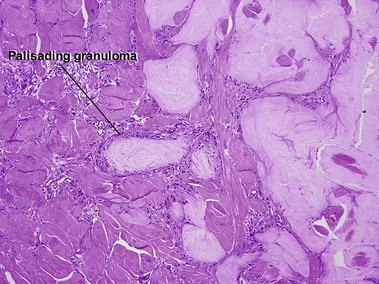
Cutaneous calcification
Blackmon, JA, Jeffy, BG, Malone, JC, et al. Oxalosis involving the skin: case report and literature review. Arch Dermatol. 2011; 147(11):1302–1305.
Girardi, M, Kay, J, Elston, DM, et al. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011; 65(6):1095–1106. e7.
Hashimoto, K, Nakayama, H, Chimenti, S, et al. Juvenile colloid milium: Immunohistochemical and ultrastructural studies. J Cutan Pathol. 1989; 16(3):164–174.
Kövary, PM, Vakilzadeh, F, Macher, E, et al. Monoclonal gammopathy in scleredema. Observations in three cases. Arch Dermatol. 1981; 117(9):536–539.
Kucher, C, Xu, X, Pasha, T, et al. Histopathologic comparison of nephrogenic fibrosing dermopathy and scleromyxedema. J Cutan Pathol. 2005; 32(7):484–490.
Markova, A, Lester, J, Wang, J, et al. Diagnosis of common dermopathies in dialysis patients: a review and update. Semin Dial. 2012; 25(4):408–418.
Masuda, C, Mohri, S, Nakajima, H. Histopathological and immunohistochemical study of amyloidosis cutis nodularis atrophicans – comparison with systemic amyloidosis. Br J Dermatol. 1988; 119(1):33–43.
Saad, AG, Zaatari, GS. Scrotal calcinosis: is it idiopathic? Urology. 2001; 57(2):365.
Yanagihara, M, Mehregan, AH, Mehregan, DR. Staining of amyloid with cotton dyes. Arch Dermatol. 1984; 120(9):1184–1185.