CHAPTER 48 Meningiomas and Neurofibromatosis Type 2
INTRODUCTION
The neurofibromatoses consist of two different, dominantly inherited disorders, neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). Although NF2 was first described in 1822 by the Scottish surgeon James Wishart and NF1 was fully delineated in the late 19th century by von Recklinghausen, until the late 1980s these two different entities were thought to represent different presentations of a single disease.1–3 After the localization of the NF1 and NF2 genes to chromosome 17 and 22, respectively, these two conditions were clearly established as separate entities.4–6 Table 48-1 demonstrates the different features of these two distinct conditions.
TABLE 48-1 Differences between neurofibromatosis type 1 and type 2
| NF1 | NF2 | |
|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant |
| Spontaneous mutations | 50% | 50% |
| Incidence | 1:3000 | 1:40,000 |
| Chromosome | 17q11.2 | 22q12.2 |
| Gene product | Neurofibromin | Merlin |
| Typical presentation | Café-au-lait macules during infancy or early childhood | Hearing loss or vestibular dysfunction in young adulthood or cataracts |
| Nerve sheath tumors | Neurofibroma, plexiform neurofibroma, MPNSTs | Schwannoma |
| Intracranial tumors | Optic pathway gliomas, other astrocytomas/gliomas | Vestibular schwannomas, meningiomas |
| Spinal tumors | Nodular plexiform neurofibromas (roots) | Schwannomas (roots), ependymomas (intramedullary) |
| Cutaneous features | Café-au-lait macules, axillary/inguinal freckling, cutaneous neurofibromas, subcutaneous neurofibromas | Cutaneous schwannomas |
| Cognitive | LD and ADHD common, IQ mildly decreased | Normal |
| Ophthalmologic | Lish nodules, congenital glaucoma | Juvenile subcapsular lenticular opacities, cataracts, corneal scarring, retinal hamartomas |
MPNST, malignant peripheral nerve sheath tumor; LD, learning disability; ADHD, attention deficit and hyperactivity disorder
Adapted from ref. 16.
NF2, also known as “central neurofibromatosis” or “bilateral acoustic neurofibromatosis,” is an autosomal dominant disease caused by mutations of the NF2 tumor suppressor gene.7,8 Accordingly, the risk to develop this disease for any offspring of an affected parent is 50%.4 Fifty percent of the cases have no family history and represent sporadic gene mutations.9,10 The birth incidence of the disease based on a study on the UK population is estimated between 33,000 and 40,000 with a prevalence of 1 in 210,000. The annual incidence rate is 1 per 2,355,000 (newly diagnosed symptomatic patients), which represents about one new case per year.9 The difference between the birth incidence and diagnostic prevalence is the result of the fact that many patients do not develop the disease until the third decade or later, and many others die before that time.1,2
Using genetic analyses of large pedigrees, the NF2 gene has been localized to the long arm of chromosome 22. The disease results from inactivating mutations of a known tumor suppressor gene. The gene is comprised of 110 kb of genomic DNA and encodes a 595-amino-acid protein termed merlin or schwannomin.2,9,11–13 Merlin is structurally related to the moesin/ezrin/radixin proteins, which link the actin cytoskeleton to cell surface glycoproteins that control growth and cellular remodeling.2,14 Merlin is widely expressed in Schwann cells, meningeal cells, peripheral nerves, and the lens. There is evidence that the main function of the NF2 gene is tumor suppression and regulation of proliferation of Schwann cells and leptomeningeal cells.2 Inactivating mutations and the loss of merlin expression were also detected in almost all sporadic schwannomas, in 50% to 70% of sporadic meningiomas, gastrointestinal stromal tumors, malignant mesothelioma, and sporadic ependymomas.15,16
CLINICAL DIAGNOSIS OF NF2
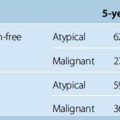
In 1987, the National Institutes of Health (NIH) Consensus Development Conference on Neurofibromatosis established clinical diagnostic guidelines to differentiate NF2 from NF1.17 These criteria diagnose NF2 in patients with bilateral vestibular schwannomas and in those with a first-degree relative with NF2 and either a unilateral vestibular schwannoma or at least two other characteristic features of NF2 (meningioma, schwannoma, glioma, neurofibroma, juvenile posterior subcapsular lenticular opacity). However, half of all patients with NF2 do not have a family history of the disease, and patients can present with other features of the disease long before the appearance of vestibular schwannomas.10,18–20 Owing to the drawbacks in the diagnosis of NF2 in patients without bilateral vestibular schwannomas and without a positive family history, these criteria were revised multiple times. A second NIH Consensus Conference in 1991, the Manchester group in 1992, and a group organized by the National Neurofibromatosis Foundation (NNFF) in 1997 revised these criteria.7 Table 48-2 demonstrates the latest diagnostic criteria for NF2. Baser and colleagues have examined the diagnostic efficiency of the four sets of criteria in patients who do not have bilateral vestibular schwannomas but who do have other signs of NF2 and concluded that although none of the sets is satisfactory for diagnosing patients without bilateral vestibular schwannomas, the Manchester criteria are the most sensitive.7
TABLE 48-2 Evolution of clinical diagnostic criteria for NF2
| 1997 NFFF criteria |
|---|
| 1991 NIH criteria |
|---|
| 1987 NIH criteria |
|---|
* In the Manchester criteria, “any two of” refers to two individual tumors or cataract, whereas in the other sets of criteria, it refers to two tumor types or cataract.
Adapted from ref. 7.
CLINICAL MANIFESTATIONS
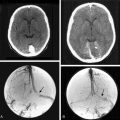
The hallmark of NF2 is bilateral vestibular schwannomas (Fig. 48-1). The penetrance of vestibular schwannomas in families with the NF2 gene is greater than 95%9 and this pathognomonic finding is virtually present in 95% of adult patients.10,20,21 Besides these tumors, the clinical presentation of the disease is quite heterogeneous. One can now classify patients into two main groups, severe and mild phenotypes, based on differing clinical course, age at onset, and the presence of other cranial and spinal tumors.19 The severity of the disease is mainly associated with type of mutation. Studies of genotype and phenotype correlations in NF2 have demonstrated that, in general, constitutional nonsense and frameshift mutations in the NF2 gene are associated with severe disease, while missense mutations, in-frame deletions, large deletions, and somatic mosaicism are associated with mild illness.10,19,22–24
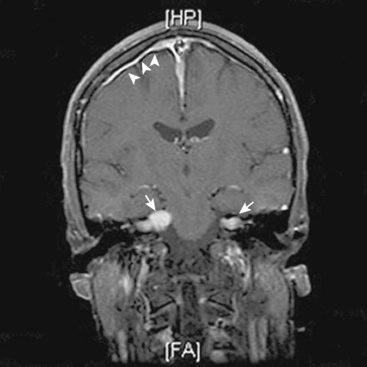
Although different authors describe cutoff points for severe and mild disease with different numbers, the mild phenotype, which is referred to as the “Gardner” form, is characterized by late onset of bilateral vestibular schwannomas (usually after the second decade of life) with limited numbers of additional tumors of the cerebrum and spine. On the other hand, the severe phenotype, which is referred as the “Wishart” form, is associated with numerous tumors of the cranium and the spine with early-onset vestibular schwannomas.9,19,25 The severe phenotype usually presents with a tumor other than a vestibular schwannoma in early childhood and is associated with a high morbidity and mortality.1,25
The average age at onset of symptoms in individuals with NF2 is 18 to 24 years; however, they can present as early as 2 years and as late as 70 years.9 The average age of mortality is 36 years.22 There is no preponderance for sex and race.
Unilateral hearing loss, symptoms related to skin tumors, focal weakness due to spinal tumors or neuropathies, tinnitus, and imbalance are the most common symptoms in affected adults. Eleven percent of the cases are asymptomatic and detected on screening because of a positive family history.1,9,19,25
Ophthalmologic features are common in NF2. About 30% of the cases have decreased visual acuity in one or both eyes. Between 60% and 80% of the patients have posterior subcapsular lenticular opacities. Retinal hamartoma, epiretinal membranes, optic nerve meningioma, optic disc glioma, intraocular schwannoma, and neurotrophic keratopathy are other manifestations of NF2. Lisch nodules were not found to be a feature of the disease.1,3,10,19,26,27
Cutaneous features in NF2 are much rarer than in NF1. Although 70% of the patients have skin tumors, the number of these tumors does not exceed 10 in most of the cases. Café au lait spots do occur in about 40% of NF2 patients, but again the size and number of these lesions are far less than in NF1. Fewer than 2% of NF2 patients harbor more than six café au lait spots, which may pose some diagnostic confusion between NF1 and NF2.12 Axillary and groin freckling is not a characteristic of the disease. Skin tumors occur in about 70% of the patients and the most common type of these tumors is intracutaneous, slightly raised plaque-like lesions. Schwannoma is the main histopathologic diagnosis, but occasionally neurofibromas occur. The back, chest, arms, and face are the most common locations.1,4,10,19,28
Although rare, recent studies have highlighted peripheral nerve involvement that cannot be attributed to tumors in NF2 patients.21,29 Evans and colleagues1 have reported peripheral nerve lesions unrelated to tumors in 6% of NF2 cases. Sperfeld and colleagues investigated definite NF2 patients and found clinical and electrophysiologic signs related to peripheral neuropathy in the majority of the cases.30
The presentation of the disease in childhood is quite different that that in adults. Although NF2 is recognized as an adult-onset disease, many patients are found to have had some signs of the disease in early childhood.18,31 Unlike the adulthood presentation, vestibular schwannoma–related symptoms occur in as few as 15% to 30% of patients as initial symptoms. Patients may present with meningiomas and spinal tumors long before vestibular schwannomas. Children present more frequently with neurologic symptoms primarily related to spinal cord compression and visual involvement.18 Cataracts can be present in the neonatal period and can affect vision in early life. In addition, there is a tendency to mononeuropathy, particularly affecting the facial nerve.1 Some children may present with oculomotor palsy or a foot or hand drop.1,18,20,31 NF2 patients who present in childhood are likely to have a more severe disease course with multiple tumors.
TUMORS ASSOCIATED WITH NEUROFIBROMATOSIS 2
As previously described, patients with NF2 almost always develop bilateral vestibular schwannomas throughout the course of their lives. Of all NF2 patients, about 10% to 20% have unilateral vestibular schwannoma at presentation.32 Schwannomas may occur in virtually all parts of the body; however, there is a high preference for the superior vestibular branch of the VIIIth cranial nerve for reasons that remain poorly understood. Both in the cranial cavity and in the spinal canal, the sensory roots are involved much more frequently than the motor roots.8 The growth rates of schwannomas are generally higher in younger patients; however, these are extremely variable both between patients and over time in the same patient.33,34 Slattery and colleagues have estimated an average growth rate of 1.3 mm per year for vestibular schwannomas in NF2 patients.35 Interestingly histologic comparison of sporadic and NF2–associated schwannomas have demonstrated that NF2 schwannomas are often lobular, tend to infiltrate the adjacent facial nerve, and have a higher proliferative potential.36 Schwannomas at other cranial nerves, spinal cord, and peripheral nerves are also very common. The second most common site for cranial schwannoma is the trigeminal nerve. As well as vestibular schwannomas, other cranial nerve schwannomas may occur bilaterally. Mautner and colleagues demonstrated that 38% of the patients have tumors of other cranial nerves including oculomotor, hypoglossal, vagal, and accessory nerve schwannomas.19,37 In their series of 83 patients with NF2, Fisher and colleagues reported 51% of the patients have at least one nonvestibular schwannoma.38 Nevertheless, it is rare for a nonvestibular nerve schwannoma to grow to a significant size necessitating surgical removal.
Although different authors report different figures, approximately 2% to 6% of NF2 patients develop ependymomas while 1% to 4% develops astrocytomas.1,10,19 Ependymomas usually occur in the cervicothoracic region.8
Spinal tumors are another important feature of NF2. Although few NF2 patients will undergo surgery for all of their intraspinal tumors, it can be roughly speculated that schwannomas, meningiomas, and neurofibromas constitute the most common extramedullary tumors, while astrocytomas and ependymomas constitute most of the intramedullary tumors.39 Mautner and colleagues have demonstrated that spinal tumors are more common than previously described. In their series, 90% of patients have radiologically confirmed spinal tumors, meaning that they are almost as common as vestibular schwannomas; however, only 25% to 30% of these tumors are symptomatic.19,28 In their retrospective review, Dow and colleagues found 67% of the NF2 cases harbored one or more spinal tumors and demonstrated that as in cranial tumors, truncating mutations that lead to severe disease are usually associated with increased numbers of spinal tumors.40 Parry and colleagues estimated the mean number of spinal tumors in NF2 patients as 8.7.10 Radiating pain, paresthesias, muscle weakness, atrophy, cramping, and numbness are among the presenting symptoms of spinal tumors.
MENINGIOMAS IN NEUROFIBROMATOSIS TYPE 2
As previously emphasized throughout this book, meningiomas are the second most common tumor of the brain, accounting for approximately 20% of all primary adult intracranial tumors. Although there is still an ongoing endeavor to increase our knowledge about underlying factors leading to meningioma formation, sporadic meningioma cases have played an important role in the localization of the NF2 gene to chromosome 22.12 After detection of the NF2 tumor suppressor gene on chromosome 22q, various studies in sporadic meningioma cases revealed that more than 60% of meningiomas exhibit a chromosome 22q abnormality.41 As more than half but not all meningiomas harbor these abnormalities on chromosome 22q, there must be other genes involved in meningioma formation. Studies have revealed various genes on other chromosomes as well as other genes on chromosome 22 that have important roles in meningioma formation.42–44 However, the genetic alterations related to the NF2 gene are widely accepted as an essential initiation step for meningioma formation.
It has been shown that molecular genetic characterization of meningiomas may be grouped into tumors with and without mutations in the NF2 gene.41,42 On the other hand, the clinical presentation of meningiomas can be divided into three categories: sporadic or single cases, multiple and nonfamilial cases, or multiple and familial cases.45–47
Sporadic meningiomas have different features than the meningiomas in NF2 patients; however, a sporadic meningioma simply represents a “focal” form of NF2 that is restricted to a specific group of meningeal cells. Sporadic meningiomas exhibit NF2 gene mutations to a certain degree, but these mutations occur at similar frequencies in all three World Health Organization (WHO) grades. These data suggest that NF2 gene inactivation must be an initiation step in meningioma formation. However, there are likely other genetic pathways both for de novo meningioma formation and for malignant progression.44 Tumors that harbor mutations in the NF2 gene are usually of the transitional or fibrous subtype, while tumors without NF2 gene mutations are generally of the meningothelial subtype.41,42,44 Further, Kros and colleagues have speculated that there is a relationship between the NF2 gene status of a meningioma and the location of the tumor. They have evaluated 42 sporadic meningioma cases and demonstrated that tumors located in the anterior parts of the skull base did not showed disruption of the NF2 gene, whereas most meningiomas occurring at the convexity, falx, tentorium, and posterior fossa were associated with NF2 gene alterations.48
Multiple meningioma cases without a familial background represent 1% to 8% of all cases, and studies have demonstrated that these tumors also exhibit NF2 gene mutations to some degree.47,49,50 Although almost all familial cases or sporadic cases presenting with multiple meningiomas are related to NF2, a few families have been reported without any obvious stigmata of the NF2 disease but with meningiomas (sporadic familial meningiomas).51 Pulst and colleagues have studied members of a family harboring multiple meningiomas and ependymoma but not vestibular schwannomas and demonstrated that none of the family members had a mutation in the NF2 gene.52 In addition, meningiomas described in a number of genetic conditions other than NF2 such as von Hipple Lindau disease, NF1, Rubinstein–Taybi syndrome, Li–Fraumeni syndrome, Werner syndrome, and Gardner syndrome.44 In addition, Asgharian and colleagues have speculated meningiomas may be a part of multiple endocrine neoplasia type 1.53
Meningiomas are the second most common tumor in NF2 patients after vestibular schwannomas. NF2-related meningiomas account for 1% of all meningiomas.54,55 On the other hand, three large series report approximately 50% of NF2 patients have at least one cranial or spinal meningioma.1,10,19 Intracranial involvement is reported to be greater than the spinal involvement.18,42
Different series of large cohorts of NF2 patients have reported rates for meningioma incidence. Mautner and colleagues19 reported the diagnosis of meningioma in NF2 patients to be as high as 58%, while Evans1 and Parry10 reported rates of 45% and 49%, respectively. Apart from these figures, a report from Finland reported that as few as 20% of patients with multiple meningiomas had NF2.54 However, this study was criticized as an underestimate because it is known that some patients will present with meningiomas before they develop classical features of NF2. Evans and colleagues have demonstrated that about 8% of patients of NF2 present with meningioma before a vestibular schwannoma.50
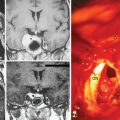
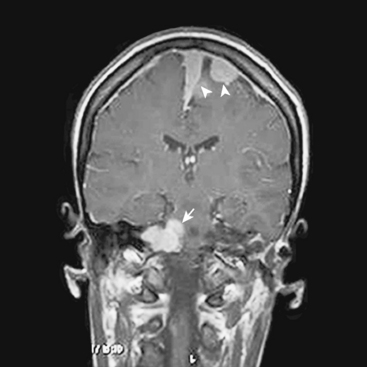
In addition, meningiomas in NF2 are more likely to be multiple in comparison with their sporadic counterparts. Mautner and colleagues reported 38% of their patients had multiple meningiomas with a mean number of 3.8 tumors per person, while 21% had solitary lesions.19 On the other hand, in their series of 63 NF2 patients, Parry and colleagues10 reported a mean number of 4.3 meningiomas per person in NF2 patients harboring meningiomas (Figs. 48-2 and 48-3). According to Parry and colleagues, many individuals with meningiomas had no related symptoms at initial presentation; however, 50% of these patients developed symptoms related to at least one of these meningiomas over time.10 The symptoms associated with meningiomas were headaches in increasing frequency and severity, decreasing visual acuity, and various focal symptoms such as dysphagia, vertigo, and personality change.
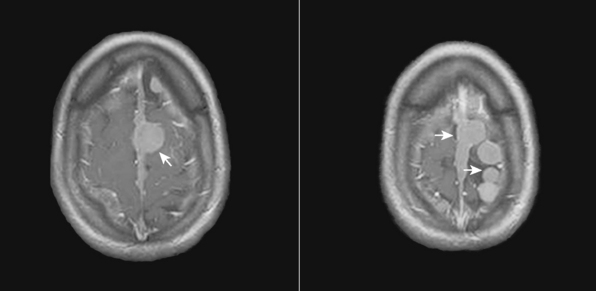
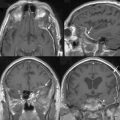
FIGURE 48-2 Axial postcontrast T1-weighted MR images of an NF2 patient with multiple intracranial meningiomas (arrows).
Another important issue about NF2-related meningiomas is that the histopathologic behavior of meningiomas in NF2 cases is also different from that in sporadic ones. Antinheimo and colleagues compared histological features and proliferation potential of the sporadic and NF2-associated meningiomas and found NF2 meningiomas have a more frequent incidence of mitotic figures and nuclear pleomorphism.36 Similarly, Perry and colleagues also demonstrated that NF2-associated meningiomas are more predisposed toward malignant progression.55
Despite these differences in presentation, meningiomas occurring in the setting of NF2 do not appear to exhibit different clinical behavior from sporadic tumors. Similarly, the radiologic appearance of these tumors does not differ from that of their sporadic counterparts except for increased multiplicity. However, one should note that, apart from classical schwannomas and meningiomas, NF2 patients can harbor unique tumors such as lesions containing both schwannomatous and meningiomatous elements in the same tumor.56 To date only 12 cases of such “mixed” or so called “collision” tumors have been reported in the literature.46 Although extremely rare, these collision tumors within the cerebellopontine angle are thought to be exclusively associated with NF2.1,56
Another important issue regarding meningiomas and NF2 is the optic nerve sheath involvement. They represent 1% to 2% of all meningioma cases.57 In their review, Bosch and colleagues58 evaluated 30 NF2 patients and demonstrated that 27% of these patients had optic nerve sheath meningiomas. Although previous series have reported optic nerve sheath meningiomas in approximately 5% of NF2 cases, it is important to note that these tumors have a higher incidence in NF2 patients than in their sporadic counterparts. Optic nerve sheath meningiomas in the NF2 background may be bilateral and/or multifocal.57 Most of these tumors present with decreased visual acuity or reduced ocular motility.
In contrast to those in adults, childhood meningiomas account for fewer than 3% of all primary central nervous system tumors.59 However, meningiomas can be the first presenting tumor in pediatric NF2 patients and it is worth highlighting that, of the pediatric patients presenting with an isolated meningioma, 10% will eventually develop other features of NF2.1,31 Parry and colleagues analyzed NF2-associated meningiomas in children and demonstrated that although they share the common molecular alterations of their sporadic counterparts in adults, a higher fraction of these tumors behave aggressively.55
MANAGEMENT
NF2 patients present difficult management issues and they optimally should be managed in experienced hands by a multidisciplinary team consisting of a neurosurgeon, otolaryngologist, audiologist, ophthalmologist, neuroradiologist, and geneticist. Individualized treatment options should be tailored according to the needs of each patient.25,60 Baser and colleagues studied NF2 patients and families and demonstrated that mortality increased with decreasing age, presence of intracranial meningiomas, and presence of mutations other than constitutional NF2 missense mutations. In addition, people who were treated at multidisciplinary centers had a more favorable progress compared with those who were not.22 This study demonstrates the importance of multidisciplinary, dedicated centers in the management of NF2 patients.
Children of affected patients should be considered to be at 50% risk of NF2 and screening should be initiated. Formal screening for vestibular schwannomas should start at 10 years, as it is significantly rare for an individual, even in severely affected families, to present with these tumors before that age.1 However, other features of the disease could be present before that age. Screening should include comprehensive neurologic and physical examinations as well as audiologic, dermatologic, and ophthalmologic evaluations. Studies demonstrate that there is a considerable delay between the age of first symptom and the diagnosis. This could be solved by meticulous follow-up.28 Any screening protocol should seriously consider spinal involvement because, although most of them are asymptomatic, 90% of NF2 patients have spinal involvement.39
On the other hand, children presenting with meningeal or Schwann cell tumor, patients presenting with multiple central nervous system tumors that are not attributable to another disease, and patients younger than 30 years of age presenting with unilateral vestibular schwannoma should be carefully monitored because it is estimated that about 6% of these patients develop other manifestations of the disease.2,4
The cornerstone of modern NF2 management is conservation of function and the maintenance of quality of life. Presence of a tumor is not sufficient for establishing an indication for surgery. Complications, which are common, should be seriously considered. Other treatment options such as radiation therapy, whether conventional or stereotactic radiosurgery, should be taken into account.25
[1] Evans D.G., Sainio M., Baser M.E. Neurofibromatosis type 2. J Med Genet. 2000;37:897.
[2] Ferner R.E. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007;6:340.
[3] Al-Otibi M., Rutka J.T. Neurosurgical implications of neurofibromatosis type I in children. Neurosurg Focus. 2006;20:E2.
[4] Martuza R.L., Eldridge R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med. 1988;318:684.
[5] Rouleau G.A., Wertelecki W., Haines J.L., et al. Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature. 1987;329:246.
[6] Seizinger B.R., Rouleau G.A., Ozelius L.J., et al. Genetic linkage of von Recklinghausen neurofibromatosis to the nerve growth factor receptor gene. Cell. 1987;49:589.
[7] Baser M.E., Friedman J.M., Wallace A.J., et al. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology. 2002;59:1759.
[8] Inoue Y., Nemoto Y., Tashiro T., et al. Neurofibromatosis type 1 and type 2: review of the central nervous system and related structures. Brain Dev. 1997;19:1.
[9] Evans D.G., Huson S.M., Donnai D., et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841.
[10] Parry D.M., Eldridge R., Kaiser-Kupfer M.I., et al. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994;52:450.
[11] Hirsch N.P., Murphy A., Radcliffe J.J. Neurofibromatosis: clinical presentations and anaesthetic implications. Br J Anaesth. 2001;86:555.
[12] Ruttledge M.H., Rouleau G.A. Role of the neurofibromatosis type 2 gene in the development of tumors of the nervous system. Neurosurg Focus. 2005;19:E6.
[13] Gusella J.F., Ramesh V., MacCollin M., et al. Merlin: the neurofibromatosis 2 tumor suppressor. Biochim Biophys Acta. 1999;1423:M29.
[14] Gutmann D.H. The neurofibromatoses: when less is more. Hum Mol Genet. 2001;10:747.
[15] Xiao G.H., Chernoff J., Testa J.R. NF2: the wizardry of merlin. Genes Chromosomes Cancer. 2003;38:389.
[16] Yohay K.H. The genetic and molecular pathogenesis of NF1 and NF2. Semin Pediatr Neurol. 2006;13:21.
[17] Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575.
[18] MacCollin M., Mautner V.F. The diagnosis and management of neurofibromatosis 2 in childhood. Semin Pediatr Neurol. 1998;5:243.
[19] Mautner V.F., Lindenau M., Baser M.E., et al. The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery. 1996;38:880.
[20] Mautner V.F., Tatagiba M., Guthoff R., et al. Neurofibromatosis 2 in the pediatric age group. Neurosurgery. 1993;33:92.
[21] Baser M.E., DG R.E., Gutmann D.H. Neurofibromatosis 2. Curr Opin Neurol. 2003;16:27.
[22] Baser M.E., Friedman J.M., Aeschliman D., et al. Predictors of the risk of mortality in neurofibromatosis 2. Am J Hum Genet. 2002;71:715.
[23] Ruttledge M.H., Andermann A.A., Phelan C.M., et al. Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am J Hum Genet. 1996;59:331.
[24] Baser M.E., Kuramoto L., Woods R., et al. The location of constitutional neurofibromatosis 2 (NF2) splice site mutations is associated with the severity of NF2. J Med Genet. 2005;42:540.
[25] Evans D.G., Baser M.E., O’Reilly B., et al. Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg. 2005;19:5.
[26] McLaughlin M.E., Pepin S.M., MacCollin M., et al. Ocular pathologic findings of neurofibromatosis type 2. Arch Ophthalmol. 2007;125:389.
[27] Mulvihill J.J., Parry D.M., Sherman J.L., et al. NIH conference. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis). An update. Ann Intern Med. 1990;113:39.
[28] Evans D.G., Huson S.M., Donnai D., et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling. J Med Genet. 1992;29:847.
[29] Gijtenbeek J.M., Gabreels-Festen A.A., Lammens M., et al. Mononeuropathy multiplex as the initial manifestation of neurofibromatosis type 2. Neurology. 2001;56:1766.
[30] Sperfeld A.D., Hein C., Schroder J.M., et al. Occurrence and characterization of peripheral nerve involvement in neurofibromatosis type 2. Brain. 2002;125:996.
[31] Evans D.G., Birch J.M., Ramsden R.T. Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child. 1999;81:496.
[32] Evans D.G., Lye R., Neary W., et al. Probability of bilateral disease in people presenting with a unilateral vestibular schwannoma. J Neurol Neurosurg Psychiatry. 1999;66:764.
[33] Baser M.E., Makariou E.V., Parry D.M. Predictors of vestibular schwannoma growth in patients with neurofibromatosis type 2. J Neurosurg. 2002;96:217.
[34] Mautner V.F., Baser M.E., Thakkar S.D., et al. Vestibular schwannoma growth in patients with neurofibromatosis type 2: a longitudinal study. J Neurosurg. 2002;96:223.
[35] Slattery 3rd W.H., Fisher L.M., Iqbal Z., et al. Vestibular schwannoma growth rates in neurofibromatosis type 2 natural history consortium subjects. Otol Neurotol. 2004;25:811.
[36] Antinheimo J., Haapasalo H., Haltia M., et al. Proliferation potential and histological features in neurofibromatosis 2-associated and sporadic meningiomas. J Neurosurg. 1997;87:610.
[37] Hanemann C.O. Magic but treatable? Tumours due to loss of Merlin. Brain. 2008;131:606-615. [Epub ahead of print]
[38] Fisher L.M., Doherty J.K., Lev M.H., et al. Distribution of nonvestibular cranial nerve schwannomas in neurofibromatosis 2. Otol Neurotol. 2007;28:1083.
[39] Mautner V.F., Tatagiba M., Lindenau M., et al. Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. AJR Am J Roentgenol. 1995;165:951.
[40] Dow G., Biggs N., Evans G., et al. Spinal tumors in neurofibromatosis type 2. Is emerging knowledge of genotype predictive of natural history. J Neurosurg Spine. 2005;2:574.
[41] Evans J.J., Jeun S.S., Lee J.H., et al. Molecular alterations in the neurofibromatosis type 2 gene and its protein rarely occurring in meningothelial meningiomas. J Neurosurg. 2001;94:111.
[42] Hansson C.M., Buckley P.G., Grigelioniene G., et al. Comprehensive genetic and epigenetic analysis of sporadic meningioma for macro-mutations on 22q and micro-mutations within the NF2 locus. BMC Genomics. 2007;8:16.
[43] Ragel B.T., Jensen R.L. Molecular genetics of meningiomas. Neurosurg Focus. 2005;19:E9.
[44] Simon M., Bostrom J.P., Hartmann C. Molecular genetics of meningiomas: from basic research to potential clinical applications. Neurosurgery. 2007;60:787.
[45] Claus E.B., Bondy M.L., Schildkraut J.M., et al. Epidemiology of intracranial meningioma. Neurosurgery. 2005;57:1088.
[46] Hoffman C.E., Amant J., Black P. Meningioma and NF2: A review. Neurosurg Q. 2007;17:128.
[47] Perry A., Gutmann D.H., Reifenberger G. Molecular pathogenesis of meningiomas. J Neurooncol. 2004;70:183.
[48] Kros J., de Greve K., van Tilborg A., et al. NF2 status of meningiomas is associated with tumour localization and histology. J Pathol. 2001;194:367.
[49] Heinrich B., Hartmann C., Stemmer-Rachamimov A.O., et al. Multiple meningiomas: Investigating the molecular basis of sporadic and familial forms. Int J Cancer. 2003;103:483.
[50] Evans D.G., Watson C., King A., et al. Multiple meningiomas: differential involvement of the NF2 gene in children and adults. J Med Genet. 2005;42:45.
[51] Maxwell M., Shih S.D., Galanopoulos T., et al. Familial meningioma: analysis of expression of neurofibromatosis 2 protein Merlin. Report of two cases. J Neurosurg. 1998;88:562.
[52] Pulst S.M., Rouleau G.A., Marineau C., et al. Familial meningioma is not allelic to neurofibromatosis 2. Neurology. 1993;43:2096.
[53] Asgharian B., Chen Y.-J., Patronas N.J., et al. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1. Clin Cancer Res. 2004;10:869.
[54] Antinheimo J., Sankila R., Carpen O., et al. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology. 2000;54:71.
[55] Perry A., Giannini C., Raghavan R., et al. Aggressive phenotypic and genotypic features in pediatric and NF2-associated meningiomas: a clinicopathologic study of 53 cases. J Neuropathol Exp Neurol. 2001;60:994.
[56] Gelal F., Rezanko T., Uyaroglu M.A., et al. Islets of meningioma in an acoustic schwannoma in a patient with neurofibromatosis-2: pathology and magnetic resonance imaging findings. Acta Radiol. 2005;46:519.
[57] Eddleman C.S., Liu J.K. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus. 2007;23:E4.
[58] Bosch M.M., Wichmann W.W., Boltshauser E., et al. Optic nerve sheath meningiomas in patients with neurofibromatosis type 2. Arch Ophthalmol. 2006;124:379.
[59] Rushing E.J., Olsen C., Mena H., et al. Central nervous system meningiomas in the first two decades of life: a clinicopathological analysis of 87 patients. J Neurosurg. 2005;103:489.
[60] Yohay K. Neurofibromatosis types 1 and 2. Neurologist. 2006;12:86.