Metabolic Bone Disease
Abnormalities of Mineralization
In the process of bone formation, osteoblasts produce organic bone matrix (osteoid), which then must be mineralized with deposition of hydroxyapatite crystals.1–4 Mineralization of cartilage and osteoid requires sufficient levels of circulating calcium and phosphate as reflected in an adequate calcium x phosphate product. Mineralization also requires alkaline phosphatase to hydrolyze pyrophosphate, which otherwise would inhibit crystal formation.5 Hence deficient mineralization of cartilage and osteoid may be due to either an insufficient calcium x phosphate product as in rickets and osteomalacia or alkaline phosphatase deficiency as in hypophosphatasia. Rickets is a complex disorder of the growth plate that involves not only deficient mineralization of cartilage and osteoid but also disruption of endochondral ossification, leading to the accumulation of excessive cartilage. This phenomenon results from failure of hypertrophic chondrocytes to undergo normal apoptosis, which is now believed to be caused by hypophosphatemia as the common metabolic pathway of all forms of rickets (in calcipenic rickets, hypophosphatemia results from secondary hyperparathyroidism).6 Osteomalacia is a pure disorder of insufficient mineralization of osteoid at sites other than the physes, hence at either sites of bone turnover or intramembranous bone formation. Forms of rickets due to insufficient calcium are mostly a result of abnormalities of vitamin D, whose major function is maintenance of a sufficient calcium x phosphate product. Vitamin D may be synthesized in the skin from 7-dehydrocholesterol upon exposure to sunlight containing ultraviolet B radiation (290 to 315 nm) or it may be provided in the diet, although natural dietary sources of vitamin D are quite limited. Vitamin D then undergoes 25-hydroxylation in the liver followed by highly regulated 1-α-hydroxylation in the kidney to produce 1,25(OH)2-vitamin D, which is the active form of vitamin D (calcitriol). The most important biological function of calcitriol is facilitation of gastrointestinal absorption of calcium by inducing transcription of the gene for calcium-binding protein. Calcitriol synthesis by 1-α-hydroxylase is promoted by parathyroid hormone (PTH) in response to hypocalcemia.
Rickets also may result from a variety of disorders that cause hypophosphatemia, in most instances from renal tubular phosphate wasting. Renal tubular phosphate wasting or retention is regulated by PTH and by a variety of other factors, most of which are produced in bone by osteocytes.7–12 The most important of these is fibroblast growth factor 23 (FGF23), which causes renal tubular phosphate wasting by downregulating the sodium-phosphate cotransporter that facilitates phosphate reabsorption. Additionally, FGF23 downregulates 1-α-hydroxylase, leading to diminished calcitriol with excessive FGF23 signaling. Similarly, insufficient FGF23 signaling increases calcitriol.
Nutritional (Vitamin D–Deficiency) Rickets
Etiology: Both inadequate exposure to sunlight and insufficient dietary intake of vitamin D must be present for rickets to occur.1–3,13–16 The first and most severe epidemic of rickets occurred during the industrial revolution as a result of urbanization and diminished exposure to sunlight from smog and the practice of staying indoors. This epidemic largely ended with food fortification after the discovery of vitamin D synthesis. However, rickets then became more prevalent in the United States during the mid 1990s with increased breast feeding in the African American and Hispanic populations, because breast milk provides relatively little vitamin D. Less commonly, nutritional rickets may result from insufficient calcium intake.17
Clinical manifestations of rickets include failure to thrive, short stature, bowing deformities, and predisposition to fractures. In addition, prominence of anterior rib ends causing a “rachitic rosary” and craniotabes (i.e., a ping-pong ball deformity of the skull) may be seen. Weakness is a clinical feature of rickets, correlating with the presence of vitamin D receptors in skeletal muscle.18
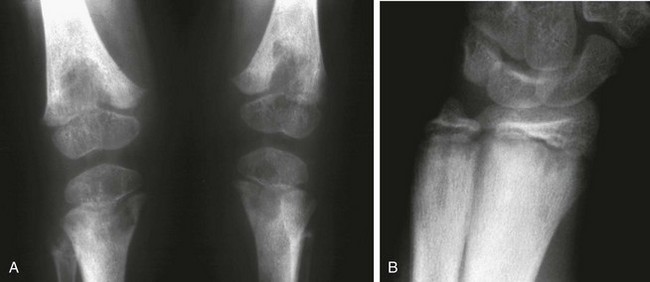
Imaging: The manifestations of rickets are most pronounced with rapid bone growth with mineralization that is unable to keep pace with new bone formation. As a result the features of rickets are most pronounced in regions of greatest bone growth, particularly the distal radius and ulna, distal femur, proximal tibia, proximal humerus, and anterior rib ends. An insufficient calcium x phosphate product causes decreased mineralization of the zone of provisional calcification and lack of normal chondrocytic terminal differentiation. As a result the initial radiographic finding is rarefaction of the normally sharply defined zone of provisional calcification on the metaphyseal side of the growth plate so that metaphyseal bone fades gradually into the lucent physeal and epiphyseal cartilage (Fig. 140-1 and e-Fig. 140-2). Also seen is loss of definition of the Laval-Jeantet collar, a short cylindrical segment of the metaphysis adjacent to the growth plate that is an indicator of the most recently formed bone in young infants.19 Deficient chondrocyte terminal differentiation and apoptosis causes accumulation of disorganized cartilage in the metaphysis in addition to nonmineralized osteoid, leading to widening of the distance between the epiphysis and metaphysis, metaphyseal fraying, and metaphyseal concavity (cupping). Metaphyseal concavity varies by site, being most pronounced in the distal forearm bones (see Fig. 140-1). However, distal ulnar metaphyseal concavity with no other abnormality should be recognized as a normal finding. Metaphyseal findings that may be recognized on chest radiographs include involvement of the proximal humeral metaphyses and rib ends, producing the rachitic rosary (Fig. 140-3), although this term more properly refers to the bulbous rib ends on physical examination. The classic metaphyseal findings of rickets are best illustrated by comparison of active and healed or healing phases (see Fig. 140-1 and e-Fig 140-2). Similar but less pronounced findings may be seen in the slower growing epiphyses and small bones with loss of definition of the zone of provisional calcification surrounding the ossification centers.19
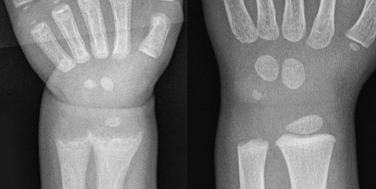
Figure 140-1 Vitamin D deficiency rickets.
A 14-month-old child with growth failure and severe rickets who responded well to vitamin D therapy. The initial image (left) shows loss of definition of the zones of provisional calcification for the distal radial and ulnar metaphyses along with metaphyseal fraying and concavity (“cupping”) and physeal widening with an increased distance between the epiphysis and visualized portion of the metaphysis. Periosteal new bone also is present that is seen best along the metacarpals but also is present along the distal radius. With healing (right), the zone of provisional calcification is well mineralized and the other findings have resolved.
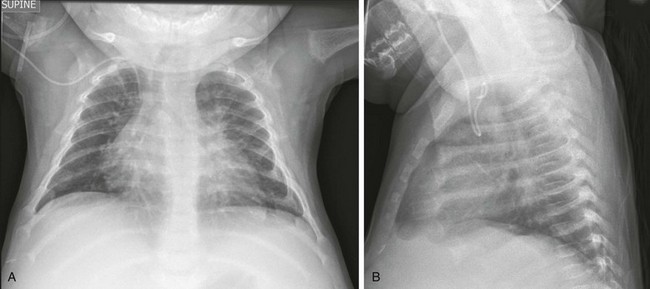
Figure 140-3 Anteroposterior (A) and lateral (B) chest radiograph views in a 5-month-old child with rickets from biliary atresia.
Note rickets in the proximal humeral metaphyses and anterior rib ends. The rib findings are best seen on the lateral view.
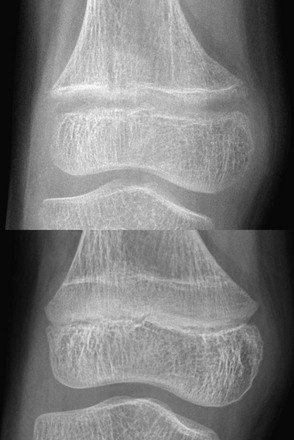
e-Figure 140-2 A less severe case of Vitamin D deficiency rickets than shown in Figure 140-1. At 9½ years of age (top), distal femoral physeal widening and metaphyseal fraying is noted that resolved several months later after treatment (bottom).
Long bone shaft findings lag behind those in the metaphysis. In rickets due to vitamin D abnormality, PTH rises in an attempt to restore a normal serum calcium concentration, producing secondary hyperparathyroidism (HPTH) with subperiosteal bone resorption, intracortical tunneling, and overall demineralization (Fig. 140-4). With ongoing bone remodeling, as nonmineralized osteoid replaces mineralized bone (osteomalacia), cortical demineralization and coarsening of the trabecular pattern in the shafts occurs, predisposing a person to insufficiency fractures. Paradoxically, periosteal new bone formation also may be seen in rickets even before healing, which likely is an anabolic effect of PTH (see Fig. 140-1).
With healing (see Fig. 140-1, e-Fig. 140-2, and Fig. 140-4), mineral deposition in the zone of provisional calcification and restoration of chondrocyte terminal differentiation occurs. The initial radiographic finding of healing is reidentification of the zone of provisional calcification as a thin opaque line that is separated from the identifiable shaft by the intervening lucent nonmineralized cartilage and osteoid. With further healing, this metaphyseal region calcifies, which may give the false impression of rapid bone growth. Less often, newly mineralized osteoid also may be seen in the metaphyseal equivalent region of the epiphysis (e-Fig. 140-5). With healing of cortical bone, the subperiosteal osteoid becomes calcified, producing either a uniform or lamellated layer.
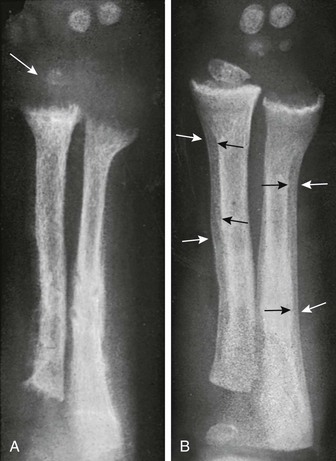
Figure 140-4 Diaphyseal findings in a patient with severe vitamin D deficiency rickets.
During the active phase (A), coarse demineralization and subperiosteal bone resorption are present, which are indicative of hyperparathyroidism as a result of rickets. Also note the severe rachitic findings in the metaphysis and poor mineralization of the distal radial epiphysis with loss of the zones of provisional calcification (arrow). With healing 3 months later (B), extensive periosteal new bone is seen (white arrows) with calcification of previously nonmineralized osteoid (black arrows) produced by periosteal osteoblasts.
Treatment and Follow-up: Prevention of vitamin D deficiency is a controversial issue because of lack of consensus regarding the level of vitamin D that is considered sufficient. The 2011 Institute of Medicine report20 concluded that a serum level of 25-hydroxy-vitamin D of at least 20 ng/mL is normal, although many experts in vitamin D research argue that higher levels are needed. Accordingly, the Institute of Medicine recommends 400 IU of vitamin D daily during the first year of life and 600 IU daily thereafter, with higher amounts suggested by researchers who believe that higher serum levels are needed.
Treatment guidelines for vitamin D deficiency are those of the Lawson Wilkins Pediatric Endocrine Society.21 Treatment is recommended for clinical manifestations of vitamin D deficiency or 25-hydroxy-vitamin D levels less than 15 ng/mL, beginning with higher doses than those used for prevention. With treatment, if radiographic evidence of healing is seen by 3 months, vitamin D dosage can be reduced to preventive levels. Because a major cause of treatment failure is noncompliance, intermittent high-dose vitamin D therapy also has been suggested.
Other Forms of Rickets
Malabsorption and Hepatobiliary Disease: Vitamin D malabsorption may cause rickets in disorders such as celiac disease and cystic fibrosis. Rickets with hepatobiliary disease is mostly a result of decreased intake and intestinal absorption of fat-soluble vitamins (A, D, E, and K); decreased hepatic 25-hydroxylation of vitamin D usually is not significant. Associated vitamin K deficiency in hepatobiliary disease may lead to recurrent hemarthrosis, most frequently involving the knee.
Vitamin D–Dependent Rickets, Types I and II: In autosomal-recessive vitamin D–dependent rickets (VDDR) type I, a defect in the renal 1-α-hydroxylase leads to undetectable or very low calcitriol levels.22,23 Rickets in VDDR I is severe, presents in the first few months of life, and has more secondary HPTH than do other forms of rickets. VDDR I responds to physiologic doses of calcitriol. Rickets also can result from a defect in the receptor for calcitriol, causing vitamin D nonresponsiveness.23,24 The designation of VDDR type II for this disorder is inappropriate because it is resistant to all forms of vitamin D, and hence the term “calcitriol-resistant rickets” is preferred. In addition to severe rickets, approximately half of the patients have manifestations of ectodermal dysplasia.
Hypophosphatemic Rickets: Hypophosphatemic forms of rickets are seen in hereditary hypophosphatemia, tumor-induced rickets and osteomalacia, and intrinsic renal tubular disease. Additionally, a major component of rickets of prematurity (i.e., metabolic bone disease of prematurity) is likely phosphate deficiency.
Hereditary Hypophosphatemic Disorders:
Etiology: X-linked hypophosphatemia (XLH), also known as familial vitamin D–resistant rickets, is the most common hereditary cause of hypophosphatemia, with rare autosomal-dominant and recessive variants.7,9,11,16 The discovery that the autosomal-dominant variant was due to a gain of function for FGF23 led to the recognition of FGF23 as a major phosphaturic factor. In XLH, FGF23 also is increased, although it is not clearly understood how this increase is caused by a mutation in the PHEX gene.
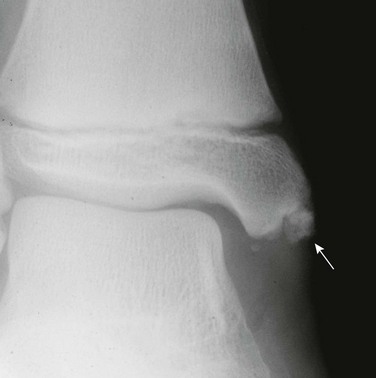
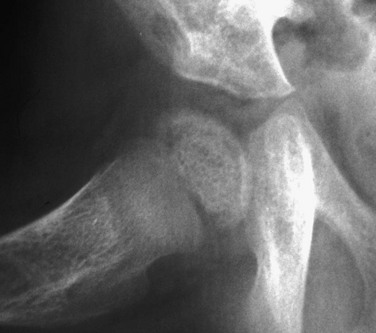
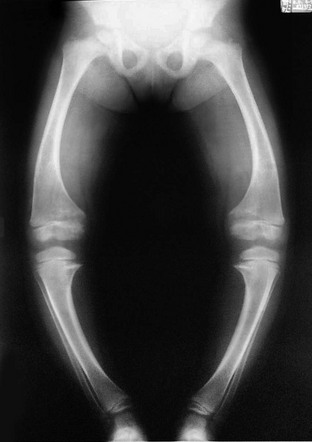
Imaging: Lower extremity bowing usually is quite prominent in XLH (Fig. 140-6), whereas the rachitic findings of XLH are often (but not always) relatively mild. Because no hypocalcemia is present, secondary HPTH is not seen in hypophosphatemic rickets. Looser zones (a feature of osteomalacia) are present more often in XLH than in nutritional rickets, likely because of the chronicity of the mineralization disorder. Looser zones, or pseudofractures, are radiolucent lines oriented perpendicular to the cortex as a result of poor mineralization of osteoid at sites of stress, which lead to increased bone remodeling. Characteristic sites include the medial aspect of the femoral neck (Fig. 140-7), the extensor surface of the ulna, the axillary border of the scapula, and the pubic ramus, with these findings often being bilateral. In infants, craniosynostosis may complicate XLH.25,26 In older patients, XLH may be associated with enthesopathy and paravertebral ossification, similar to diffuse idiopathic skeletal hyperostosis.
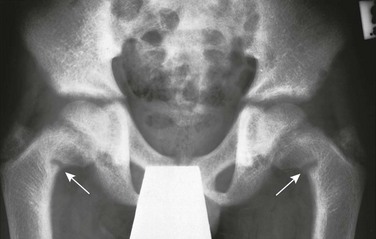
Treatment and Follow-up: Treatment of XLH includes oral phosphate replacement and calcitriol because phosphate replacement alone often leads to secondary HPTH. However, excessive calcitriol may lead to hypercalcemia, nephrocalcinosis, and nephrolithiasis; renal ultrasonography often is used to screen for this complication.27
Tumor-Induced Rickets and Osteomalacia (Oncologic Osteomalacia):
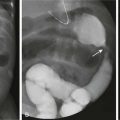
Etiology: Hypophosphatemic rickets and osteomalacia may be caused by certain tumors and tumorlike conditions that are capable of producing FGF23, with healing of rickets after removal of the tumor.28,29 TIRO may occur at any age but is quite uncommon in children. Although a large variety of benign and malignant mesenchymal neoplasms initially were described, many of these neoplasms more recently have been recategorized pathologically as “phosphaturic mesenchymal tumor, mixed connective tissue variant.”30 Other conditions causing rickets that are considered to be within the spectrum of TIRO include neurofibromatosis, polyostotic fibrous dysplasia (with or without other features of McCune-Albright syndrome), epidermal nevus syndrome, and Gorham massive osteolysis syndrome (Fig. 140-8).
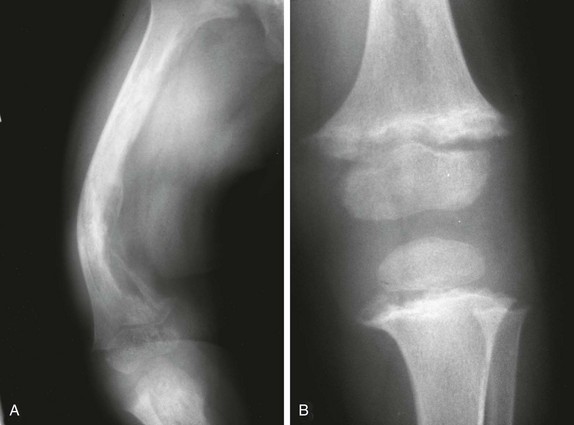
Imaging: The radiographic findings of TIRO are similar to those of other forms of chronic rickets such as XLH. Imaging also plays a role in the search for the causative tumor, which usually is small and grows slowly. Because phosphaturic mesenchymal tumors often express somatostatin receptors, In-111 octreotide scintigraphy has been used.31 Reports also indicate that fluorine-18-deoxyglucose positron emission tomography/computed tomography may be helpful.32
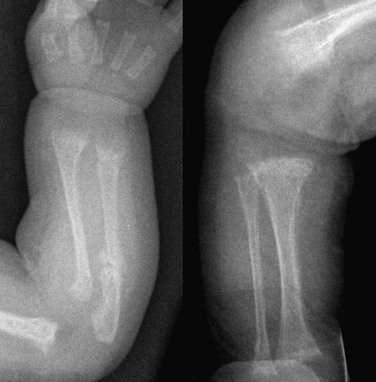
Etiology: Rickets of prematurity, also known as metabolic bone disease of prematurity, is most common in infants with a birth weight below 1 kg or gestational age younger than 28 weeks.33,34 Normally, 80% of gestational bone mineral accretion occurs during the third trimester,34 and thus infants born prematurely must accumulate mineral at a high rate to make up for this deficit. Although dietary mineral deficiency is rare in term infants and older children, inadequate intake of calcium and especially phosphorus is a major cause of metabolic bone disease of prematurity. Breast milk and infant formulas do not supply sufficient calcium and phosphorus for premature infants, and this deficiency becomes most pronounced during the period of “catch-up” growth, often near the time of hospital discharge.
Imaging: Radiographic features include generalized osteopenia, fractures, and rachitic changes in the metaphyses (Fig. 140-9). However, the rachitic findings may be masked when the infant is not growing. The fractures often are not recognized acutely and, if first discovered after discharge, may be confused with nonaccidental trauma. Extremity fractures are common and usually do not lead to long-term morbidity because ample opportunity exists for remodeling.
Hypophosphatasia
Etiology: Hypophosphatasia is a rare inherited condition in which deficient mineralization of cartilage and osteoid are attributed to alkaline phosphatase deficiency rather than an insufficient calcium x phosphate product (which characterizes rickets).5,16,35,36 In hypophosphatasia, a defect in the gene encoding alkaline phosphatase results in accumulation of pyrophosphate, which interferes with the formation of hydroxyapatite crystals. The severe perinatal and infantile forms are autosomal recessive, with autosomal-recessive and dominant inheritance for the milder forms.
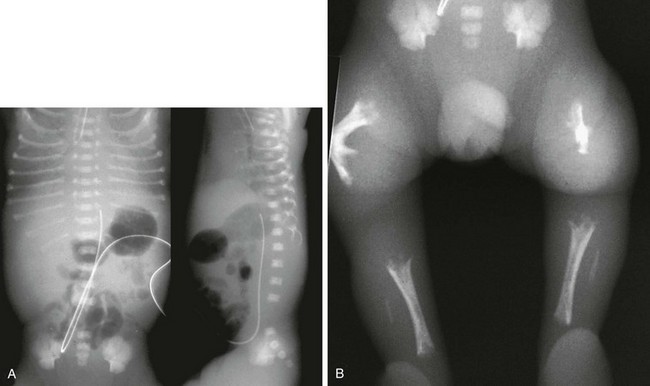
Imaging: The perinatal form (Fig. 140-10) is characterized by severe failure of mineralization of much of the skeleton. Although the skull base usually is mineralized, the calvarium may be nearly completely nonmineralized. The remainder of the axial skeleton has varying mineralization. The extremities are short and incompletely mineralized. The overall skeletal deficiency in perinatal hypophosphatasia is worse than that in the severe in utero form of osteogenesis imperfecta. In infantile and childhood hypophosphatasia, the diaphyses appear normal and the metaphyses are poorly mineralized, suggesting a process similar to rickets. However, the “rachitic” changes of hypophosphatasia are not as uniformly distributed across the growth plate. Rather, it involves a more localized portion of the growth plate with deficient mineralization extending into the metaphysis, creating a “chewed out” appearance of the metaphyses (Fig. 140-11). Because of poor calvarial mineralization, the sutures may appear widened despite actual premature closure. The adult form simulates osteomalacia with a coarse trabecular pattern, Looser zones, and metatarsal fractures.
Treatment and Follow-up: Initial attempts to treat severe forms of hypophosphatasia with alkaline phosphatase infusion were ineffective, although more recent work with a bone-targeted human recombinant enzyme is encouraging.37 Bone marrow transplantation also has been used in the treatment of infantile hypophosphatasia.38 The milder forms are treated symptomatically.
Hyperphosphatemic Disorders
Etiology: The major conditions causing hyperphosphatemia include renal insufficiency, tumoral calcinosis (TC), and hyperostosis hyperphosphatemia syndrome (HHS). TC and HHS are now considered to be different manifestations of the same disorder, with some patients showing features of each.39 TC is an autosomal-recessive disorder characterized by prominent calcific deposits and hyperphosphatemia. It usually is seen in adolescents and young adults but may present earlier. In the United States, TC is most common in African Americans. HHS is most common in the Middle East and usually presents in childhood with extremity pain and swelling along with imaging findings that mimic osteomyelitis. Recognition of hyperphosphatemia and knowledge of this condition are essential in offering an alternative diagnosis. TC and HHS biochemically mirror image hypophosphatemic rickets with hyperphosphatemia and elevated or inappropriately normal calcitriol levels. These effects are secondary to decreased FGF23 signaling, which in turn is caused by mutations affecting either FGF23 an enzyme that stabilizes it, or its receptor.40–43
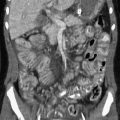
Imaging: The major finding in TC is the deposition of large calcific masses in the paraarticular soft tissues (Fig. 140-12, A).44–46 In descending order, the hips, elbows, shoulders, and feet are most frequently involved. These masses often are cystic and may show layering of chalky fluid on cross-sectional imaging or even horizontal beam radiography. Some patients with TC may also have manifestations of HHS, including medullary sclerosis, extensive periosteal new bone, abnormalities on skeletal scintigraphy, and bone marrow edema on magnetic resonance imaging (Fig. 140-12, B). These features are related to bone marrow involvement and may mimic osteomyelitis or other infiltrative disorders.
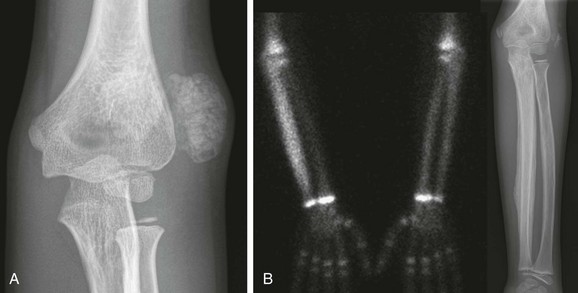
Figure 140-12 Tumoral calcinosis/hyperostosis hyperphosphatemia syndrome.
A, A left elbow radiograph at age 7 years shows a multiloculated calcified mass lateral to the left elbow. A similar mass also was present lateral to the right hip (not shown). These masses were excised with confirmation of tumoral calcinosis. B, At age 9 years, evaluation for left forearm and mandibular pain included a bone scan showing increased localization of technicium-99m diphosphonate throughout the left ulna with extensive periosteal new bone on the corresponding radiograph. The mandible and maxilla also were abnormal on the bone scan (not shown).
Treatment and Follow-up: The calcified masses of TC are amenable to surgical excision if they become painful or cause deformity or functional impairment. Many forms of therapy have been used to diminish or prevent these deposits, although they all have limited supporting data. Most often used is dietary phosphate restriction, which often is combined with aluminum-based phosphate binders. No forms of therapy have been found to be useful in patients with hyperostosis HHS, a disorder that usually shows spontaneous subsidence, often followed by recurrence.
Abnormalities of Bone Matrix Formation
Scurvy
Etiology: Scurvy is caused by dietary deficiency of vitamin C.47–49 Although infantile scurvy is now quite rare, it may be underdiagnosed. Adult scurvy remains a significant problem, particularly in elderly persons.50 Historically, infantile scurvy occurred almost exclusively in babies fed pasteurized or boiled milk, because of heat destruction of vitamin C. Scurvy nearly always presents after 6 months of age; when similar findings are seen earlier, alternative diagnoses such as congenital syphilis should be considered.
Imaging: Decreased osteoid formation in scurvy causes an imbalance between bone production and resorption, leading to generalized atrophy of the cortex and spongiosa. However, chondrocyte terminal differentiation and cartilage calcification are normal, forming a well-mineralized zone of provisional calcification. On the diaphyseal side of the physis, resorption of calcified cartilage is diminished, leading to a thickened zone of provisional calcification. Radiographically, this thickened zone is seen as a prominent opaque line (Fig. 140-13), designated as the white line of scurvy (white line of Fränkel). Despite its thickness, this zone is brittle and may fracture. The bone just beneath the thickened provisional zone is demineralized and also brittle with a sparse trabecular pattern, producing a lucent zone adjacent to the zone of provisional calcification (see Fig. 140-13) known as the scurvy zone (the Trümmerfeld zone). Later, transverse fractures may develop through the brittle zone of provisional calcification and the demineralized metaphyseal scurvy zone. If this fracture is limited to a peripheral portion of the scurvy zone, a subphyseal lucent cleft will be seen (Fig. 140-14). With transverse displacement of the epiphysis and zone of provisional calcification from the rest of the shaft, the heavily mineralized zone of provisional calcification projects peripherally, forming a metaphyseal spur (Pelkan spur), another characteristic radiographic feature of scurvy. These changes should not be confused with metaphyseal fractures of child abuse. Similar changes in the epiphyses lead to a thickened peripheral shell of calcified cartilage surrounding rarefied trabecular bone within the epiphysis, producing the Wimberger ring (see Fig. 140-13), one of the most characteristic findings of scurvy. This appearance is opposite that of rickets, in which the peripheral ring is lost with relative sparing of the central trabecular bone. In the shaft, the findings are those of generalized osteopenia with cortical thinning, although diaphyseal fractures are uncommon compared with those through the scurvy and provisional calcification zones. Subperiosteal hemorrhage due to capillary fragility is a frequent manifestation of scurvy and is most common in the larger tubular bones such as the femur, tibia, and humerus. Initially, subperiosteal hemorrhage is seen as increased soft tissue opacity. Subsequently, the periosteum produces a cloak of new bone surrounding the shaft, which eventually will become the new cortex (Fig. 140-15).
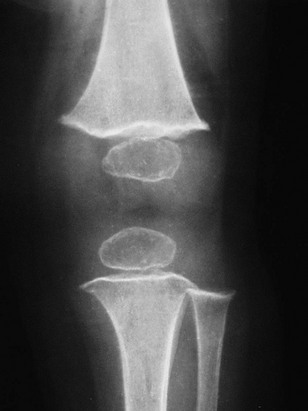
Figure 140-13 Scurvy.
The zones of provisional calcification are prominent and form the white line of scurvy. These zones stand out in high contrast compared with the subjacent demineralized scurvy zone, which is better seen for the proximal tibia than for the distal femur. Similar findings also are present at the margins of the epiphyses where the epiphyseal zone of provisional calcification forms the Wimberger ring surrounding the centrally lucent epiphysis. A small spur is present at the lateral aspect of the distal femoral metaphysis, and a proximal tibial spur is partially obscured by the fibula.
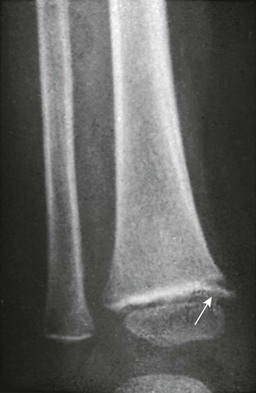
Figure 140-14 A peripheral metaphyseal cleft in a 14-month-old child with scurvy.
An eccentric lucent cortical and trabecular defect produces a cleft (arrow) just beneath the zone of provisional calcification, with the peripheral aspect of the zone of provisional calcification separated from the shaft and tilted off the shaft toward the epiphysis.
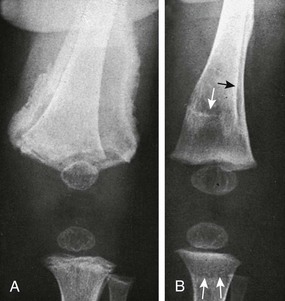
Figure 140-15 Scurvy with displaced fracture and a subperiosteal hematoma.
A, The left distal femoral epiphysis is displaced laterally relative to the metaphysis with a fracture through the brittle zone of provisional calcification. A large subperiosteal hematoma is present with periosteal calcification. Also note a nondisplaced fracture through the proximal tibial zone of provisional calcification and Wimberger rings for the distal femur and proximal tibia. B, Four months later the periosteal new bone is producing a new cortex with remodeling and eventual anatomic alignment of the prior epiphyseal displacement. Arrows indicate the initial cortex and zone of provisional calcification.
Treatment and Follow-up: The diagnosis of scurvy usually is based on a combination of the clinical and radiographic findings; an appropriate dietary history also may be helpful. Laboratory testing usually is not helpful. Although orange juice or tomato juice may be used, treatment with ascorbic acid usually is provided. The clinical response is usually rapid, confirming the diagnosis.
Copper Deficiency and Menkes Disease
Ascorbic acid oxidase is a copper-dependent enzyme, and hence copper deficiency results in deficient matrix formation and osteopenia. These findings are nonspecific, and copper deficiency has been confused with metabolic bone disease of prematurity, although neutropenia and anemia serve as other markers of copper deficiency. Menkes disease is an X-linked condition that is characterized by kinky hair, osteopenia and bone fragility, mental retardation, seizures, and intracranial hemorrhage as a result of widespread arterial abnormalities.36,51 Menkes disease is a result of deficiency of a cation-transporting enzyme needed for transport of copper from the gut and its intracellular delivery to copper-dependent enzymes, including lysyl oxidase, which is needed for proper cross-linking of collagen and elastin. This deficiency results in abnormal collagen and elastin, leading to osteopenia and increased bone fragility. Fractures in Menkes disease may follow minor trauma; in addition, metaphyseal fractures with spurring may appear similar to the metaphyseal fractures of nonaccidental trauma,52 which in some cases erroneously has led to that diagnosis (e-Fig. 140-16).
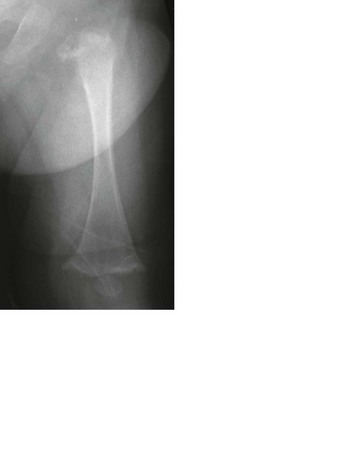
e-Figure 140-16 A patient with Menkes disease.
The left femur shows diffuse demineralization and a proximal femoral metaphyseal fracture. Healing rib fractures also were present. The infant presented with subdural hygromas related to the associated central nervous system disease and initially was suspected to be a victim of child abuse.
Hypervitaminosis A
Etiology: Vitamin A is a fat-soluble vitamin needed for vision and many other essential processes. In addition to its role in light reception, it is essential for normal epithelial differentiation of mucous membranes, and hence vitamin A deficiency causes xerophthalmia (dry ulcerating eye), the leading cause of blindness worldwide. Historically, fish liver oil and other preparations used for prevention and treatment xerophthalmia had been the major cause of vitamin A toxicity in young children, with cases often blamed on overly diligent caregivers. With discontinuation of these preparations, infantile hypervitaminosis A has become quite rare. More recently, toxicity has been from high-potency vitamin A analogs used for their pharmacologic rather than physiologic effects in dermatologic disorders.
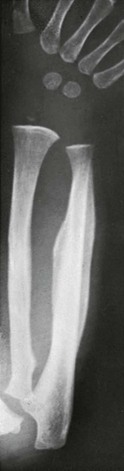
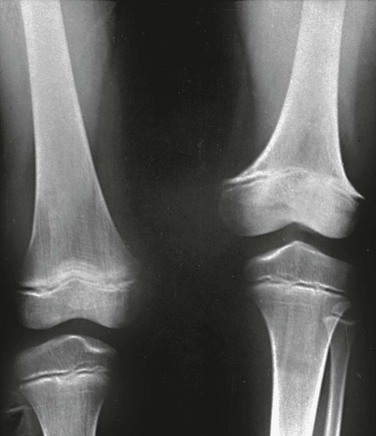
Imaging: The skeletal manifestation of hypervitaminosis A may be extremely helpful in establishing this diagnosis in a child with a confusing clinical picture.53,54 Long bone hyperostosis is the most frequent and most well-recognized finding, with periosteal new bone formation leading to undulating diaphyseal cortical thickening, most frequently involving the ulnae (Fig. 140-17) and metatarsals. Radiographically, these findings have been confused with infantile cortical hyperostosis (Caffey disease). However, age and distribution usually differentiate these, with presentation after 11 months and metatarsal involvement suggesting hypervitaminosis A, whereas presentation before 5 months and mandibular involvement suggests infantile cortical hyperostosis.53 Premature central growth plate closure also may be seen in hypervitaminosis A,55,56 likely because of an effect of retinoic acid on promoting chondrocyte terminal differentiation, thus accelerating endochondral ossification.57 This effect may lead to cone-shaped epiphyses that are invaginated into the metaphyses, mimicking the appearance of prior meningococcemia (Fig. 140-18). High-potency synthetic vitamin A analogs (e.g., isotretinoin) that are used for prolonged treatment of keratinizing disorders such as ichthyosis also may cause hyperostosis, although the pattern is different with involvement of the axial skeleton, resembling diffuse idiopathic skeletal hyperostosis.58 Beaklike ossifications also occur at the calcaneus and near the ends of the long bones compared with the diaphyseal involvement from vitamin A toxicity.
Hypervitaminosis D
Etiology: Production of calcitriol, the most active vitamin D metabolite, is a highly regulated process that provides inherent protection against vitamin D toxicity, which would require either excessive calcitriol ingestion or massive doses of less potent forms of vitamin D. Iatrogenic vitamin D poisoning has occurred in patients with physiologic bowlegs who mistakenly were thought to have XLH (vitamin D–resistant rickets) and were given escalating doses of vitamin D. The Schmid type of metaphyseal chondrodysplasia also can mimic XLH, and large doses of vitamin D will not “cure” the bowing or metaphyseal irregularity.
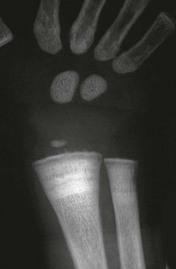
Imaging: Skeletal findings include cortical thickening, osteoporosis, and alternating bands of increased and decreased opacity (Fig. 140-19). Metastatic calcifications also may be present in blood vessels, the flax cerebri, and many organs, including the kidneys, stomach, lungs, bronchi, and adrenal gland.59
Heavy Metal Poisoning
Etiology: Lead poisoning is the most common form of heavy metal poisoning. In children, lead poisoning usually occurs as a result of the ingestion of lead that is contained in paint chips in older housing; in the United States, the sale of house paint containing lead was discontinued in 1978. Although the incidence of lead poisoning has declined with public health and education efforts, it remains a significant problem, particularly in older neighborhoods. Other environmental sources of lead are less common. Lead bullets result in toxicity only if they are within a serous space such as a synovium-lined joint, where lead will cause both systemic toxicity and synovitis, resulting in lead arthropathy.
Imaging: Chronic lead toxicity causes bands of increased opacity (lead lines) in the metaphyses. The amount of lead in these regions is far below the amount that would be needed to produce opaque bands. Rather, lead toxicity causes defective resorption of calcified cartilage in the primary spongiosa, which builds up, producing the opaque bands. When they are first formed, these bands are located along the zone of provisional calcification of long bone metaphyses and other metaphyseal equivalent regions, such as the iliac crests. The major differential diagnostic consideration is normal-variant opaque metaphyseal bands, and correlation with lead levels often is needed. Usually, the more opaque the metaphyseal band, the more likely it is to be pathologic. However, lead lines can be clearly distinguished from normal opaque metaphyseal bands once interval bone growth separates the abnormally sclerotic bone from the zone of provisional calcification. Normal opaque metaphyseal bands are always contiguous with the zone of provisional calcification, with the sclerotic bone being resorbed by the time that it “migrates away” from the physis (Fig. 140-20).
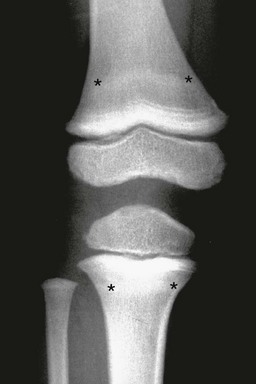
Figure 140-20 Lead toxicity.
In addition to the opaque bands at the zones of provisional calcification, faint opaque bands also are present (between asterisks) that have “migrated away,” clearly establishing that they are abnormal rather than normal-variant opaque metaphyseal bands, which are always in continuity with the zone of provisional calcification.
Treatment and Follow-up: Steps to prevent further exposure to lead are mandatory. Removal of radiographically identified lead chips from the stomach by gastric lavage has been performed to prevent subsequent absorption. However, use of this technique is controversial because the lead chips usually are poorly absorbed, and the American Academy of Clinical Toxicology does not support its use. In addition, no evidence exists to support the use of charcoal as a gastrointestinal binder to prevent absorption. For patients with particularly high lead levels, chelation therapy is indicated and is best managed by an expert in clinical toxicology who is familiar with the criteria for treatment, the agents used, and potential complications.
Alizadeh Naderi, AS, Reilly, RF. Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol. 2010;6:657–665.
Holick, MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281.
Pettifor, JM. Rickets and vitamin D deficiency in children and adolescents. Endocrinol Metab Clin North Am. 2005;34:537–553.
Rajakumar, K, Thomas, SB. Reemerging nutritional rickets: a historical perspective. Arch Pediatr Adolesc Med. 2005;159:335–341.
Whyte, MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci. 2010;1192:190–200.
References
1. Bikle, D, Adams, J, Christakos, S. Vitamin D: production, metabolism, mechanism of action, and clinical requirements. In Rosen CJ, ed.: Primer on the metabolic bone diseases and disorders of mineral metabolism, 7th ed, Washington, DC: American Society for Bone and Mineral Research, 2008.
2. DeLuca, HF. Overview of general physiological features and functions of vitamin D. Am J Clin Nutr. 2004;80:1689S–1696S.
3. Lips, P, van Schoor, NM, Bravenboer, N. Vitamin D-related disorders. In Rosen CJ, ed.: Primer on the metabolic bone diseases and disorders of mineral metabolism, 7th ed, Washington, DC: American Society for Bone and Mineral Research, 2008.
4. Resnick, D, Manolagas, SC, Niwayama, G, et al. Histogenesis, anatomy, and physiology of bone. In Resnick D, ed.: Diagnosis of bone and joint disorders, 4th ed, Philadelphia: Saunders, 2002.
5. Whyte, MP. Hypophosphatasia: nature’s window on alkaline phosphatase function in man. In Bilezikian JP, Raisz LG, Rodan GA, eds.: Principles of bone biology, 2nd ed, San Diego: Academic Press, 2002.
6. Tiosano, D, Hochberg, Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab. 2009;27:392–401.
7. Bastepe, M, Jüppner, H. Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord. 2008;9(2):171–180.
8. Farrow, EG, White, KE. Recent advances in renal phosphate handling. Nat Rev Nephrol. 2010;6(4):207–217.
9. Glorieux, FH. Hypophosphatemic vitamin-D resistant rickets. In Favis MJ, ed.: Primer on the metabolic bone diseases and disorders of mineral metabolism, 5th ed, Washington, DC: American Society for Bone and Mineral Research, 2003.
10. Levine, BS, Kleeman, CR, Felsenfeld, AJ. The journey from vitamin D-resistant rickets to the regulation of renal phosphate transport. Clin J Am Soc Nephrol. 2009;4(11):1866–1877.
11. Liu, S, Quarles, LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007;18:1637–1647.
12. Ruppe, MD, Jan de Beur, SM. Disorders of phosphate homeostasis. In Rosen CJ, ed.: Primer on the metabolic bone diseases and disorders of mineral metabolism, 7th ed, Washington, DC: American Society for Bone and Mineral Research, 2008.
13. Chesney, RW. Rickets: an old form for a new century. Pediatr Int. 2003;45:509–511.
14. Misra, M, Pacaud, D, Petryk, A, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Pediatrics. 2008;122:398–417.
15. Parfitt, AM. Vitamin D and the pathogenesis of rickets and osteomalacia. In Feldman D, Pike JW, Glorieux FH, eds.: Vitamin D, 2nd ed, San Diego: Elsevier Academic Press, 2005.
16. Pitt, MJ. Rickets and osteomalacia. In Resnick D, ed.: Diagnosis of bone and joint disorders, 4th ed, Philadelphia: Saunders, 2002.
17. Pettifor, JM. Nutritional rickets: deficiency of vitamin D, calcium, or both? Am J Clin Nutr. 2004;80(suppl 6):1725S–1729S.
18. Simpson, RU, Thomas, GA, Arnold, AJ. Identification of 1,25-dihydroxyvitamin D3 receptors and activities in muscle. J Biol Chem. 1985;260:8882–8891.
19. Oestreich, AE. The acrophysis: a unifying concept for understanding enchondral bone growth and its disorders. II. Abnormal growth. Skeletal Radiol. 2004;33:119–128.
20. Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Washington, DC: National Academies Press; 2011.
21. Misra, M, Pacaud, D, Petryk, A, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Pediatrics. 2008;122:398–417.
22. Fraser, D, Kooh, SW, Kind, HP, et al. Pathogenesis of hereditary vitamin-D-dependent rickets. An inborn error of vitamin D metabolism involving defective conversion of 25-hydroxyvitamin D to 1-alpha, 25-dihydroxyvitamin D. N Eng J Med. 1973;289:817–822.
23. Malloy, PJ, Feldman, D. Genetic disorders and defects in vitamin D action. Endocrinol Metab Clin North Am. 2010;39:333–346.
24. Brooks, MH, Bell, NH, Love, L, et al. Vitamin-D-dependent rickets type II. Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298:996–999.
25. Currarino, G. Sagittal synostosis in X-linked hypophosphatemic rickets and related diseases. Pediatr Radiol. 2007;37:805–812.
26. Murthy, AS. X-linked hypophosphatemic rickets and craniosynostosis. J Craniofac Surg. 2009;20:439–442.
27. Carpenter, TO, Imel, EA, Holm, IA, et al. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26:1381–1388.
28. Drezner, MK. Tumor-induced osteomalacia. Rev Endocr Metab Disord. 2001;2:175–186.
29. Farrow, EG, White, KE. Tumor-induced osteomalacia. Expert Rev Endocrinol Metab. 2009;4:435–442.
30. Folpe, AL, Fanburg-Smith, JC, Billings, SD, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
31. Duet, M, Kerkeni, S, Sfar, R, et al. Clinical impact of somatostatin receptor scintigraphy in the management of tumor-induced osteomalacia. Clin Nucl Med. 2008;33:752–756.
32. Roarke, MC, Nguyen, BD. PET/CT localization of phosphaturic mesenchymal neoplasm causing tumor-induced osteomalacia. Clin Nucl Med. 2007;32:300–301.
33. Bishop, N. Bone disease in preterm infants. Arch Dis Child. 1989;64:1403–1409.
34. Sharp, M. Bone disease of prematurity. Early Hum Dev. 2007;83:653–658.
35. Whyte, MP. Hypophosphatasia. In Favis MJ, ed.: Primer on the metabolic bone diseases and disorders of mineral metabolism, 5th ed, Washington, DC: American Society for Bone and Mineral Research, 2003.
36. Whyte, MP. Enzyme defects and the skeleton. In Rosen CJ, ed.: Primer on the metabolic bone diseases and disorders of mineral metabolism, 7th ed, Washington, DC: American Society for Bone and Mineral Research, 2008.
37. Whyte, M, Greenberg, CR, Wenkert, D, et al, Hypophosphatasia: enzyme replacement therapy for children using bone-targeted, tissue-nonspecific alkaline phosphatase. J Bone Miner Res, 2010;25(suppl 1). Available at, http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=bb726d8a-643b-4fb9-9282-0ddfc214bfce. [2011 Accessed January 13].
38. Whyte, MP, Kurtzberg, J, McAlister, WH, et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003;18:624–636.
39. Narchi, H. Hyperostosis with hyperphosphatemia: evidence of familial occurrence and association with tumoral calcinosis. Pediatrics. 1997;99:745–748.
40. Chefetz, I, Sprecher, E. Familial tumoral calcinosis and the role of O-glycosylation in the maintenance of phosphate homeostasis. Biochim Biophys Acta. 2009;1792:847–852.
41. Gok, F, Chefetz, I, Indelman, M, et al. Newly discovered mutations in the GALNT3 gene causing autosomal recessive hyperostosis-hyperphosphatemia syndrome. Acta Orthop. 2009;80:131–134.
42. Ichikawa, S, Imel, EA, Kreiter, ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691.
43. Ichikawa, S, Baujat, G, Seyahi, A, et al. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010;152A:896–903.
44. Clarke, E, Swischuk, LE, Hayden, CK, Jr. Tumoral calcinosis, diaphysitis, and hyperphosphatemia. Radiology. 1984;151:643–646.
45. Martinez, S, Vogler, JB, 3rd., Harrelson, JM, et al. Imaging of tumoral calcinosis: new observations. Radiology. 1990;174:215–222.
46. Olsen, KM, Chew, FS. Tumoral calcinosis: pearls, polemics, and alternative possibilities. Radiographics. 2006;26:871–885.
47. Grewar, O. Infantile scurvy. Clin Pediatr. 1965;4:82–89.
48. Park, EA, Guild, HG, Jackson, D. Recognition of scurvy with especial reference to early x-ray changes. Arch Dis Child. 1935;10:265–294.
49. Resnick, D. Hypervitaminosis and hypovitaminosis. In Resnick D, ed.: Diagnosis of bone and joint disorders, 4th ed, Philadelphia: Saunders, 2002.
50. Fain, O. Musculoskeletal manifestations of scurvy. Joint Bone Spine. 2005;72:124–128.
51. Lachman, RS. Taybi and Lachman’s radiology of syndromes, metabolic disorders, and skeletal dysplasias, 5th ed. Philadelphia: Mosby Elsevier; 2007.
52. Brill, PW, Winchester, P. Differential diagnosis of child abuse. In: Kleinman PK, ed. Diagnostic imaging of child abuse. Baltimore: Williams and Wilkins, 1987.
53. Caffey, J. Chronic poisoning due to excess of vitamin A. Am J Roentgenol Radium Ther. 1951;65:12–26.
54. Resnick, D. Disorders due to medications and other chemical agents. In Resnick D, ed.: Diagnosis of bone and joint disorders, 4th ed, Philadelphia: Saunders, 2002.
55. Pease, CN. Focal retardation and arrestment of growth of bones due to vitamin A intoxication. JAMA. 1962;182:980–985.
56. Woodard, JC, Donovan, GA, Fisher, LW. Pathogenesis of vitamin (A and D)-induced premature growth-plate closure in calves. Bone. 1997;21:171–182.
57. Wang, W, Xu, J, Kirsch, T. Annexin-mediated Ca2+ influx regulates growth plate chondrocyte maturation and apoptosis. J Biol Chem. 2003;278:3762–3769.
58. DiGiovanna, JJ. Isotretinoin effects on bone. J Am Acad Dermatol. 2001;45:S176–S182.
59. Resnick, D. Hypervitaminosis and hypovitaminosis. In Resnick D, ed.: Diagnosis of bone and joint disorders, 4th ed, Philadelphia: Saunders, 2002.