CHAPTER 11 Membrane Physiology
This chapter describes how pumps, carriers, and channels cooperate in living systems. These three components often work together in circuits or cycles. Pumps establish gradients of ions across membranes (see Chapter 8). Channels regulate membrane permeability to these ions to maintain the electrical potential (see Chapter 10) required for membrane excitability. Carriers use ion gradients as a source of energy to drive transport as well as to do other work (see Chapter 9). Coupling ion fluxes through pumps and carriers to do work is called a chemiosmotic cycle.
Chemiosmotic Cycles
A simple chemiosmotic cycle couples a cation transporting pump to solute transport by a carrier (Fig. 11-1). The membrane could be a plasma membrane or an organelle membrane. The driving reaction is called the primary transport step, indicating an input of energy and, in most cases, some chemical reaction. Other reactions are called secondary transport reactions, indicating that they depend on ion gradients. The transported substrate is the same chemically on both sides of the mem-brane. Although they are simple in concept, the importance and power of chemiosmotic cycles should not be underestimated. They operate in every membrane of every cell.
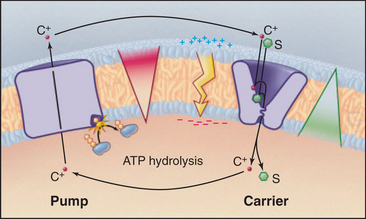
Pumps use energy derived from ATP hydrolysis, light absorption, or another chemical reaction (see Table 8-1) to move ions in one direction across a membrane. This raises the concentration of a cation (C+) on one side and depletes it on the other side of a membrane-bounded compartment. An ion gradient is characterized by both a chemical term, the concentration gradient, and an electrical gradient (the membrane potential explained in Fig. 10-17). The electro-chemical potential across a membrane represents a reservoir of power and a capacity to do work, also known as an ion-motive force. A mechanical analog would be using a pump to fill an elevated reservoir with fluid.
Carriers and other membrane proteins use the potential energy of ion gradients to drive other processes. This is analogous to using fluid flow out of a reservoir to drive a turbine, which puts the energy to use for other types of work. Many carriers use energy derived from the downhill passage of one substrate to transport one or more other substances up their concentration gradients across the same membrane barrier. In Figure 11-1, the carrier links the transport of solute S to the movement of cation C+ down its gradient. Recirculation of cations allows a cell to accumulate solute against its concentration gradient. In addition to the osmotic work illustrated in the figure, chemiosmotic cycles can do chemical work. During both oxidative and photosynthetic phosphorylation, proton cycles drive ATP synthesis by F0F1-ATP synthases (see Fig. 19-5). Chemiosmotic cycles can also perform mechanical work. The electrochemical gradient of protons across the plasma membrane drives rotation of bacterial flagella (see Fig. 38-24).
Epithelial Transport
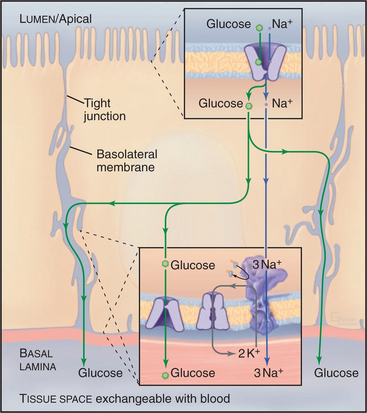
Net transport across an epithelium depends on tight junctions (see Fig. 31-2) that seal the extracellular space between the cells (Fig. 11-2). These junctions separate two extracellular compartments. The apical compartment is the free surface (e.g., the skin) or the lumen of the organ (e.g., the intestine, respiratory tract, or kidney tubules). The basolateral compartment lies between epithelial cells and is continuous with the underlying connective tissue and its blood vessels. Tight junctions seal the extracellular space, inhibiting diffusion of solutes, between the apical and basolateral compartments of the extracellular space. The extent of this seal varies from very tight to leaky. Tight junctions also separate the plasma membrane into apical and basolateral domains, restricting the movement of integral membrane proteins between these domains.
Glucose Transport in the Intestine, Kidney, Fat, and Muscle
A chemiosmotic cycle transports glucose uphill from the lumen of the intestine to the blood (Fig. 11-2). Tight junctions restrict movement of glucose between the epithelial cells, so all of the glucose must move through the cytoplasm. Glucose transport across the epithelial cells requires the following components:
Glucose uptake by fat and muscle cells offers a different perspective. These tissues are designed to take up glucose from the blood when it is plentiful following a meal. Mammals have genes for six isoforms of the classical D-glucose uniporter. Insulin in the blood regulates the availability of the GLUT4 isoform in the plasma membrane. Muscle and fat express GLUT4 but store it internally in membrane vesicles. After a meal, high blood glucose stimulates secretion of insulin into blood. Signal transduction mechanisms (see Fig. 27-7) lead to fusion of these GLUT4 vesicles with the plasma membrane. That increases the rate of glucose transport into fat and muscle by 5-fold to 20-fold, lowering the blood glucose concentration and providing these cells with glucose, which they then convert to glycogen and triglycerides for storage.
Salt and Water Transport in the Kidney
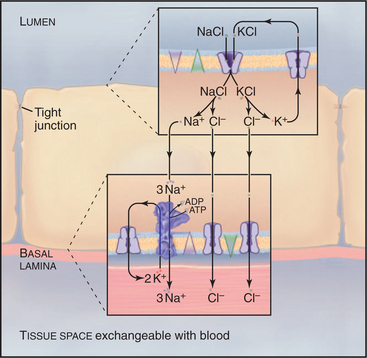
In a section of the kidney tubule called the loop of Henle, the epithelium uses Na+K+-ATPase pumps and Na+/K+/2Cl− symporters to reabsorb NaCl that is filtered from blood into the excretory pathway (Fig. 11-3). Without this provision, salt would be lost in urine. Tight junctions seal this epithelium, so that salt must pass through the cells to return to the blood. Na+/K+/2Cl− symporters in the apical plasma membrane provide a way for NaCl to enter the cell down its concentration gradient. Abundant Na+K+-ATPases in the basolateral plasma membrane (5000/mm2) create a Na+ gradient to drive the symporter and to clear the cytoplasm of Na+ accumulated from the tubule lumen. KCl that enters with Na+ through the Na+/K+/2Cl− symporter leaves the cell through channels: K+ channels in apical and basolateral membranes and Cl− channels in basolateral membranes.
Cystic Fibrosis as a Transporter Disease
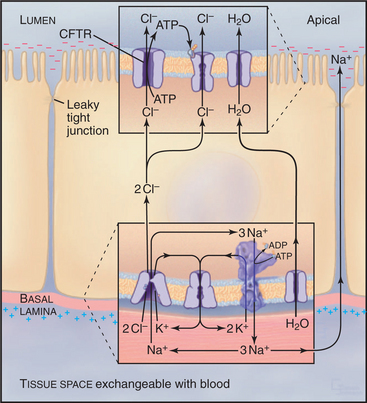
Normally, cells in the lung and gastrointestinal tract use a complicated selection of familiar pumps and carriers to secrete salt and water at their apical sur-faces (Fig. 11-4). Na+K+-ATPases in the basolateral membrane set up an electrochemical gradient of Na+, which is exploited by basolateral membrane Na+/K+/2Cl− symporters to take in Na+, along with K+ and Cl− anions. The inward movement of Na+ down its electrochemical gradient drives the entry of K+ and Cl− up their gradients. This brings excess potassium chloride into the cell. (The K+ brought in by both the Na+K+-ATPase and the Na+/K+/2Cl− symporter recycles after exiting from the cell by way of channels in the basolateral plasma membrane. Thus, K+ is merely catalytic.) Excess Cl− is left inside the cell. The cystic fibrosis transmembrane regulator (CFTR) protein, an ABC pump, in the apical plasma membrane acts as a Cl− channel. When protein kinase phosphorylates the regulatory domain and ATP binds to the cytoplasmic domains, a conformational change opens a Cl− channel across the membrane. Cl− moves down its electrochemical gradient out of the cell, carrying charge to the outside. The whole epithelium becomes polarized, with the lumen electrically negative relative to the extracellular fluid compartment. This electrical driving force allows Na+ to move between cells, from the extracellular fluid compartment through leaky tight junctions to the surface of the epithelium. Sodium chloride on the apical surface creates an osmotic force that draws water down its concentration gradient across the cells to the outside through water channels (see Fig. 10-15). CFTR also appears to inhibit the transport mechanisms that reabsorb fluid from the lumen of the epithelium. A balance between this fluid secretion and fluid reabsorption normally keeps the surface of the epithelium properly hydrated, allowing the cilia to clear the lung of bacteria and secretions and the ducts of the pancreas to secrete digestive enzymes.
Cellular Volume Regulation
Cells employ both short- and long-term strategies involving pumps, carriers, and channels to maintain a constant volume (Fig. 11-5). These compensatory mechanisms are required because water moves across the plasma membrane through water channels and slowly through lipid bilayers if the osmotic strength of the environment differs even slightly from that inside the cell. Water moves to maintain an osmotic equilibrium, as is illustrated for red blood cells in Figure 7-6. In a hypotonic medium, water moves into a cell to dilute the cytoplasm. In a hypertonic medium, water moves out to concentrate the cytoplasm. Mechanisms that are employed to compensate for these volume changes are well defined, but the mechanisms that sense volume changes and trigger these responses are still being investigated.
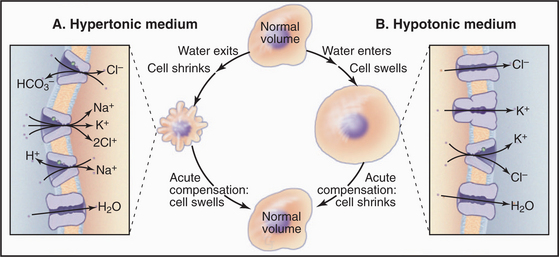
Animal cells respond acutely to loss of water by activating Na+/H+ antiporters, Cl−/HCO3− antiporters, and/or Na+/K+/2Cl− symporters that bring potassium chloride and sodium chloride into the cell. Water follows, returning the cell to its original volume in minutes. The acute response to swelling activates K+ channels, Cl− channels (ClC-3), and/or a K+/Cl− symporter, taking potassium chloride and water out of the cell. Compensation by moving inorganic ions works in the short run but is not an acceptable long-term solution because changes in the internal concentrations of K+, Na+, and Cl− affect the membrane potential and other physiological processes.
Excitable Membranes
Electrical excitability is not limited to nerves and muscles. Eggs use a form of action potential as an early step in blocking fertilization by more than one sperm. Chemotaxis by macrophages and secretion of insulin and other hormones both depend on electrical excitability. The reader should be familiar with the appendixes in Chapter 10 to appreciate the following material.
Description of an Action Potential
If a microelectrode (see Fig. 10-16A) drives a small positive or negative current into a cell, a second microelectrode a short distance away detects a small voltage response. These electrotonic potentials decline rapidly with distance if the cell in nonexcitable.
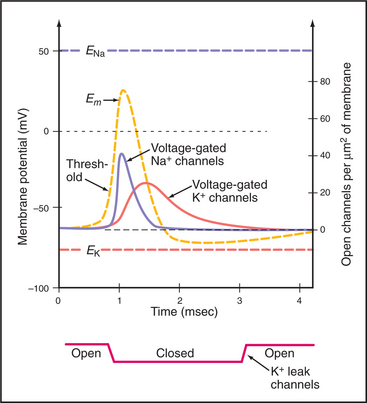
In striking contrast to these small local currents, when the plasma membrane of an excitable cell, such as neuron or muscle, is depolarized beyond a certain level, called a threshold, the membrane responds over a few milliseconds with a large, stereotyped change in membrane potential, called an action potential (Fig. 11-6). Voltage-gated ion channels (see Fig. 10-7) generate this powerful electrical signal that spreads rapidly (10 m/sec) over the entire plasma membrane. During an action potential, the membrane potential can reach a peak of +40 to 50 mV before repolarizing to the resting potential. Because action potentials are self-triggering, they travel without dissipation over long distances. This high-speed transmission is very efficient, requiring movement of very few ions across the membrane.
The molecular events during an action potential were first characterized around 1950 in squid giant axons using microelectrodes coupled to an electronic feedback circuit. This clever “voltage clamp” holds the membrane potential constant by providing the cell with electrical current to compensate for changes in ion currents. Investigators discovered that changes in permeability to Na+ and K+ ions produced action potentials. Changing one variable at a time, they determined the time and voltage dependence of ion-specific conductance. They also determined the relationship between conductance and voltage. From these relationships, measured under controlled conditions, they could calculate the membrane response to virtually any experimental condition. To explain these changes in permeability, they postulated the existence of ion channels. The voltage clamp provided a direct measure of this channel activity. This approach also revealed the behavior of channels held at a potential more positive than their resting potential.
Three Channels Generating Action Potentials
Voltage-gated Na+ and K+ channels open and close in sequence to produce action potentials. Depending on the type of open channel, the membrane potential varies in time between the K+ equilibrium potential (EK) and the Na+ equilibrium potential (ENa) (see Fig. 10-18). Because membrane depolarization activates these ion channels, and because the response spreads this depolarization, triggering an action potential initiates a cascade of reactions that moves over the membrane, first to depolarize and then to repolarize the membrane. In nerves, just three types of voltage-gated channels are required to generate action potentials:
The properties of these channels explain the time course of an action potential as follows:
During an action potential, the membrane voltage changes by 100 to 150 mV in 1 to 2 msec. The membrane bilayer is approximately 7 nm thick, so this voltage corresponds to a field variation on the order of 150,000 volts/cm in 1 to 2 msec. Such strong forces elicit conformational changes in membrane proteins, such as voltage-gated ion channels.
Synaptic Transmission
Most neurons use chemical messengers called neurotransmitters (Fig. 11-7) to communicate rapidly with each other and with effector cells, such as skeletal muscle and glands. This chemical communication occurs at sites called synapses (Figs. 11-8 and 11-9), where the sending cell is specialized to secrete a particular neurotransmitter and the receiving cell is specialized to respond to that neurotransmitter. The sending side of a synapse is referred to as presynaptic, whereas the receiving side is designated postsynaptic. Small vesicles containing neurotransmitter pack the presynaptic nerve terminal. Neurotransmitter receptors concentrate in the postsynaptic plasma membrane. Modest changes in either the presynaptic release of neurotransmitter or postsynaptic receptor activation can profoundly influence how a neuron processes this information. Analysis of synaptic transmission has revealed much about the mechanisms of secretion (see Chapter 22), signal transduction, and psychoactive drugs that affect behavior. Not all synapses use chemical transmitters. In special cases, gap junctions (see Fig. 31-6) connect neurons at “electrical synapses,” where current moves directly between the two cells.
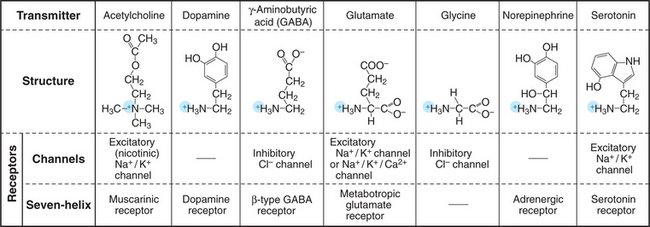
Neurotransmitters are generally small organic molecules with an amino group. These include acetylcholine, norepinephrine, 5-hydroxytryptamine (serotonin), and the amino acids glycine and glutamic acid (Fig. 11-7). Secretory mechanisms are similar at all synapses, but each neurotransmitter requires its own biochemical machinery for synthesis, packaging in synaptic vesicles, and reception by postsynaptic cells. Such distinctive features of synapses using a particular transmitter make it possible to modify synaptic transmission selectively, such as in treatment with psychoactive drugs.
In addition to activating ligand-gated ion channels, most neurotransmitters also stimulate particular seven-helix receptors (Fig. 11-7; see also Fig. 24-3). For example, acetylcholine stimulates the seven-helix muscarinic acetylcholine receptor, which uses a trimeric G-protein intermediary to activate Kir3.1 K+ channels (Fig. 11-12). Glutamate stimulates seven-helix “metabotropic” receptors, which also act through trimeric G proteins. Disruption of the gene for metabotropic glutamate receptors leaves mice with defects in coordination and learning, and overstimulation of these receptors might contribute to some forms of mental retardation in humans.
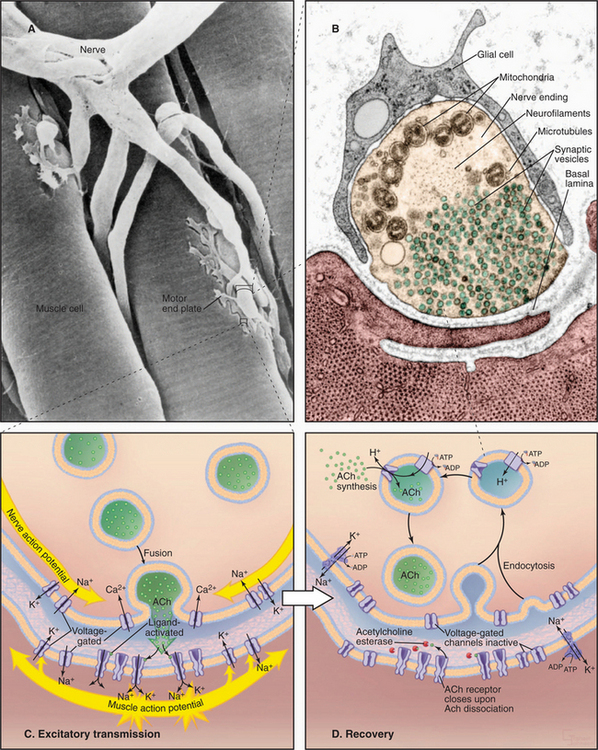
Neuromuscular Junction
Motor neurons in the spinal cord and brainstem control contraction of skeletal muscle cells (see Fig. 39-14). Long axons from these neurons terminate in synapses on skeletal muscle cells, called neuromuscular junctions (Fig. 11-8A–B). Every neuronal action potential that reaches a neuromuscular junction evokes an action potential that spreads over the postsynaptic surface of the muscle cell and initiates contraction. This highly reliable, one-to-one communication depends on chemical transmission by acetylcholine between the nerve and muscle. Highly concentrated nicotinic acetylcholine receptors in the postsynaptic membrane (˜20,000/mm2) transduce the arrival of extracellular acetylcholine into membrane depolarization.
Figure 11-8C illustrates the membrane proteins required for neuromuscular transmission. Both the nerve terminal and muscle depend on Na+K+-ATPase and Ca2+-ATPase pumps to maintain gradients of Na+, K+, and Ca2+ across their plasma membranes. Both presynaptic and postsynaptic cells need voltage-gated Na+ channels and K+ channels for action potentials. Additionally, the presynaptic membrane requires voltage-gated Ca2+ channels to trigger secretion of acetylcholine.
Weak but cooperative binding of acetylcholine to two subunits of the acetylcholine receptor (see Fig. 10-12) opens a nonselective cation channel. The open pore is about equally permeable to K+ and Na+ and less permeable to Ca2+, so the membrane potential collapses toward a reversal potential (see the section titled “Consequence of Multiple Channel Types Opening Simultaneously”) of about 0 mV. This is above threshold for triggering a self-propagating action potential in the muscle plasma membrane, which occurs with nearly 100% efficiency. The action potential traveling over the muscle plasma membrane activates voltage-sensitive Ca2+ channels that trigger Ca2+ release from smooth endoplasmic reticulum, resulting in contraction (see Fig. 39-15).
Nerve terminals retrieve synaptic vesicle membrane by endocytosis (see Chapter 22). Cytoplasmic enzymes synthesize new acetylcholine. A V-type ATPase proton pump (see Fig. 8-5C) acidifies the lumen of synaptic vesicles, providing an electrochemical potential to drive an acetylcholine/H+ antiporter, which concentrates acetylcholine in vesicles.
Central Nervous System Synapses
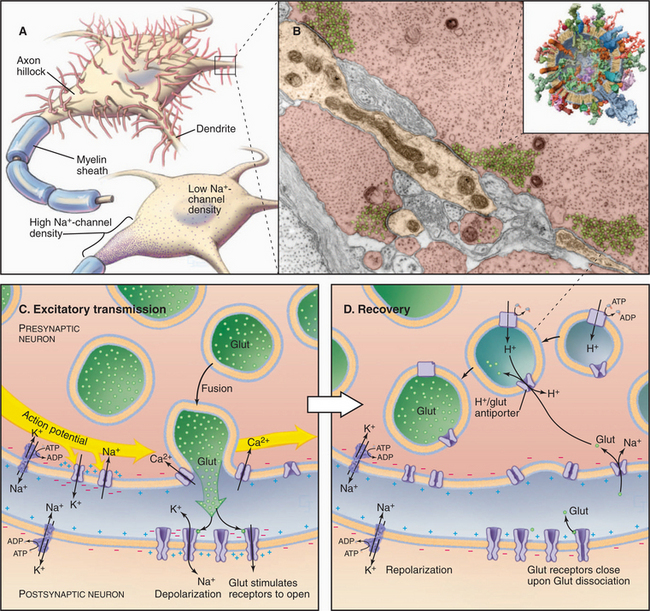
Synaptic transmission between neurons in the CNS (Fig. 11-9) differs fundamentally from the efficient, one-to-one coupling at neuromuscular junctions, where every presynaptic action potential triggers a postsynaptic action potential. The approximately 100 billion (1011) neurons in the human brain receive synaptic inputs from many neurons, forming about 1015 synapses. Synapses cover the surface of dendrites and the cell body (Fig. 11-9A). Some synapses excite the postsynaptic cell by opening ligand-gated cation channels that depolarize the membrane locally. Such small, local changes tend to push the membrane toward threshold for an action potential. Other synapses are inhibitory, hyperpolarizing the postsynaptic membrane locally by opening ligand-gated Cl− channels. These changes are inhibitory, since they drive the membrane potential away from threshold (see Fig. 10-18). From moment to moment, neurons spatially average excitatory and inhibitory stimuli and fire action potentials when the combined effects of these opposing stimuli exceed threshold potential in the proximal part of the axon, called the axon hillock. Both the pattern and frequency of action potentials carry information in the brain.
Transmission at chemical synapses in the CNS depends on cooperation of pumps, carriers, and channels (Fig. 11-9C–D). ATPase pumps maintain concentration gradients of Na+, K+, and Ca2+ across both presynaptic and postsynaptic plasma membranes. Potassium channels establish the resting membrane potential, and voltage-gated K+ and Na+ channels fire action potentials. Neurotransmitters secreted by the presynaptic cell activate ligand-gated channels that control the postsynaptic membrane potential. Carriers in the presynaptic membrane and adjacent supporting cells terminate transmission by removing neurotransmitter from the synaptic cleft.
Transmitters activate ligand-gated channels that cause a local, short-lived change in membrane potential, called a postsynaptic potential (PSP). At excitatory synapses, the neurotransmitter glutamate activates receptors (see Fig. 10-11) that open cation channels that depolarize the membrane. However, in contrast to the neuromuscular junction, individual PSPs do not fire action potentials. First, individual PSPs raise the membrane potential only a few millivolts, so they do not bring the postsynaptic membrane to threshold. Second, dendritic and cell body plasma membranes contain few voltage-gated Na+ channels. Furthermore, inhibitory synapses on the same cell counteract excitatory synapses by secreting glycine or γ-aminobutyric acid (GABA) to activate Cl− channels that hyperpolarize the membrane, taking it farther from threshold.
Excitatory and inhibitory PSPs spread passively over the postsynaptic membrane and generate an action potential only when their sum at a particular time brings the membrane potential at the axon hillock to threshold (Fig. 11-9A). The axon hillock is located at the base of each axon. This part of the plasma membrane is particularly sensitive to voltage, owing to a high concentration of voltage-gated Na+ channels. At threshold, they open, depolarizing the membrane (Fig. 11-6). Delayed-rectifier K+ channels then repolarize the membrane in preparation for subsequent action potentials. Each action potential is identical and propagates down the axon.
Removal of neurotransmitters from the synaptic cleft terminates activation of postsynaptic receptors (Fig. 11-8D). Na+K+-ATPase pumps provide a Na+ gradient to drive symporters that return neurotransmitters to their presynaptic cells. Within the presynaptic cell, a second proton-driven chemiosmotic cycle concentrates transmitter in synaptic vesicles. A V-type, proton-translocating ATPase acidifies the lumen of the synaptic vesicle and establishes the proton electrochemical gradient across the vesicle membrane to drive the antiporter.
Modification of CNS Synapses by Drugs and Disease
Acetylcholine secreted by neurons and nicotine from tobacco modulate synaptic transmission in the CNS by activating the same neurotransmitter receptor. In the CNS, acetylcholine acts on presynaptic terminals rather than participating directly in fast synaptic transmission as it does at the neuromuscular junction. Nicotinic acetylcholine receptors in the presynaptic plasma membrane are highly permeable to Ca2+, so their stimulation admits Ca2+ into the presynaptic terminal. This enhances both the spontaneous release of neurotransmitter and release in response to action potentials. The isoform composition of CNS acetylcholine receptors differs from that of muscles (see Fig. 10-12). Some are homopentamers of α-subunits. Others are heteropentamers of α- and β-subunits. Activation of these ligand-gated channels in different regions of the brain may account for the enhancing effects of nicotine on learning and memory but also for tobacco addiction. Loss of CNS neurons that secrete acetylcholine might contribute to dementia in Alzheimer’s disease.
Modification of CNS Synapses by Use
Memories are thought to be laid down in structural changes that modify the strength or numbers of synapses between neurons in the brain. Particular patterns of stimulation can produce long-term changes that enhance or reduce the efficiency of transmission of various glutamate-mediated synapses (Fig. 11-10). The hippocampus, a region of the vertebrate cerebral cortex that is known to participate in some forms of learning and memory, is favorable for observing a simple form of cellular learning. Intense stimulation of excitatory glutamate synapses (20 pulses over a period of 200 msec) can increase synaptic strength for days or weeks. This is called long-term potentiation (LTP). Conversely, slow, prolonged stimulation of glutamate synapses reduces the response for hours. This is called long-term depression (LTD). The mechanisms of LTP and LTD are under intense investigation, because high-order brain functions, such as learning and memory, depend on changes in the flow of impulses through neural circuits, and the changes in transmission during LTP and LTD occur on an appropriate time scale.
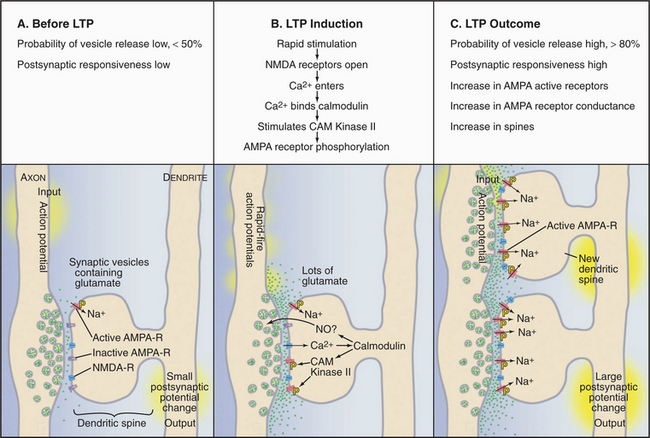
Induction of LTP typically involves two types of glutamate receptors (see Fig. 10-11): AMPA receptors and NMDA receptors found in postsynaptic specializations called dendritic spines (Fig. 11-10). AMPA receptors open and close rapidly in response to glutamate. When open, AMPA receptors admit Na+ and NMDA receptors admit Ca2+ to depolarize the postsynaptic plasma membrane. The slow response of NMDA receptors to glutamate depends on the membrane potential, as partial depolarization is required to displace an extracellular Mg2+ ion blocking the channel. This dual dependence on glutamate and membrane potential makes NMDA receptors coincidence detectors, responsive to rapid stimulation or stimulation at nearby excitatory synapses. A synapse with only NMDA receptors is functionally “silent.” Such silent synapses can be aroused when presynaptic release of glutamate is coordinated with sufficient membrane depolarization from neighboring synaptic activation. This results in the insertion of AMPA receptors, waking up the synapse.
In principle, LTP and LTD might alter the efficiency of synaptic transmission by changing glutamate release from the presynaptic cell or responsiveness of the postsynaptic cell to glutamate. In fact, a wide range of experiments suggests that both presynaptic and postsynaptic processes contribute. The best-documented presynaptic change is an increase in the probability that an action potential will stimulate the fusion of a glutamate-containing synaptic vesicle with the plasma membrane. In the resting state, exocytosis of these vesicles is unreliable. LTP increases the probability of exocytosis from less than 0.5 to greater than 0.8. In addition, the postsynaptic side responds more robustly to glutamate for two reasons: a higher concentration of active AMPA receptors with enhanced conductance when open and formation of additional synapses with the stimulating axon.
The mechanisms that bring about these changes are incompletely understood, but the following is well established. LTP depends on stimulation of NMDA receptors and Ca2+ entry. Within the postsynaptic dendritic spine, Ca2+ activates processes that initiate and maintain LTP. Within seconds, Ca2+ binds calmodulin (see Fig. 3-12C) and triggers events that depend on calcium-calmodulin, including activation of protein kinases, such as CAM-kinase II (see Fig. 25-4A). Phosphorylation of AMPA receptors by CAM-kinase II increases their responsiveness to glutamate, perhaps “waking up silent synapses.” AMPA receptors divide their time between the postsynaptic membrane and intracellular recycling endosomes. LTP shifts more AMPA receptors from endosomes to the postsynaptic membrane. No consensus has been reached on the nature of the extracellular messengers that provide feedback to the presynaptic terminal to modify its exocytosis efficiency.
Within minutes, induction of LTP triggers signaling cascades that maintain the increased efficacy, leading to structural changes and increased protein synthesis. These changes may induce dendrites to stabilized existing spines or sprout new filopodia and spines; these are presumed to account for the increased number of synapses observed after an hour or so. Extension of these processes and remodeling of the shape of dendritic spines depend on actin filament assembly (see Fig. 38-8). Growth of axons and formation of new synapses provide mechanisms to generate novel connections in response to use. Over the longer term, the postsynaptic cell initiates gene transcription and protein synthesis, bringing about further changes that stabilize enhanced synaptic transmission.
Cardiac Membrane Physiology
Spontaneous Action Potentials of Pacemaker Cells
Intrinsically excitable pacemaker cells in the sinoatrial node drive rhythmic contractions of the heart (see Fig. 39-19). The membrane potential of these cells drifts spontaneously toward threshold, setting off action potentials about once each second (Fig. 11-11). Cardiac action potentials spread via gap junctions (see Fig. 31-6) from cell to cell throughout the heart, activating contraction in a reproducible pattern.
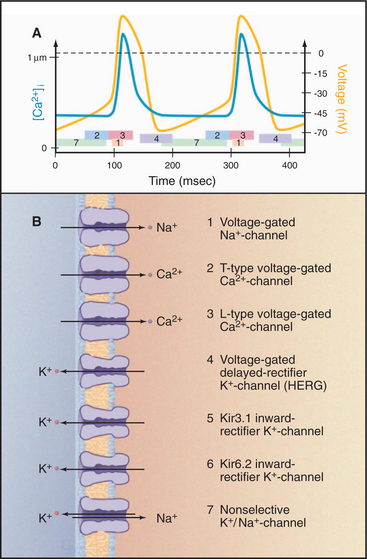
Acting together, these channels produce a spontaneous cycle of pacemaker action potentials. At the threshold potential (about -40 mV), voltage-gated Na+ channels open synchronously and rapidly depolarize the membrane. As they inactivate, L-type Ca2+ channels open, prolonging the depolarization and admitting Ca2+; this, in turn, triggers contraction by releasing more Ca2+ from internal stores (see Fig. 39-15B). As these Ca2+ channels slowly inactivate, delayed-rectifier K+ channels open and drive the membrane potential toward EK, the K+ equilibrium potential. As the membrane potential reaches a minimum, delayed-rectifier K+ channels inactivate, but the two Kir channels open. In the absence of other channel activity, the membrane potential would remain near EK, but the nonselective Na+/K+ channels open and the membrane slowly depolarizes, drifting toward threshold. T-type Ca2+ channels contribute to the slow, spontaneous depolarization. At threshold, the cycle repeats.
Regulation of Heart Rate by G Proteins and Phosphorylation
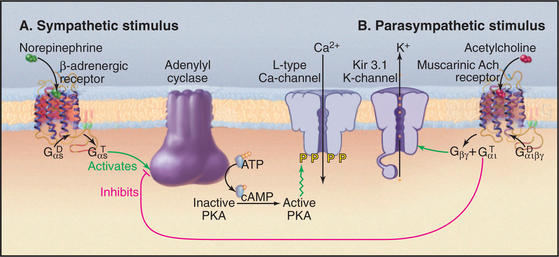
Regulation of pacemaker cells by neurotransmitters secreted by autonomic nerves is an example of the widespread regulation of channels by G proteins and phosphorylation (Fig. 11-12). Neurotransmitters from the two parts of the autonomic nervous system have opposite effects on the frequency of cardiac contraction. The resting rate reflects a compromise in the competition between these two inputs. Acetylcholine from parasympathetic nerves slows the heartbeat, whereas norepinephrine from sympathetic nerves speeds the rate and increases the strength of contraction. These neurotransmitters modify their target channels indirectly by activating two different seven-helix receptors and their associated trimeric G-proteins (see Fig. 25-9).
Norepinephrine increases the heart rate by modulating L-type Ca2+ channels. Norepinephrine that binds to plasma membrane β-adrenergic receptors activates trimeric G proteins, which stimulate adenylyl cyclase, the enzyme that makes cyclic adenosine monophosphate (cAMP) (see Fig. 26-2). This second messenger stimulates cyclic AMP–dependent protein kinase (see Fig. 25-3) to phosphorylate cytoplasmic residues of L-type, voltage-gated Ca2+ channels in the plasma membrane. Phosphorylated Ca2+ channels are more likely to open in response to membrane depolarization than are unphosphorylated channels. Phosphorylation increases the rate at which the membrane potential drifts toward threshold. This increases the frequency of action potentials of pacemaker cells and the heart rate. In a parallel pathway, cAMP stimulates HCN cation channels, which push the membrane potential toward threshold and increase the heart rate.
Regulation of Cardiac Contractility
A set of channels similar to those in the sinoatrial node generate action potentials in cardiac muscle cells and stimulate contraction. Cardiac muscle cells can generate spontaneous action potentials, but they have fewer T-type Ca2+ channels and more Kir K+ channels, so the rate of spontaneous action potentials is lower than that of pacemaker cells. Except in disease, pacemaker cells drive action potentials throughout the rest of the heart. Sympathetic nervous stimulation, acting through cyclic AMP–dependent protein kinase, strengthens cardiac contraction. Phosphorylated L-type Ca2+ channels admit more Ca2+ to activate the contractile machinery more fully. The same kinase activates delayed-rectifier K+ channels, which prevent activated Ca2+ channels from prolonging the action potential. This allows heart muscle cells to keep up with stimuli generated at a higher rate from pacemaker cells. Cyclic AMP–dependent protein kinase also enhances contractility by phosphorylating proteins of the contractile apparatus and smooth endoplasmic reticulum (see Chapter 39).
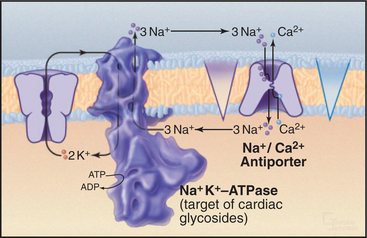
Therapeutic Effect of Digitalis in Congestive Heart Failure
In congestive heart failure, cardiac contraction fails to produce enough force to maintain adequate circulation of blood. Cardiac glycosides, such as digitalis (from the foxglove plant), ameliorate this common human condition by inhibiting an isoform of Na+K+ATPase in the plasma membrane of heart cells (Fig. 11-13). Plasma membrane L-type Ca2+ channels and endoplasmic reticulum Ca2+ release channels activate contraction by transiently increasing cytoplasmic Ca2+. Calcium ATPase pumps (see Fig. 8-8) in smooth endoplasmic reticulum clear most of this Ca2+ from cytoplasm, but plasma membrane Na+/Ca2+ antiporters, driven by the plasma membrane Na+ gradient, contribute as well.
Chklovskii DB, Mel BW, Svoboda K. Cortical rewiring and information storage. Nature. 2004;431:782-788.
Cohen-Cory S. The developing synapse: Construction and modulation of synaptic structures and circuits. Science. 2002;298:770-776.
Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1-105.
Dorwart M, Thibodeau P, Thomas P. Cystic fibrosis: Recent structural insights. J Cystic Fibrosis. 2004;3:91-94.
Fernández-Alfonso T, Ryan TA. The efficiency of the synaptic vesicle cycle at central nervous system synapses. Trends Cell Biol. 2006;16:413-420.
Hogg RC, Bertrand D. What genes tell us about nicotine addiction. Science. 2004;306:983-984.
Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569-580.
Malenka RC, Bear MF. LTP and LDP: An embarrassment of riches. Neuron. 2004;44:5-21.
Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103-126.
Record MT, Courtenay ES, Cayley DS, Guttman HJ. Responses of E. coli to osmotic stress: Large changes in amounts of cytoplasmic solutes and water. Trends Biochem Sci. 1998;23:143-150.
Schuske K, Jorgensen EM. Vesicular glutamate transporter: Shooting blanks. Science. 2004;304:1750-1752.
Severs NJ. The cardiac muscle cell. Bioessays. 2000;22:188-199.
Strange K. Cellular volume homeostasis. Adv Physiol Educ. 2004;28:155-159.
Vankeerberghen A, Cuppens H, Cassiman J-J. The cystic fibrosis transmembrane conductance regulator: An intriguing protein with pleiotropic functions. J Cystic Fibrosis. 2002;1:13-29.
Vincent GM. The long QT syndrome: Bedside to bench to bedside. New Engl J Med. 2003;348:1837-1838.
Zagotta WN, Olivier NB, Black KD, et al. Structural basis of modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200-205.