[level-membership-for-neurosurgery-category]
CHAPTER 132 Meningeal Sarcoma and Meningeal Hemangiopericytoma
Meningeal Sarcoma
History
Sarcomas are tumors arising from mesenchymal tissue. Sarcoma is derived from the Greek word sar, which denotes a fleshy character related to the gross appearance of these tumors.1 Sarcomatous tumors of the leptomeninges were described as early as 1929.2 Although Virchow had reported CNS sarcomas before this, he actually may have been describing high-grade gliomas.3 In 1953, Christensen and Lara reported 24 cases of intracranial sarcoma, which they grouped into three distinct entities: fibrous sarcoma, spindle cell sarcoma, and polymorphocellular sarcoma.4 The distinctions were based on light microscopy findings. This system is not part of the 2007 World Health Organization (WHO) tumor classification scheme, but it did have prognostic significance. Median survival for these three subtypes is quite different: fibrous, 74 months; spindle cell, 27 months; and polymorphocellular, less than 1 year. The 2007 WHO classification of tumors of the CNS separates intracranial sarcomas according to their pattern of differentiation rather than their cell of origin (Table 132-1).5
Adapted from Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
Clinical Significance
Sarcoma progenitors cells are derived from mesenchymal tissue. The nervous system has multiple tissues of mesenchymal origin and thus may serve as the origin for sarcomas: dura, pia-arachnoid, stroma of the choroid plexus, adventitial fibroblasts associated with blood vessels, and tela choroidea.3,6 Intracranial sarcomas may arise in continuity with the meninges or as intraparenchymal masses without a dural base when the origin is from blood vessels, the tela choroidea, or the choroid plexus stroma.
One can classify intracranial sarcomatous lesions into seven distinct groups7,8:
Lesions in the last group are not true sarcomas but do share some common histologic findings. The first three entities are discussed in this chapter. Gliosarcoma is classified as a subtype of glioblastoma, an astrocytic tumor of neuroepithelial origin.5 The entity sarcoglioma, or the development of a glial neoplasm associated with a primary intracranial sarcoma, is discussed later. Sarcomatous metastases are similar in clinical behavior to other brain metastases and are discussed elsewhere. It is important to note that these metastases are histologically indistinguishable from primary CNS sarcomas, although new molecular techniques may enable us to elucidate the etiology of these lesions. Given the rarity of primary intracranial sarcomas, the finding of sarcomatous tissue in the brain should lead to a thorough search for a primary source outside the CNS. Scalp and skull tumors are discussed in separate chapters of this text.
Sarcomas can occur at any age. They account for just 0.1% to 3% of all intracranial tumors in both adult and pediatric series.7,9 The less well-differentiated sarcomas (Christensen and Lara’s polymorphocellular sarcomas) usually occur in infants and young children. Meningeal sarcomatosis, or diffuse infiltration of the dura by sarcomatous cells in the absence of a clear mass, also tends to occur in very young children. Most authors have reported that the incidence is equal in males and females.7,10 Sarcomas can occur at any site in the brain. In Paulus and coworkers’ series, 12 of 19 tumors (63%) were in direct contact with the dura.7 The remainder arose within the parenchyma of the brain, presumably from the mesenchymal tissue associated with blood vessels, the tela choroidea, or the choroid plexus. With the exception of rhabdomyosarcomas, which reportedly occur more frequently in the posterior fossa11 and in the midline,12 sarcomas occur with equal likelihood throughout the cranium.12
Pathology and Pathogenesis
The most recent revision of the WHO classification of brain tumors includes an expanded section on the wide range of mesenchymal tumors occurring in the CNS. The committee based this classification on the system for soft tissue tumors occurring outside the CNS.13 Differentiation of sarcomas by pathology is based on light microscopy (such as islands of cartilage in chondrosarcoma and osseous areas in osteosarcoma), immunohistochemistry (desmin and myoglobulin positivity in rhabdomyosarcoma), or ultrastructural features (Z bands in rhabdomyosarcoma).
The macroscopic appearance of these lesions is heterogeneous, but they tend to be large. Those arising from the dura are often firm, whereas those arising without clear dural attachment are often softer. In contrast to gliomas, sarcomas tend to be more distinct, with clearer macroscopic borders between the tumor and surrounding brain. They also tend to be firmer than gliomas. Hemorrhage, cysts, and necrosis can be seen within sarcomas.11,12,14
Angiosarcoma
These rare tumors constitute less than 1% of all sarcomas and usually arise in the skin or superficial soft tissues.11 Occasionally, they arise from intracranial vessels. They typically have a spongy appearance because of abundant vessels. In more differentiated areas of the tumor, vascular channels may be seen. The basement membrane of the vessels can be demonstrated with a reticulum stain. Brain invasion is frequently seen with these lesions despite a circumscribed radiographic appearance.11
Chondrosarcoma
These tumors arise from the dura, from the skull base, or rarely, from intraparenchymal sources.13 Chondrosarcomas arising in the fourth ventricle15 or in the falx16 have been reported. Some of these tumors exhibit a lower grade of malignancy than other sarcomas, but local recurrence and local aggressive behavior are still seen frequently.12 Histologically, the cartilage component is less well developed, and the tumors are more cellular and pleomorphic than chondromas.14 Intracranial chondrosarcomas represent 0.15% of all primary intracranial lesions.14
Mesenchymal Chondrosarcoma
These lesions are usually attached to the dura and may occur in either the cranial vault or the spinal canal. They are often found in the first or second year of life. Mesenchymal chondrosarcomas exhibit a pattern of pale staining and atypical chondroid areas alternating with compact cellular aggregates.12 Other histologic characteristics include thin-walled vascular spaces and limited areas of bone formation. These sarcomas have a moderate tendency to recur locally9 and may metastasize distantly.17
Rhabdomyosarcoma
These malignant tumors are composed of rhabdomyoblasts. They can arise either in the brain parenchyma or from the leptomeninges. They occur more frequently in children, often in the posterior fossa,7,11,13 where they can be difficult to differentiate from medullomyoblastoma. Rhabdomyosarcoma can also be seen as part of germ cell tumors of the pineal and suprasellar region.13 Histologically, these tumors are highly cellular and have myoblastic components. The diagnosis is aided by immunohistochemistry for myoglobin, myosin, muscle-specific actin, and desmin.13 Ultrastructure observations include the presence of Z bands and disorganized thick and thin filaments.13
Fibrosarcoma
Fibrosarcomas are the most common intracranial sarcomas (Fig. 132-1)11–13 and are more frequently seen in adults.7 They develop as a component of gliosarcoma, after radiotherapy (particularly treatment of the sella for pituitary adenoma), or as a primary intracranial sarcoma. These tumors arise from the dura or leptomeningeal infolds and can extend along the Virchow-Robin spaces.13 The tumors appear similar to fibrosarcomas occurring elsewhere in the body. Thus, a careful search for another primary site should be made if the diagnosis of intracranial fibrosarcoma is entertained. Given the higher frequency of gliosarcoma than fibrosarcoma, careful staining of the specimen with glial fibrillary acidic protein should be performed to exclude a glial component and the diagnosis of gliosarcoma.13
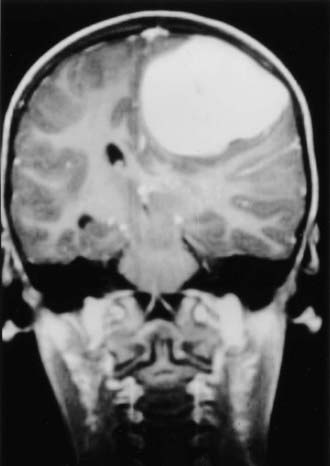
It may be difficult to differentiate between a true fibrosarcoma and sarcomatous degeneration of a meningioma. Burger and Scheithauer identified the following as useful methods for making that distinction.13 First, the well-known herringbone appearance of fibrosarcoma is not usually seen after sarcomatous degeneration of a meningioma. Second, meningioma stains for epithelial membrane antigen (EMA), whereas fibrosarcoma does not. Unfortunately, not all malignant meningiomas stain for EMA. Thus, the presence of EMA immunostaining supports the diagnosis of a malignant meningioma, but its absence does not exclude it. Third, a past history of meningioma aids in the diagnosis, as does the presence of whorls or psammoma bodies.
Malignant Fibrous Histiocytoma
This rare intracranial tumor is histologically similar to its extracranial counterpart. Histologic findings include high cellularity, nuclear polymorphism, and varying inflammation.13 These tumors can arise in the dura18 or, less commonly, intraparenchymally.19 In Paulus and coworkers’ series, malignant fibrous histiocytomas were more common in adults.7
Primary Meningeal Osteosarcoma
Primary meningeal osteosarcoma is an extremely rare entity described in the literature only via case reports. Review of the literature demonstrates six cases. These tumors arise from the meninges and are within the differential diagnosis for dural-based lesions.20 They are typically treated by aggressive resection and subsequent radiation therapy.
Other Sarcomas
Primary meningeal sarcomatosis refers to diffuse spread of sarcomatous tissue throughout the meninges without a focal mass.11 Most cases are reported in young children. The tumor in the subarachnoid space covers the surface of the brain and may grow along penetrating vessels into the brain, as well as into the spinal and cranial nerves. Additionally, tumor can surround the spinal cord.9 Histologically, small, round tumor cells are usually seen.
Extraskeletal lesions with histologic findings identical to those of the bony lesions of Ewing’s sarcoma are rare but well described.21–25 The most frequent site involving the nervous system is the epidural space (particularly in the spine), but intraparenchymal lesions have also been described. Distant metastases from these lesions can subsequently be seen.12 It has been suggested that these tumors are a type of the small, round cell tumor derived from neuroendocrine tissue12 rather than from mesenchymal tissue. If so, they are not truly sarcomas. Recent evidence has classified these tumors as central and peripheral primitive neuroectodermal tumors, but they are still classified as mesenchymal tumors in the 2007 classification.
The cause of intracranial sarcomas is often unknown. Sarcoma develops in some patients after radiotherapy for a brain tumor26 and has developed in at least one patient after chemotherapy for a brain tumor.27 Of the seven cases reported by Chang and colleagues, four were fibrosarcomas and three were malignant fibrous histiocytomas. Radiation doses ranged from 1600 to 6000 Gy.26 The median time to diagnosis of the tumor was 8 years after radiation therapy. Sarcomas have been reported most frequently after irradiation of the sella for pituitary adenoma.8,28 The median time to development of sarcoma was 10 years, the minimum dose was 2000 Gy, and multiple courses of radiation increased the risk. Histologically, there may be evidence of recurrent adenoma within the sarcoma.13,23
Clinical Findings and Evaluation
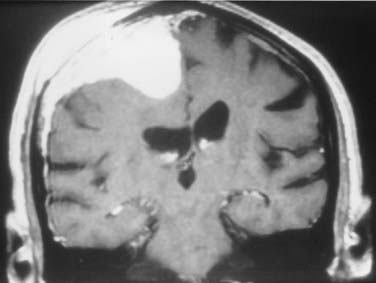
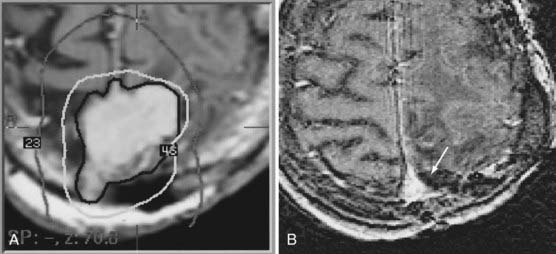
The clinical features of patients with meningeal sarcomas are similar to those of other patients with malignant intracranial tumors. Symptoms are due to mass effect from the tumor or peritumoral edema (Fig. 132-2). Clinical symptoms include headaches, seizures, weakness, mental status changes, or symptoms of hydrocephalus (Table 132-2). Patients seen in this fashion are usually stabilized with therapy for increased intracranial pressure, such as steroids. If the tumor erodes through the skull into the soft tissues of the scalp, it may be manifested as a palpable mass (Fig. 132-3). Imaging should be obtained immediately.
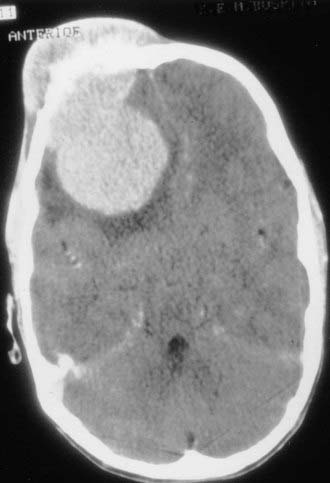
TABLE 132-2 Symptoms of Dural-Based Lesions Vary According to Location
| LOCATION | SYMPTOMS |
|---|---|
| Frontal/parietal | Affective, hemiparesis, neglect, sensory disturbance, seizures; may be asymptomatic |
| Temporal | Seizures, speech disturbance |
| Frontal fossa | Behavioral disorder, olfactory loss |
| Sellar/parasellar | Visual loss, pituitary disturbance |
| Sphenoid wing | Visual disturbance, psychiatric disturbance, seizures, speech disorders |
| Cavernous sinus | Eye movement disturbance, facial pain or numbness |
| Posterior fossa | Ataxia, hydrocephalus, cranial nerve dysfunction |
Intracranial sarcomas have no typical imaging features. When they arise from the dura, they may simulate the more common meningioma (see Fig. 132-1). The presence of bone erosion overlying the mass suggests a more malignant tumor, but this finding may also be seen with typical and atypical meningioma. Extension into the scalp is more common with sarcoma than with meningioma. Underlying edema is also nearly universal with sarcomas but may also be seen in up to 50% of typical meningiomas. Invasion of the superficial venous structures may occur with both types of tumors. Although most adult CNS sarcomas are extra-axial masses, sarcomas and meningiomas in children commonly arise as intra-axial or intraventricular masses. Intraparenchymal sarcomas have no specific imaging features.
Imaging
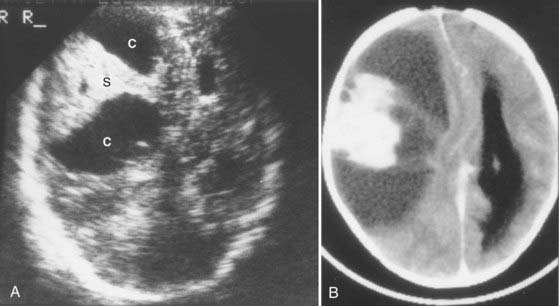
Angiography plays a limited role in the evaluation of these tumors. It is no longer used for tumor localization but may be used to map blood supply to the lesion. In sarcomas, blood supply can be derived from the internal carotid artery, the external carotid artery, or both.1 Hypervascular tumors may benefit from presurgical embolization to decrease their blood supply. Angiography may also show vessel encasement by tumor, but MRI is a better means of visualizing encasement. Sonography has a very limited role in the evaluation of these tumors. It may be used for diagnosis of tumors in utero,29 for screening of newborns with enlarging heads (Fig. 132-4), and as an aid to intraoperative localization of sarcomas arising in the brain parenchyma.
Treatment
Treatment options include surgery, radiotherapy, and chemotherapy. The goal of surgical therapy, as with all sarcomas, is complete surgical excision. Unfortunately, brain invasion is common with these lesions despite their circumscribed appearance on imaging studies. This often prevents obtaining a tumor-free margin. There are numerous case reports of long-term survivors after aggressive surgical resection with or without subsequent radiotherapy.10 The best outcome usually follows radical resection of a well-circumscribed tumor (fibrosarcoma or mesenchymal chondrosarcoma).3
The exact role of radiotherapy and chemotherapy for CNS sarcomas is not yet established. Clearly, these modalities play a significant role in the treatment of sarcomas that occur elsewhere in the body. Because sarcomas of the CNS are histologically similar to those occurring elsewhere, it has been suggested that these therapies may have a role in treating CNS sarcomas as well, but supportive data for this hypothesis are lacking. However, current treatment strategies include radiation therapy for subtotal resection either in the form of conformal radiation therapy or stereotactic radiosurgery for residual or recurrent disease.30
In general, patients with these tumors have short survival periods. Frequently, the tumors recur locally despite aggressive resection. Furthermore, metastatic disease is not uncommon. For determining prognosis, the histologic subtype of the tumor is a strong predictor. Patients with fibrosarcoma often have rapid progression of their disease and a median survival of 6 to 9 months. Local recurrence is common, and metastases can also be seen.12,31,32 Local high-dose radiation therapy is used frequently in these patients. Subarachnoid spread can also occur. Treatment of patients with rhabdomyosarcoma is difficult. Despite aggressive chemotherapy and radiation therapy, most studies report median survival of less than 24 months.32 Children with meningeal sarcomatosis have a poor prognosis. Their clinical course can mimic that of meningoencephalitis.12 Surgical resection is not possible because of the diffuse nature of the tumor, and the condition is usually fatal within a few weeks.33
Meningeal Hemangiopericytoma
History
Meningeal hemangiopericytoma is a malignant neoplasm that occurs within the CNS and has the biologic capability to escape from the CNS and metastasize widely. The originating cells are thought to be meningeal capillary pericytes, Zimmerman pericytes, or precursor cells with angioblastic tendencies.34,35 This lesion was dubbed hemangiopericytoma by Stout and Murray in 1942, although it was extracranial in location.34 Information about this tumor comes from cases reported variably as angioblastic meningioma within large series of meningiomas36–40 and series devoted to meningeal hemangiopericytomas.41–51 Its biologic behavior, as well as molecular and genetic profile, suggests that this lesion may be accurately described as a dural-based sarcoma.52
In 1938, Cushing and Eisenhardt identified a vascular form of meningioma and termed it angioblastic meningioma.53 One variant behaved in a particularly malignant fashion, and it is to this tumor that the term angioblastic is generally applied.53,54 Shortly thereafter, a soft tissue sarcoma called hemangiopericytoma was recognized to be histologically identical to angioblastic meningioma.34,55,56 Begg and Garret, who gave the first report of a primary intracranial hemangiopericytoma in 1954, proposed that Cushing’s angioblastic meningioma was actually a primary meningeal hemangiopericytoma,57 which was compatible with the known aggressive behavior of this tumor, and it was histologically identical to the tumor described by Stout and Murray.
The 2007 WHO classification differentiates meningeal hemangiopericytoma from meningioma and groups hemangiopericytoma in the category of mesenchymal tumors.5 Certainly, meningeal hemangiopericytoma must be recognized as a clinical entity distinct from meningioma, and management must reflect its much more aggressive behavior.
Clinical Significance
Hemangiopericytomas constitute 2% to 4% of large series of meningeal tumors.36,38,41,43,44,47,49,58,59 About 10% of meningeal hemangiopericytomas occur in children.60 They can be misdiagnosed as meningioma and may be more common than reported.
Several studies have shown that unlike meningiomas, meningeal hemangiopericytomas are more common in males (56% to 75%) than females, even in the spinal location,42,47,49,61 although a more recent study showed a slight female preponderance.50 The average age at diagnosis is about 40 years.48,60,61 Hemangiopericytomas are located similar to meningiomas, with 15% in the posterior fossa and 15% in the spine.48,61,62 Of the spinal tumors, about half are in the cervical region.61 As with meningiomas, there is the occasional report of a nonmeningeal cerebral location for this tumor.63–65 Primary multifocal meningeal hemangiopericytomas have not been reported.49,61 These tumors are thought to account for approximately 1% of all CNS tumors.66
Pathology and Pathogenesis
Grossly, meningeal hemangiopericytomas are firm and lobulated and vary from pink-gray to red in color. They are extremely vascular, are often adherent to the dura, but do not usually invade the brain.47,49 They do not spread en plaque and rarely contain calcification.
The tumors are very cellular, with round to oval cells (Fig. 132-5A). The architecture varies from field to field, often resembling forms of ordinary meningioma.35,49 The cells are arranged around thin-walled vascular spaces lined by a non-neoplastic endothelium. These “staghorn” capillaries are a distinguishing feature and can be quite numerous.35,48
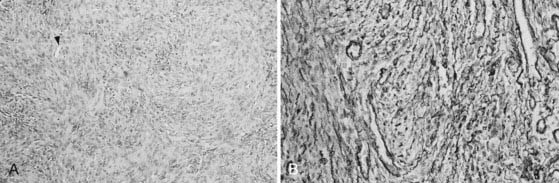
Mitoses vary from one to several per high-powered field.48,49 Microcysts, necrosis, and papillary architecture may be seen49 and have been reported in up to 50% of the tumors.48 Whorls and psammoma bodies are not seen.48,49 Reticulin envelops individual cells (Fig. 132-5B) (thus differentiating it from meningioma, in which reticulin is scarcer and segregates the tumor into lobules). Hemangiopericytoma cells exhibit leiomyoblastic differentiation and secretion of basement membrane–like material, but surface membrane specializations (interdigitations, desmosomes, zonulae adherentes, and gap junctions) are absent.51,52,67
Meningeal hemangiopericytomas usually contain subpopulations of cells that express factor XIIIa, which distinguishes them from meningioma and other soft tissue tumors, although this marker has been found to be positive in anaplastic meningioma as well.52,68–70 Other markers include vimentin, HLA-DR, CD34, Leu-7, S-100 protein, CD99, and BCL-2.52,68 Tumor cells are negative for the immunohistochemical markers factor VIII-RA and glial fibrillary acidic protein; EMA can be variable.71,72
Hemangiopericytomas are genetically distinct from meningiomas.73 Rearrangements of chromosome 12q13 are common in hemangiopericytoma, and several oncogenes are located in this region, including MDM2, CDK4, and CHOP/GADD153.74,75 Cytogenic alterations in chromosomes 12q13, 19q13, 6p21, and 7p15 are not common in meningiomas.76 Mutation of the NF2 tumor suppressor gene, which is frequently associated with meningiomas, is not found in hemangiopericytomas.77 Loss of 1p32, 14q32, and 4.1B is also common in meningiomas but rarely seen in hemangiopericytoma.52 The biologic behavior of individual hemangiopericytomas cannot be correlated with histologic features such as mitoses, MIB-1, Ki-67, or DNA ploidy.56,68,77
Recurrent tumors retain their histologic features, and metastatic hemangiopericytomas are histologically identical to the primary tumor.49 Meningeal hemangiopericytomas are histologically and biologically distinct from atypical or malignant meningiomas.78
Clinical Findings and Evaluation
Patients tend to be symptomatic for shorter periods before initial evaluation than is the case with meningioma patients, perhaps because of the more rapid growth rate of these tumors.16 The initial symptoms are related to tumor location.47,49,61 The most frequent initial symptom is headache or focal signs related to tumor location (see Table 132-2).42,50 Seizures are initially present in only about 16% of patients with supratentorial tumors,49 which is compatible with the fact that they do not microscopically infiltrate the brain and grow rather rapidly. Intracranial hemorrhage as the initial symptom has also been described.79
Imaging
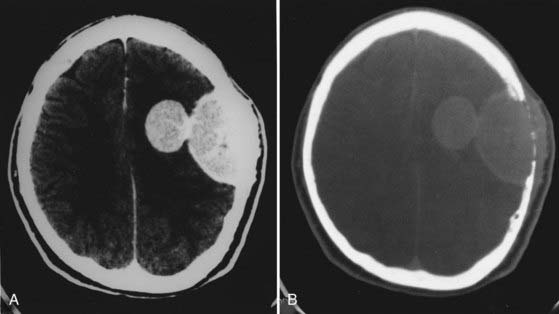
Meningeal hemangiopericytomas may resemble meningiomas on imaging studies. CT typically shows a narrow or broad-based meningeal attachment. Most often the tumors appear hyperdense with focal areas of hypodensity on unenhanced CT scans and exhibit areas of heterogeneous enhancement with the administration of contrast material (Fig. 132-6).50,80 They may have features suggesting malignancy, such as macroscopic brain invasion, or “mushrooming” inhomogeneous contrast enhancement and irregular borders.81 Bone erosion is seen in more than 50% of hemangiopericytomas,50 and hyperostosis is not a feature of this tumor.47,49,80,82
Hemangiopericytomas are usually isointense with gray matter and display prominent vascular flow voids on T1- and T2-weighted MRI (Fig. 132-7).50 Heterogeneous enhancement is the most common appearance on T1-weighted gadolinium-enhanced images. About half the tumors have a dural tail sign. CT and MRI characteristics that may help differentiate hemangiopericytomas from meningiomas include a narrow-based dural attachment (seen more frequently in hemangiopericytomas) and hyperostosis of adjacent bone (seen in meningiomas but not in hemangiopericytomas).50
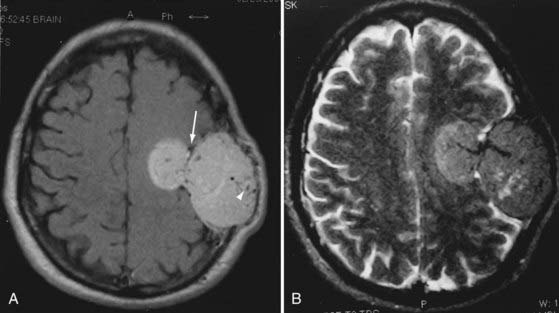
FIGURE 132-7 A and B, Magnetic resonance images of the hemangiopericytoma in Figure 132-6 show that the tumor is arising from the convexity meninges and growing through the skull, as well as through the dura into the brain (arrow). Note the large vascular voids characteristic of these tumors (arrowhead). Homogeneous enhancement is not unusual.
The tumors frequently exhibit characteristic arteriographic features, including a corkscrew vascular configuration, shunting, and a long-lasting venous stain (Fig. 132-8).83 As many as half have a significant internal carotid blood supply,47,49,83 and few show early venous drainage, another factor that distinguishes them from ordinary meningiomas.47,49 Hemangiopericytomas frequently have a dual blood supply from the internal carotid or vertebral arteries in addition to the external carotid supply.80
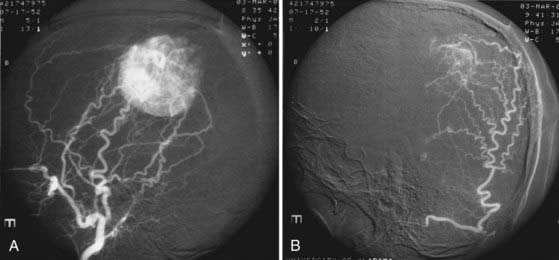
FIGURE 132-8 A pre-embolization arteriogram of the lesion presented in Figures 132-6 and 132-7 shows very large meningeal feeders from the external carotid circulation along with rapid filling of the tumor (A). Selective embolization of the feeders resulted in marked devascularization (B), which greatly facilitated surgical hemostasis.
Treatment
The principles of surgical resection are similar to those for meningioma. Surgery with the aim of gross total resection, Simpson grade I, is the primary modality for treatment of hemangiopericytoma. This entails resection of dura, bone, and if required, brain or vascular structures if able to do so without producing an unacceptable neurological deficit. Every effort should be made to achieve complete removal at the initial resection because surgery for recurrence is often more difficult and is less effective. In large series reporting surgery for these tumors, complete resection was possible in only 50% to 67% of cases.47,49,61 If complete removal is not possible, adjuvant radiation therapy is recommended. Disease-free survival is demonstrably longer after complete resection, and recurrence is higher after subtotal resection.66,84,85
Preoperative embolization can be effective in reducing the vascularity of these tumors. In cases in which hemangiopericytoma is highly suspected or known, preoperative angiography and embolization can reduce blood loss and facilitate tumor removal by significantly reducing the blood supply to the lesion.66 Fountas and coauthors reported 50% lower average blood loss with embolization.66
Radiosurgery
Radiosurgery is reportedly effective in controlling meningioma,86 so it would seem to be a reasonable therapeutic option for meningeal hemangiopericytoma as well. Multiple series have described the experience of stereotactic radiosurgery for residual or recurrent disease.85,87–89 Lunsford’s group reported a series in which stereotactic radiosurgery provided local control of 80% of lesions.88 Galanis and coworkers reported that 17 of 20 lesions in 20 patients responded favorably.87 Chang and Sakamoto reported a good response in 75% of patients.89 Even though all these studies had a small sample size, the evidence clearly supports the use of stereotactic radiosurgery for the treatment of recurrence or residual disease, and variable numbers of these patients also received conventional radiotherapy. Disease progression and recurrence are assessed radiographically, and response to treatment can be quite dramatic (Fig. 132-9).
Recurrence
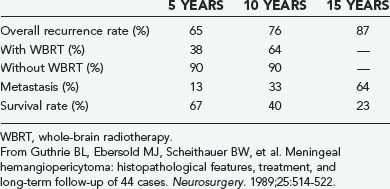
Meningeal hemangiopericytoma has a relentless tendency to recur, even after what is considered complete surgical resection. Allowing for variable definitions of “extent of resection” and “recurrence,” median recurrence-free intervals after resection have ranged from 40 to 78 months.47,49,61 Many years of recurrence-free survival are not uncommon.49 After reviewing the literature, Schroder and colleagues found the 5-year recurrence rate to be around 60%.61 From an actuarial standpoint, Guthrie and coworkers calculated 5-, 10-, and 15-year recurrence rates of 65%, 76%, and 87%, respectively (Table 132-3).49 Kim and coauthors reported a 5-year recurrence-free rate of 59.2%, 72.7% in the total excision group and 20% in the incomplete excision group.84 From these data it is obvious that meningeal hemangiopericytomas are much more aggressive than meningiomas and that recurrence is likely if a patient survives for 5 years; it becomes more likely with extended survival. The fact is that if the patient lives long enough, the tumor will probably recur, thus requiring close follow-up and aggressive treatment at the initial appearance of signs and symptoms.
After the first recurrence, meningeal hemangiopericytomas tend to recur at shorter intervals. In a review of 44 patients operated on 79 times, the average interval to subsequent recurrence tended to shorten.49 The average time to a second, third, and fourth operation for recurrence was 38, 35, and 17 months, respectively. In addition, 53% of patients improved and 3% worsened after the first operation, whereas only 33% improved and 13% worsened after subsequent operations, thus again suggesting that the time for optimal patient benefit is at the first operation.
Metastasis
Meningeal hemangiopericytomas are unique in that they frequently metastasize outside the CNS. The most common sites of metastasis in descending frequency are bone, lung, and liver.35,36,39,41,45,47,49,61,90 The rate of metastasis of meningeal hemangiopericytoma at 5, 10, and 15 years is 13%, 33%, and 64%, respectively (see Table 132-3),84 thus making metastasis likely with prolonged survival. Realization that extraneural metastasis can occur after years of apparent tumor-free survival is critical for the long-term management of these patients. Intra-axial seeding of meningeal hemangiopericytomas is extremely rare. Treatment of metastasis usually requires a similar multimodality approach tailored to the location of the disease.
Survival
Guthrie and coworkers found that the median survival after the first operation was 60 months, with actuarial 5-, 10-, and 15-year survival rates of 67%, 40%, and 23%, respectively (see Table 132-3).49 This is comparable to the cumulative survival rates of approximately 65%, 45%, and 15%, respectively, compiled by Schroder and colleagues from a review of 118 cases in the literature up to 1985.61 Fountas and coauthors reported a mean survival of 5.5 years.66
Extraneural metastasis is a devastating occurrence and significantly shortens survival. In the Mayo Clinic series, 10 of 44 patients experienced extraneural metastasis at an average time of 99 months. The average length of survival after metastasis was 24 months. Five patients alive at 99 months in whom metastasis did not develop survived an additional 76 months.49
Other than extraneural metastasis, the factor most clearly related to survival is postoperative radiotherapy. Guthrie and coworkers found that patients who underwent radiotherapy after the first operation experienced recurrence at an average of 74 months, with 5- and 10-year recurrence rates of 38% and 64%, respectively (see Table 132-3).49 Patients not irradiated suffered recurrence at an average of 29 months, with 5- and 10-year recurrence rates of 90% (Table 132-3).49 These authors found a dose effect in that patients receiving less than 4500 cGy tended to experience recurrence sooner than did those receiving 5000 cGy or more. In this same series, patients who underwent radiotherapy after the first operation survived an average of 92 months, and those who did not lived 62 months. Others have observed this response for meningeal as well as extraneural locations of this tumor.61,91–94
Discussion
Meningeal hemangiopericytoma is a malignant primary neoplasm of the meninges that has the biologic behavior and characteristics of a sarcoma and is far more aggressive than its common counterpart meningioma. These tumors have a slight male preponderance, with an average age at diagnosis of about 40 years.37,38,58,59,95 Symptoms are usually present for a shorter time before initial evaluation than with meningioma, and seizures are less common than with meningioma.
These aggressive tumors have 5- and 10-year recurrence rates of 65% and 76%, respectively,49 as compared with 20% and 30%, respectively, for meningioma.59,95 Hemangiopericytomas are relatively responsive to radiation.49,96,97 Perhaps the most striking difference from meningioma is the tendency for meningeal hemangiopericytoma to metastasize. Kepes found very few metastatic ordinary meningiomas.39 In contrast, the rate of metastasis of meningeal hemangiopericytomas at 5, 10, and 15 years was 13%, 33%, and 64%, respectively,49 thus making metastasis likely with prolonged survival (see Table 132-3).
Meningeal hemangiopericytoma should not be equated with atypical or malignant meningioma. The latter tumor shows loss of architecture, increased cellularity, nuclear atypia, and mitoses but remains recognizable as meningioma. Jaaskelainen and associates studied a series of atypical and anaplastic meningiomas and found them to be more aggressive than ordinary meningiomas, but less so than meningeal hemangiopericytomas.78 Malignant meningiomas have little or no tendency to metastasize.
Conclusion
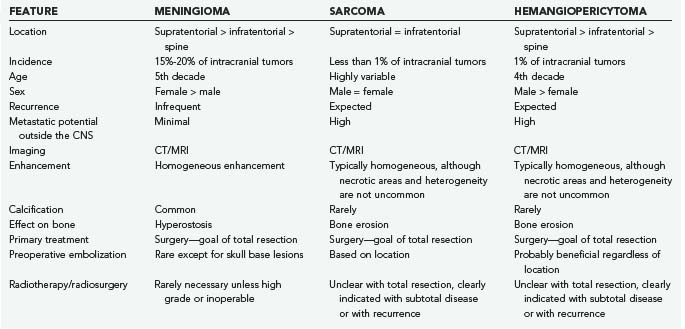
Meningeal sarcoma and hemangiopericytoma are two exceptionally aggressive lesions that should be in the differential diagnosis for any dural-based lesion (Table 132-4). Certain imaging or historical features may increase the preoperative suspicion for these lesions. Surgical resection is the mainstay of treatment of both entities, and aggressive primary resection leads to the best outcomes. Preoperative embolization can be helpful when the diagnosis is known or suspected before resection. Patients should be assessed for metastatic lesions because intracranial lesions could represent a primary site or metastatic disease. Radiosurgery is proving to be an effective tool for the treatment of residual or recurrent disease. Follow-up should include not only imaging of the craniospinal axis but also assessment for metastatic disease because both of these lesions can and do metastasize outside the CNS.
Fountas Fountas KN, Kapsalaki E, Kassam M, et al. Management of intracranial meningeal hemangiopericytomas: outcome and experience. Neurosurg Rev. 2006 Apr;29(2):145-153. Epub 2006 Jan 4
Haddad Haddad G, Al-Mefty O. Meningeal sarcoma. In: Kaye A, Laws E, editors. Brain tumors: an encyclopedic approach. Edinburgh: Churchill Livingstone; 1995:713-721.
Chang Chang SM, Barker FG2nd, Larson DA, et al. Sarcomas subsequent to cranial irradiation. Neurosurgery. 1995;36:685-690.
1 Haddad G, Al-Mefty O. Meningeal sarcoma. In: Kaye A, Laws E, editors. Brain Tumors: An Encyclopedic Approach. Edinburgh: Churchill Livingstone; 1995:713-721.
2 Bailey P. Intracranial sarcomatous tumors of leptomeningeal origin. Arch Surg. 1929;18:1359-1402.
3 Rubinstein L. Sarcomas of the nervous system. In: Minckler J, editor. Pathology of the Nervous System. New York: McGraw-Hill; 1971:2144-2164.
4 Christensen E, Lara D. Intracranial sarcomas. J Neuropathol Exp Neurol. 1953;12:41-56.
5 Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
6 Burger M, Kros J. Sarcomas and neoplasms of blood vessels. In: Youmans J, editor. Neurological Surgery. 4th ed. Philadelphia: WB Saunders; 1996:2700-2714.
7 Paulus W, Slowik L, Jellinger K. Primary intracranial sarcoma: histopathologic features of 19 cases. Histopathology. 1991;18:395-402.
8 Bernstein M, Deck J, Fleming J. Intracranial sarcomas. In: Wilkins R, Rengachary S, editors. Neurosurgery. New York: McGraw-Hill; 1996:1725-1731.
9 Tomita T, Gonzalez-Crussi F. Intracranial primary nonlymphomatous sarcomas in children: experience with eight cases and review of the literature. Neurosurgery. 1984;14:529-540.
10 Zulch K. Histiologic Typing of Tumours of the Central Nervous System. Geneva: World Health Organization; 1979. 53-58
11 Burger P, Scheithauer B, Vogel F. Surgical Pathology of the Nervous System and Its Coverings. New York: Churchill Livingstone; 1991. 67-142
12 Bruner J, Tien R, Enterline D. Tumors of the meninges and related tissues. In: Bigner D, McLendon R, Bruner J, editors. Russell and Rubinstein’s Pathology of the Tumors of the Nervous System. London: Arnold; 1998:67-140.
13 Burger P, Scheithauer B. Atlas of Tumor Pathology: Tumors of the Central Nervous System. Washington, DC: Armed Forces Institute of Pathology; 1994. 452
14 Chandler JP, Yashar P, Laskin WB, et al. Intracranial chondrosarcoma: a case report and review of the literature. J Neurooncol. 2004;68:33-39.
15 Scott R, Dickersin G, Wolpert S, et al. Myxochondrosarcoma of the fourth ventricle. J Neurosurg. 1976;44:386-389.
16 Cybulski G, Russel E, D’Angelo C, et al. Falcine chondrosarcoma: case report and literature review. Neurosurgery. 1985;16:412-415.
17 Rollo J, Green W, Kahn L. Primary meningeal mesenchymal chondrosarcoma. Arch Pathol Lab Med. 1979;103:239-243.
18 Swamy K, Shankar S, Asha T, et al. Malignant fibrous histiocytoma arising from the meninges of the posterior fossa. Surg Neurol. 1986;25:18-24.
19 Roosen N, Cras P, Paquier P, et al. Primary thalamic malignant fibrous histiocytoma of the dominant hemisphere causing severe neuropsychological symptoms. Clin Neuropathol. 1989;8:16-21.
20 Setzer M, Lang J, Turowski B, Marquardt G. Primary meningeal osteosarcoma: case report and review of the literature. Neurosurgery. 2002;51:488-492.
21 Angervall L, Enzinger FM. Extraskeletal neoplasm resembling Ewing’s sarcoma. Cancer. 1975;36:240-251.
22 Mehta Y, Hendrickson FR. CNS involvement in Ewing’s sarcoma. Cancer. 1974;33:859-862.
23 Scheithauer BW, Egbert BM. Ewing’s sarcoma of the spinal epidural space: report of two cases. J Neurol Neurosurg Psychiatry. 1978;41:1031-1035.
24 Simonati A, Vio M, Iannucci AM, et al. Lumbar epidural Ewing sarcoma: light and electron microscopic investigation. J Neurol. 1981;225:67-72.
25 Sonntag VK, Stein BM. Intracerebral Ewing’s sarcoma: case report. Neurosurgery. 1978;2:55-57.
26 Chang SM, Barker FG2nd, Larson DA, et al. Sarcomas subsequent to cranial irradiation. Neurosurgery. 1995;36:685-690.
27 Martinez-Salazar A, Supler M, Rojiani AM. Primary intracerebral malignant fibrous histiocytoma: immunohistochemical findings and etiopathogenetic considerations. Mod Pathol. 1997;10:149-154.
28 Waltz T, Brownell B. Sarcoma: a possible late result of effective radiation therapy for pituitary adenoma. J Neurosurg. 1966;24:901-907.
29 van Vliet MA, Bravenboer B, Kock HC, et al. Congenital meningeal sarcoma—case report. J Perinat Med. 1983;11:249-254.
30 Tzortzidis F, Elahi F, Wright DC, et al. Patient outcome at long-term follow-up after aggressive microsurgical resection of cranial base chondrosarcomas. Neurosurgery. 2006;58:1090-1098.
31 Aung TH, Tse CH. Bifrontal meningeal fibrosarcoma in a patient with metastases to the liver, kidneys and suprarenal glands. Aust N Z J Surg. 1993;63:746-748.
32 Olsen J, Menezez A, Godersky J, et al. Primary intracranial rhabdomyosarcoma. Neurosurgery. 1985;17:25-34.
33 Budka H, Pilz P, Guseo A. Primary leptomeningeal sarcomatosis: clinicopathological report of six cases. J Neurol. 1975;211:77-93.
34 Stout AP, Murray MR. Hemangiopericytoma: a vascular tumor featuring Zimmerman’s pericyte. Ann Surg. 1942;116:26-33.
35 Horten BC, Urich H, Rubinstein LJ, et al. The angioblastic meningioma: a reappraisal of nosological problem. J Neurol Sci. 1977;31:387-410.
36 Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22-39.
37 Skullerud K, Loken AC. The prognosis in meningiomas. Acta Neuropathol (Berl). 1974;29:337-344.
38 Jellinger K, Slowik F. Histological subtypes and prognostic problems in meningiomas. J Neurol. 1975;208:279-298.
39 Kepes JJ. Meningiomas: Biology, Pathology and Differential Diagnosis. New York: Masson; 1982.
40 de la Monte SM, Fickinger J, Linggood RM. Histopathologic resection. Am Surg Pathol. 1986;10:836-843.
41 Pitkethly DT, Hardman JM, Kempe LG, et al. Angioblastic meningiomas: clinicopathologic study of 81 cases. J Neurosurg. 1970;32:539-544.
42 Goellner JR, Laws ERJr, Soule EH, et al. Hemangiopericytoma of the meninges: Mayo Clinic experience. J Clin Pathol. 1978;70:375-380.
43 Fabiani A, Favero M, Trebini F. On the primary meningeal tumors with special concern to the hemangiopericytoma pathology and biology. Zentralbl Neurochir. 1980;41:273-284.
44 Wara WM, Sheline GE, Newman H, et al. Radiation therapy of meningiomas. Am J Roentgenol Radium Ther Nucl Med. 1975;123:453-458.
45 Thomas HG, Dolman CL, Berry K. Malignant meningioma: clinical and pathological features. J Neurosurg. 1981;55:929-934.
46 Kruse FJr. Hemangiopericytoma of the meninges (angioblastic meningioma of Cushing and Eisenhardt): clinicopathologic aspects and follow-up studies in 8 cases. Neurology. 1961;11:771-777.
47 Jaaskelainen J, Servo A, Haltia M, et al. Intracranial hemangiopericytoma: radiology, surgery, radiotherapy, and outcome in 21 patients. Surg Neurol. 1985;23:227-236.
48 Kochanek S, Schroder R, Firsching R. Hemangiopericytoma of the meninges: histopathological variability and differential diagnosis. Zentralbl Neurochir. 1986;47:183-190.
49 Guthrie BL, Ebersold MJ, Scheithauer BW, et al. Meningeal hemangiopericytoma: histopathological features, treatment, and long-term follow-up of 44 cases. Neurosurgery. 1989;25:514-522.
50 Chiechi M, Smirniotopoulos J, Mena H. Intracranial hemangiopericytomas: MR and CT features. AJNR Am J Neuroradiol. 1996;17:1365-1371.
51 Brunori A, Delitala A, Oddi G, et al. Recent experience in the management of meningeal hemangiopericytomas. Tumori. 1997;83:856-860.
52 Rajaram V, Brat DJ, Perry A. Anaplastic meningioma versus meningeal hemangiopericytoma: immunohistochemical and genetic markers. Hum Pathol. 2004;35:1413-1418.
53 Cushing H, Eisenhardt L. Meningiomas: Their Classification, Regional Behavior, Life History, and Surgical Results. Springfield, IL: Charles C Thomas; 1938.
54 Bailey P, Cushing H, Eisenhardt L. Angioblastic meningiomas. Arch Pathol. 1928;6:953-990.
55 Stout AP, Murray MR. Hemangiopericytoma occurring in the meninges. Cancer. 1942;2:1027-1035.
56 Enzinger F, Smith B. Hemangiopericytoma: an analysis of 106 cases. Hum Pathol. 1976;7:61-82.
57 Begg CF, Garret R. Hemangiopericytoma occurring in the meninges. Cancer. 1954;7:602-606.
58 Chan RC, Thompson GB. Morbidity, mortality, and quality of life following surgery for intracranial meningiomas: a retrospective study in 257 cases. J Neurosurg. 1984;60:52-60.
59 Mirimanoff RO, Dosoretz DE, Linggood RM, et al. Meningioma: analysis of recurrence and progression following neurosurgical resection. J Neurosurg. 1985;62:18-24.
60 Herzog C, Leeds N, Bruner J, et al. Intracranial hemangiopericytomas in children. Pediatr Neurosurg. 1995;22:274-279.
61 Schroder R, Firsching R, Kochanek S. Hemangiopericytoma of the meninges. II. General and clinical data. Zentralbal Neurochir. 1986;47:191-199.
62 Cappabianca P, Mauri F, Pettinato G, et al. Hemangiopericytoma of the spinal canal. Surg Neurol. 1981;15:298-302.
63 Stone JL, Cybulski GR, Rhee HL, et al. Excision of a large pineal region hemangiopericytoma (angioblastic meningioma, hemangiopericytoma type). Surg Neurol. 1983;19:181-189.
64 Lesion F, Bouchez B, Krivosic I, et al. Hemangiopericytic meningioma of the pineal region: case report. Eur Neurol. 1984;23:274-277.
65 Sell J, Hart B, Rael J. Hemangiopericytoma: a rare pineal mass. Neuroradiology. 1996;38:782-784.
66 Fountas KN, Kapsalaki E, Kassam M, et al. Management of intracranial meningeal hemangiopericytomas: outcome and experience. Neurosurg Rev. 2006;29:145-153.
67 Pena CE. Meningioma and intracranial hemangiopericytoma: a comparative electron microscopic study. Acta Neuropathol (Berl). 1977;39:69-74.
68 Probst-Cousin S, Bergmann M, Schroder R, et al. Ki-67 and biological behavior in meningeal hemangiopericytomas. Histopathology. 1996;29:57-61.
69 Nemes Z. Differentiation markers in hemangiopericytoma. Cancer. 1992;69:133-140.
70 Molnar P, Nemes Z. Hemangiopericytoma of the cerebellopontine angle: diagnostic pitfalls and the diagnostic value of the subunit A of factor XIII as a tumor marker. Clin Neuropathol. 1995;14:19-24.
71 Nakamura M, Inoue H, Ono N, et al. Analysis of hemangiopericytic meningiomas by immunohistochemistry, electron microscopy and cell culture. J Neuropathol Exp Neurol. 1987;46:57-71.
72 Hahn HP, Bundock EA, Hornick JL. Immunohistochemical staining for claudin-1 can help distinguish meningiomas from histologic mimics. Am J Clin Pathol. 2006;125:203-208.
73 Ono Y, Ueki K, Joseph J, et al. Homozygous deletions of the CDKN2/p16 gene in dural hemangiopericytomas. Acta Neuropathol (Berl). 1996;91:221-225.
74 Mandahl N, Orndal C, Heim S, et al. Aberrations of chromosome segment 12q13-15 characterize a subgroup of hemangiopericytomas. Cancer. 1993;71:3001-3013.
75 Henn W, Wullich B, Thonnes M, et al. Recurrent t(12;19)(q13; q13.3) in intracranial and extracranial hemangiopericytoma. Cancer Genet Cytogenet. 1993;71:151-154.
76 Joseph J, Lisle D, Jacoby L, et al. NF2 gene analysis distinguishes hemangiopericytoma from meningioma. Am J Pathol. 1995;147:1450-1455.
77 Zellner A, Meixensberger J, Roggendorf W, et al. DNA ploidy and cell-cycle analysis in intracranial meningiomas and hemangiopericytomas: a study with high-resolution DNA flow cytometry. Int J Cancer. 1998;79:116-120.
78 Jaaskelainen J, Haltia M, Servo A. Atypical and anaplastic meningiomas: radiology, surgery, radiotherapy and outcome. Surg Neurol. 1986;25:233-242.
79 Maruya J, Seki Y, Morita K, et al. Meningeal hemangiopericytoma manifesting as massive intracranial hemorrhage—two case reports. Neurol Med Chir (Tokyo). 2006;46:92-97.
80 Sibtain NA, Butt S, Connor SE. Imaging features of central nervous system haemangiopericytomas. Eur Radiol. 2007;17:1685-1693.
81 New PFJ, Hesselink JR, O’Caroll CP, et al. Malignant meningiomas: CT and histologic criteria, including a new CT sign. AJNR Am J Neuroradiol. 1982;3:267-276.
82 Osborne DR, Dubois P, Drayer B, et al. Primary intracranial meningeal and spinal hemangiopericytoma: radiologic manifestations. AJNR Am J Neuroradiol. 1981;2:69-74.
83 Marc JA, Takei Y, Schecter MM, et al. Intracranial hemangiopericytomas: angiography, pathology, and differential diagnosis. AJR Am J Roentgenol. 1975;125:823-832.
84 Kim JH, Jung HW, Kim YS, et al. Meningeal hemangiopericytomas: long-term outcome and biological behavior. Surg Neurol. 2003;59:47-53.
85 Soyuer S, Chang EL, Selek U, et al. Intracranial meningeal hemangiopericytoma: the role of radiotherapy: report of 29 cases and review of the literature. Cancer. 2004;100:1491-1497.
86 Duma CM, Lunsford LD, Kondziolka D, et al. Stereotactic radiosurgery of cavernous sinus meningiomas as an addition or alternative to microsurgery. Neurosurgery. 1983;32:699-705.
87 Galanis E, Buckner JC, Scheithauer BW, et al. Management of recurrent meningeal hemangiopericytomas. Cancer. 1998;82:1915-1920.
88 Sheehan J, Kondziolka D, Flickinger J, et al. Radiosurgery for treatment of recurrent intracranial hemangiopericytomas. Neurosurgery. 2002;51:905-910.
89 Chang SD, Sakamoto GT. The role of radiosurgery for hemangiopericytomas. Neurosurg Focus. 2003;14(5):e14.
90 Inoue H, Tamura M, Koizumi H, et al. Clinical pathology of malignant meningiomas. Acta Neurochir (Wien). 1984;73:179-191.
91 Friedman M, Egan JW. Irradiation of hemangiopericytoma of Stout. Radiology. 1960;74:721-729.
92 Lal H, Sanyal B, Pant CG, et al. Hemangiopericytoma: report of three cases regarding role of radiation therapy. AJR Am J Roentgenol. 1976;126:887-891.
93 Mira JG, Chu FCH, Fornter JF. The role of radiotherapy in the management of malignant hemangiopericytoma: report of eleven new cases and review of the literature. Cancer. 1977;39:1254-1259.
94 Fuki M, Kitamura K, Nakagaki H, et al. Irradiated meningiomas: a clinical evaluation. Acta Neurochir (Wien). 1980;54:33-34.
95 Adegbite AB, Khan MI, Paine KWE, et al. The recurrence of intracranial meningiomas after surgical treatment. J Neurosurg. 1983;58:51-56.
96 King DL, Chang CH, Pook JL. Radiotherapy in the management of meningiomas. Acta Radiol. 1966;5:26-33.
97 Carella RJ, Ransohoff J, Newal J. Role of radiation therapy in the management of meningioma. Neurosurgery. 1982;10:332-339.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 132 Meningeal Sarcoma and Meningeal Hemangiopericytoma
Meningeal Sarcoma
History
Sarcomas are tumors arising from mesenchymal tissue. Sarcoma is derived from the Greek word sar, which denotes a fleshy character related to the gross appearance of these tumors.1 Sarcomatous tumors of the leptomeninges were described as early as 1929.2 Although Virchow had reported CNS sarcomas before this, he actually may have been describing high-grade gliomas.3 In 1953, Christensen and Lara reported 24 cases of intracranial sarcoma, which they grouped into three distinct entities: fibrous sarcoma, spindle cell sarcoma, and polymorphocellular sarcoma.4 The distinctions were based on light microscopy findings. This system is not part of the 2007 World Health Organization (WHO) tumor classification scheme, but it did have prognostic significance. Median survival for these three subtypes is quite different: fibrous, 74 months; spindle cell, 27 months; and polymorphocellular, less than 1 year. The 2007 WHO classification of tumors of the CNS separates intracranial sarcomas according to their pattern of differentiation rather than their cell of origin (Table 132-1).5
Adapted from Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
Clinical Significance
Sarcoma progenitors cells are derived from mesenchymal tissue. The nervous system has multiple tissues of mesenchymal origin and thus may serve as the origin for sarcomas: dura, pia-arachnoid, stroma of the choroid plexus, adventitial fibroblasts associated with blood vessels, and tela choroidea.3,6 Intracranial sarcomas may arise in continuity with the meninges or as intraparenchymal masses without a dural base when the origin is from blood vessels, the tela choroidea, or the choroid plexus stroma.
One can classify intracranial sarcomatous lesions into seven distinct groups7,8:
Lesions in the last group are not true sarcomas but do share some common histologic findings. The first three entities are discussed in this chapter. Gliosarcoma is classified as a subtype of glioblastoma, an astrocytic tumor of neuroepithelial origin.5 The entity sarcoglioma, or the development of a glial neoplasm associated with a primary intracranial sarcoma, is discussed later. Sarcomatous metastases are similar in clinical behavior to other brain metastases and are discussed elsewhere. It is important to note that these metastases are histologically indistinguishable from primary CNS sarcomas, although new molecular techniques may enable us to elucidate the etiology of these lesions. Given the rarity of primary intracranial sarcomas, the finding of sarcomatous tissue in the brain should lead to a thorough search for a primary source outside the CNS. Scalp and skull tumors are discussed in separate chapters of this text.
Sarcomas can occur at any age. They account for just 0.1% to 3% of all intracranial tumors in both adult and pediatric series.7,9 The less well-differentiated sarcomas (Christensen and Lara’s polymorphocellular sarcomas) usually occur in infants and young children. Meningeal sarcomatosis, or diffuse infiltration of the dura by sarcomatous cells in the absence of a clear mass, also tends to occur in very young children. Most authors have reported that the incidence is equal in males and females.7,10 Sarcomas can occur at any site in the brain. In Paulus and coworkers’ series, 12 of 19 tumors (63%) were in direct contact with the dura.7 The remainder arose within the parenchyma of the brain, presumably from the mesenchymal tissue associated with blood vessels, the tela choroidea, or the choroid plexus. With the exception of rhabdomyosarcomas, which reportedly occur more frequently in the posterior fossa11 and in the midline,12 sarcomas occur with equal likelihood throughout the cranium.12
Pathology and Pathogenesis
The most recent revision of the WHO classification of brain tumors includes an expanded section on the wide range of mesenchymal tumors occurring in the CNS. The committee based this classification on the system for soft tissue tumors occurring outside the CNS.13 Differentiation of sarcomas by pathology is based on light microscopy (such as islands of cartilage in chondrosarcoma and osseous areas in osteosarcoma), immunohistochemistry (desmin and myoglobulin positivity in rhabdomyosarcoma), or ultrastructural features (Z bands in rhabdomyosarcoma).
The macroscopic appearance of these lesions is heterogeneous, but they tend to be large. Those arising from the dura are often firm, whereas those arising without clear dural attachment are often softer. In contrast to gliomas, sarcomas tend to be more distinct, with clearer macroscopic borders between the tumor and surrounding brain. They also tend to be firmer than gliomas. Hemorrhage, cysts, and necrosis can be seen within sarcomas.11,12,14
Angiosarcoma
These rare tumors constitute less than 1% of all sarcomas and usually arise in the skin or superficial soft tissues.11 Occasionally, they arise from intracranial vessels. They typically have a spongy appearance because of abundant vessels. In more differentiated areas of the tumor, vascular channels may be seen. The basement membrane of the vessels can be demonstrated with a reticulum stain. Brain invasion is frequently seen with these lesions despite a circumscribed radiographic appearance.11
Chondrosarcoma
These tumors arise from the dura, from the skull base, or rarely, from intraparenchymal sources.13 Chondrosarcomas arising in the fourth ventricle15 or in the falx16 have been reported. Some of these tumors exhibit a lower grade of malignancy than other sarcomas, but local recurrence and local aggressive behavior are still seen frequently.12 Histologically, the cartilage component is less well developed, and the tumors are more cellular and pleomorphic than chondromas.14 Intracranial chondrosarcomas represent 0.15% of all primary intracranial lesions.14
Mesenchymal Chondrosarcoma
These lesions are usually attached to the dura and may occur in either the cranial vault or the spinal canal. They are often found in the first or second year of life. Mesenchymal chondrosarcomas exhibit a pattern of pale staining and atypical chondroid areas alternating with compact cellular aggregates.12 Other histologic characteristics include thin-walled vascular spaces and limited areas of bone formation. These sarcomas have a moderate tendency to recur locally9 and may metastasize distantly.17
Rhabdomyosarcoma
These malignant tumors are composed of rhabdomyoblasts. They can arise either in the brain parenchyma or from the leptomeninges. They occur more frequently in children, often in the posterior fossa,7,11,13 where they can be difficult to differentiate from medullomyoblastoma. Rhabdomyosarcoma can also be seen as part of germ cell tumors of the pineal and suprasellar region.13 Histologically, these tumors are highly cellular and have myoblastic components. The diagnosis is aided by immunohistochemistry for myoglobin, myosin, muscle-specific actin, and desmin.13 Ultrastructure observations include the presence of Z bands and disorganized thick and thin filaments.13
Fibrosarcoma
Fibrosarcomas are the most common intracranial sarcomas (Fig. 132-1)11–13 and are more frequently seen in adults.7 They develop as a component of gliosarcoma, after radiotherapy (particularly treatment of the sella for pituitary adenoma), or as a primary intracranial sarcoma. These tumors arise from the dura or leptomeningeal infolds and can extend along the Virchow-Robin spaces.13 The tumors appear similar to fibrosarcomas occurring elsewhere in the body. Thus, a careful search for another primary site should be made if the diagnosis of intracranial fibrosarcoma is entertained. Given the higher frequency of gliosarcoma than fibrosarcoma, careful staining of the specimen with glial fibrillary acidic protein should be performed to exclude a glial component and the diagnosis of gliosarcoma.13
It may be difficult to differentiate between a true fibrosarcoma and sarcomatous degeneration of a meningioma. Burger and Scheithauer identified the following as useful methods for making that distinction.13 First, the well-known herringbone appearance of fibrosarcoma is not usually seen after sarcomatous degeneration of a meningioma. Second, meningioma stains for epithelial membrane antigen (EMA), whereas fibrosarcoma does not. Unfortunately, not all malignant meningiomas stain for EMA. Thus, the presence of EMA immunostaining supports the diagnosis of a malignant meningioma, but its absence does not exclude it. Third, a past history of meningioma aids in the diagnosis, as does the presence of whorls or psammoma bodies.
Malignant Fibrous Histiocytoma
This rare intracranial tumor is histologically similar to its extracranial counterpart. Histologic findings include high cellularity, nuclear polymorphism, and varying inflammation.13 These tumors can arise in the dura18 or, less commonly, intraparenchymally.19 In Paulus and coworkers’ series, malignant fibrous histiocytomas were more common in adults.7
Primary Meningeal Osteosarcoma
Primary meningeal osteosarcoma is an extremely rare entity described in the literature only via case reports. Review of the literature demonstrates six cases. These tumors arise from the meninges and are within the differential diagnosis for dural-based lesions.20 They are typically treated by aggressive resection and subsequent radiation therapy.
Other Sarcomas
Primary meningeal sarcomatosis refers to diffuse spread of sarcomatous tissue throughout the meninges without a focal mass.11 Most cases are reported in young children. The tumor in the subarachnoid space covers the surface of the brain and may grow along penetrating vessels into the brain, as well as into the spinal and cranial nerves. Additionally, tumor can surround the spinal cord.9 Histologically, small, round tumor cells are usually seen.
Extraskeletal lesions with histologic findings identical to those of the bony lesions of Ewing’s sarcoma are rare but well described.21–25 The most frequent site involving the nervous system is the epidural space (particularly in the spine), but intraparenchymal lesions have also been described. Distant metastases from these lesions can subsequently be seen.12 It has been suggested that these tumors are a type of the small, round cell tumor derived from neuroendocrine tissue12 rather than from mesenchymal tissue. If so, they are not truly sarcomas. Recent evidence has classified these tumors as central and peripheral primitive neuroectodermal tumors, but they are still classified as mesenchymal tumors in the 2007 classification.
The cause of intracranial sarcomas is often unknown. Sarcoma develops in some patients after radiotherapy for a brain tumor26 and has developed in at least one patient after chemotherapy for a brain tumor.27 Of the seven cases reported by Chang and colleagues, four were fibrosarcomas and three were malignant fibrous histiocytomas. Radiation doses ranged from 1600 to 6000 Gy.26 The median time to diagnosis of the tumor was 8 years after radiation therapy. Sarcomas have been reported most frequently after irradiation of the sella for pituitary adenoma.8,28 The median time to development of sarcoma was 10 years, the minimum dose was 2000 Gy, and multiple courses of radiation increased the risk. Histologically, there may be evidence of recurrent adenoma within the sarcoma.13,23
Clinical Findings and Evaluation
The clinical features of patients with meningeal sarcomas are similar to those of other patients with malignant intracranial tumors. Symptoms are due to mass effect from the tumor or peritumoral edema (Fig. 132-2). Clinical symptoms include headaches, seizures, weakness, mental status changes, or symptoms of hydrocephalus (Table 132-2). Patients seen in this fashion are usually stabilized with therapy for increased intracranial pressure, such as steroids. If the tumor erodes through the skull into the soft tissues of the scalp, it may be manifested as a palpable mass (Fig. 132-3). Imaging should be obtained immediately.
TABLE 132-2 Symptoms of Dural-Based Lesions Vary According to Location
| LOCATION | SYMPTOMS |
|---|---|
| Frontal/parietal | Affective, hemiparesis, neglect, sensory disturbance, seizures; may be asymptomatic |
| Temporal | Seizures, speech disturbance |
| Frontal fossa | Behavioral disorder, olfactory loss |
| Sellar/parasellar | Visual loss, pituitary disturbance |
| Sphenoid wing | Visual disturbance, psychiatric disturbance, seizures, speech disorders |
| Cavernous sinus | Eye movement disturbance, facial pain or numbness |
| Posterior fossa | Ataxia, hydrocephalus, cranial nerve dysfunction |
Intracranial sarcomas have no typical imaging features. When they arise from the dura, they may simulate the more common meningioma (see Fig. 132-1). The presence of bone erosion overlying the mass suggests a more malignant tumor, but this finding may also be seen with typical and atypical meningioma. Extension into the scalp is more common with sarcoma than with meningioma. Underlying edema is also nearly universal with sarcomas but may also be seen in up to 50% of typical meningiomas. Invasion of the superficial venous structures may occur with both types of tumors. Although most adult CNS sarcomas are extra-axial masses, sarcomas and meningiomas in children commonly arise as intra-axial or intraventricular masses. Intraparenchymal sarcomas have no specific imaging features.
Imaging
Angiography plays a limited role in the evaluation of these tumors. It is no longer used for tumor localization but may be used to map blood supply to the lesion. In sarcomas, blood supply can be derived from the internal carotid artery, the external carotid artery, or both.1 Hypervascular tumors may benefit from presurgical embolization to decrease their blood supply. Angiography may also show vessel encasement by tumor, but MRI is a better means of visualizing encasement. Sonography has a very limited role in the evaluation of these tumors. It may be used for diagnosis of tumors in utero,29 for screening of newborns with enlarging heads (Fig. 132-4), and as an aid to intraoperative localization of sarcomas arising in the brain parenchyma.
[/not-level-membership-for-neurosurgery-category]
