Medullary Thyroid Carcinoma and Multiple Endocrine Neoplasia Type 2
Molecular Pathogenesis: RET Proto-Oncogene
Clinical Relevance of Germline RET Mutations in MEN2A, FMTC, and MEN2B
Specific Genotype/Phenotype Correlations
Clinical Presentation of MTC and MEN2
Management Implications of Genetic Results
The Role of Somatic RET Mutations
Natural Progression and Behavior of MTC
Prevalence and Epidemiology
Medullary thyroid carcinoma (MTC) accounts for only 5% to 10% of all thyroid cancers, but its management can be very challenging, and early diagnosis is of vital importance. MTC arises from the calcitonin-secreting parafollicular cells of the thyroid known as the C cells. Seventy-five percent of MTC cases are sporadic, while the remaining 25% occur in several well-defined familial endocrine syndromes with autosomal dominant inheritance. Multiple endocrine neoplasia type 2A (MEN2A) refers to the occurrence of MTC in association with pheochromocytoma in 50% of cases, and in association with hyperparathyroidism in 20% to 30% of cases.1 Multiple endocrine neoplasia type 2B (MEN2B) consists of MTC in conjunction with pheochromocytoma and several additional features, including mucosal neuromas, ganglioneuromatoses of the gastrointestinal tract, and a marfanoid habitus. Parathyroid disease is very rare in MEN2B. Familial medullary thyroid carcinoma (FMTC) describes the autosomal dominant inheritance of medullary thyroid carcinoma alone.
The overall prevalence of MEN2 is approximately 1:25,000.2 The penetrance is approximately 70% at 70 years of age.3 It has been reported that 90% of MEN2 carriers will eventually show evidence of MTC.4 This contrasts with MEN1, in which penetrance is 94% by the age of 50 years.5
Molecular Pathogenesis: RET Proto-Oncogene
The causative gene for the MEN2 syndromes was identified in 1993.6,7 The gene is the RET (REarranged during Transfection) proto-oncogene located on chromosome 10q11.2.8
The RET proto-oncogene encodes a tyrosine kinase cell surface receptor. Point mutations in RET segregating with the disease phenotype were identified in MEN2 and FMTC in 1993.7 Germline mutations of RET were subsequently identified in MEN2B, at a different site.9 Genetic screening has revolutionized the management of MEN2 families.
The International RET Mutation Consortium performed an extensive study of 477 MEN2 families around the world and established important phenotype/genotype correlations.10 RET mutations have been characterized in approximately 97% of MEN2 families overall. In MEN2A and FMTC, approximately 96% of families will have an identifiable RET germline mutation, and in MEN2B, approximately 98% of families are mutation positive. The most common RET mutations are summarized in Table 19-1.11
Table 19-1
The Most Common Germline RET Mutations
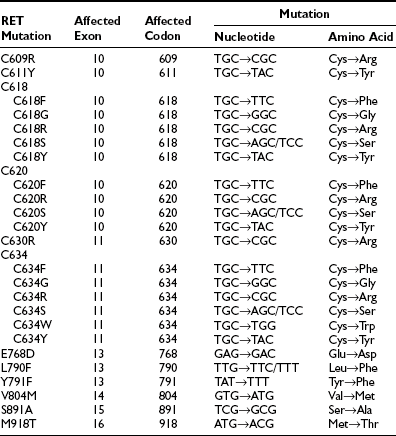
From Machens A, Niccoli-Sire P, Hoegel J et al: Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 349:1517–1525, 2003.
RET Structure and Function
The RET gene consists of over 20 exons spanning a minimum of 30 kilobases of genomic DNA. The protein product of the gene encodes a cell surface receptor. The gene consists of an extracellular domain (exons 1 to 10), a transmembrane domain (exon 11), and an intracellular domain (exons 12 onwards) (Fig. 19-1).12
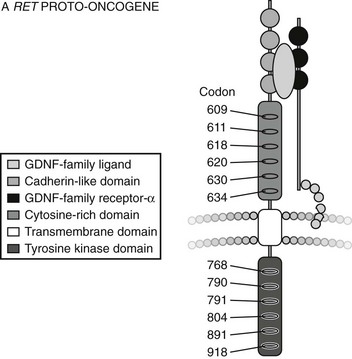
FIGURE 19-1 Structure of RET Proto-oncogene. (From Cote GJ, Gagel RF: Lessons learned from the management of a rare genetic cancer. N Engl J Med 349:1566–1568, 2003.)
The RET protein is membrane-bound, with its intracellular portion having tyrosine kinase activity. The extracellular domain extends for the initial 635 amino acids and includes an area of cadherin homology and a conserved cysteine-rich region immediately adjacent to the transmembrane domain.13 The cysteine-rich domain is typical of the transforming growth factor beta (TGF-β) superfamily.14 The protein is glycosylated to produce receptors with molecular weights of 150 and 170 kilodaltons. The 170-kD form is present in the cell membrane, while the 150-kD form is an immature form present in the cell cytoplasm.15 Phosphorylation of RET tyrosine residues results in activation of several intracellular second messenger systems and signaling pathways.16 These include phospholipase Cγ/protein kinase C (PLCγ/PKC), c-Jun N-terminal kinases (c-Jun/JNK), products of the proto-oncogene Src-related kinases, and nuclear factor κB (NF-κB). Other downstream targets include the Ras/Raf/ERK (extracellular regulated kinase) and phosphatidylinositol-3-kinase (P13-kinase)/Akt pathways. Through these signaling cascades, RET appears to have a central role in cellular proliferation, differentiation, and migration.17–19 Interestingly, in addition to its role in endocrine neoplasia, aberrant RET signaling has been associated with distinct phenotypes. Hirschsprung’s disease occurring with MEN2 is described in subjects with codon 609, 618, and 620 mutations,20 and cutaneous lichen amyloidosis with codon 634 mutations.21
RET Ligands
There was intense interest in the early 1990s in identifying the ligand for RET. In 1996 the first ligand, glial cell line–derived neurotrophic factor (GDNF), was identified.22 Other ligands were soon identified, including neurturin (NTN),23 artemin,24 and persephin.25
RET activation by ligand appears to occur via membrane-bound proteins that function as the ligand-binding domain of a ligand/receptor complex. The link between RET and its first ligand, GDNF, was suggested by the study of mice that were deficient in GDNF expression. These mice were very similar to RET knockout mice, with absent or limited development of the kidney and an absence of enteric neurons in the colon and small intestine.26
GDNF-family ligands bind to specific GDNF-family receptor proteins, all of which form receptor complexes and signal through the RET receptor, tyrosine kinase. Additional proteins, GDNF receptor alpha (GFRα) or GDNF-family receptor alpha 1 (GFRα-1), were found to be essential for the high-affinity binding of GDNF to RET and for its consequent functional effects.27 A single molecule of ligand binds two GFRα-1 molecules, which interact with two molecules of RET and lead to activation of RET tyrosine kinase. Similarly, the other ligands such as neurturin, persephin, and artemin bind to different GFRαs before eventual receptor activation occurs, with consequent effects. It was suggested that MEN2 families with no identifiable RET mutations may have GDNF mutations, but so far no such mutations have been described in MEN2. However, somatic GDNF mutations have been found in sporadic pheochromocytomas.28
Functional Effects of RET Mutations
Although the genotypes do overlap, in general, different codons are affected in either MEN2A or MEN2B. These mutations in different regions of the RET proto-oncogene increase the transforming activity of the kinase by distinct mechanisms. In MEN2A, the extracellular cysteine-rich or transmembrane domains are predominantly affected. Such mutations lead to a loss of cysteine residues. As a result, two RET molecules may undergo ligand-independent dimerization, with resultant constitutive activation of tyrosine kinase activity.29 A mutation in codon 634, usually a cysteine-to-arginine substitution (TGC to CGC), occurs in 85% of cases of MEN2A.17 In contrast, MEN2B is usually caused by a mutation in the intracellular tyrosine kinase domain. Codon 918 is involved in 95% of cases, in a methionine-to-threonine substitution (ATG to ACG).17 This alters the kinase specificity, with alternative substrate phosphorylation and altered downstream signaling.30 The rarer intracellular RET mutations in codons 768 and 804 have been shown to produce “gain of function” of the receptor, although the precise mechanism by which tumor formation occurs is unclear.31
Histogenesis
The calcitonin-secreting C cells of the thyroid originate from the neural crest. They are located mainly in the upper and middle thirds of the thyroid gland. They are members of the amine-precursor uptake and decarboxylation (APUD) family and express a variety of neuroendocrine markers, including calcitonin, chromogranin A, neuron-specific enolase, and serotonin.32 Calcitonin is a very effective tumor marker for the monitoring of MTC. Carcinoembryonic antigen (CEA) is also a C-cell marker used in clinical practice. C cells usually display an endocrine phenotype but may revert to a neuronal phenotype following malignant transformation.32 The alternately spliced calcitonin gene-related peptide (CGRP) is then the predominant protein expressed by the cells.
C-Cell Hyperplasia
C-cell hyperplasia (CCH) is the pathologic description of an increase in C-cell numbers that can precede malignancy. Criteria for the diagnosis of CCH are variable in the literature, although consensus suggests that there should be more than 50 C cells per field at ×100 magnification.33 C cells may increase in number with age, and CCH is a common finding in normal thyroids at autopsy.34 CCH is no longer considered to be a specific marker of familial MTC. Multifocal CCH is regarded as a precursor lesion to hereditary MTC. Its progression to microscopic MTC is variable and may take many years.35
Clinical Relevance of Germline RET Mutations in MEN2A, FMTC, and MEN2B
Extensive studies of MEN2A families from around the world have now clarified that 97% will have a germline RET mutation, usually in the cysteine-encoding codons 609, 611, 618, 620, or 634. The codon 634 mutation in exon 10 predominates, usually with an arginine substitution, although other substitutions such as by tyrosine or glutamine are described.10 In FMTC, 88% of families have a germline mutation in one of the listed cysteine codons or, more rarely, in intracellular codons including 768, 790, and 791. The common MEN2A C634R mutation of cysteine to arginine (TGC to CGC) is not found in FMTC.36 RET mutations in the intracellular exons 13, 14, and 15 are less common in MEN2A, although they will be detected by routine sequencing of these exons. Isolated cases of kindreds with germline mutations in other exons (including exons 5 and 8) have been reported, and these may account for cases of apparently mutation-negative FMTC.17
MEN2B
In 95% of MEN2B families, the germline RET mutation at codon 918 in exon 16 occurs, causing mutation of methionine to threonine (ATG to ACG).37 This mutation is in the tyrosine kinase domain and is specific to MEN2B.9 Of note, this codon 918 RET mutation is also found somatically in the tumor tissue in some sporadic MTCs. A germline RET codon 918 mutation can also occur de novo, where MEN2B patients have no family history of the disease. However, they still have a 50% chance of passing this mutation on to their offspring.38 Rarer tyrosine kinase domain mutations occur in codons 88339 and 891.12
Specific Genotype/Phenotype Correlations
There has been much interest in the prediction of phenotypic behavior from genotype analysis. As published by the International RET Consortium, the presence of any codon 634 RET mutation correlates significantly with the presence of both pheochromocytoma and hyperparathyroidism. One study suggested more aggressive behavior of MTC with the common arginine substitution at codon 634 (C634R), compared to the C634Y mutation.40 The C634R mutation has not been associated with FMTC. It has been suggested that FMTC is a milder variant of MEN2A, and that pheochromocytoma may eventually occur if the patient survives long enough.10 In support of this concept, it had previously been believed that the codon 804 mutation was specific to FMTC. Subsequent reports have described pheochromocytoma occurring in these families, albeit at an older age than in other MEN2A kindreds.41,42 The occurrence of different phenotypes in different families with the same germline RET mutation suggest a role for more complex gene interactions. The rarer phenotypes associated with MEN2 include Hirschsprung’s disease with RET codon 609, 618, and 620 mutations43 and cutaneous lichen amyloidosis with codon 634 mutations.44 The codon 918 mutation is MEN2B specific.17
Clinical Presentation of MTC and MEN2
Patients with MTC may present with a palpable thyroid lump or with the symptoms of calcitonin excess—in particular, diarrhea and flushing. Diarrhea does not usually occur until the calcitonin level is very high (at least 10-fold above the upper limit of the normal range). Unfortunately it is very likely that the MTC will have spread to regional lymph nodes by the time diarrhea occurs. Familial cases of MTC will have different presentations depending on the specific RET mutation, as described earlier. Patients with MEN2B will have a more aggressive phenotype and may present in early childhood with diarrhea from very high calcitonin levels or with the other features of the MEN2B syndrome.1 Mucosal neuromas can occur on the tongue, in the lips, and on the eyelids, giving a distinctive facial appearance. Neuromas in the gastrointestinal tract can cause abdominal pain, gaseous distension, and even pseudo-obstruction. Hirschsprung’s disease can have a similar presentation in children and is due to inactivating mutations in the extracellular domain of RET, as distinct from the activating mutations of the tyrosine kinase domain seen in MEN2B patients.1 MEN2B patients may also have a marfanoid body habitus and corneal nerve thickening. Some patients with MEN2A will also show corneal nerve thickening. Pheochromocytoma occurs in 50% of MEN2B patients, sometimes at a young age, but hyperparathyroidism is very rare.
Screening in MEN2 and FMTC
The pentagastrin (PG) stimulation test was used for many years to identify biochemically affected members of MEN2 families. The advent of genetic screening has dramatically reduced the need for this test, although it still has a role in follow-up and management. A number of limitations of the PG test have been uncovered since RET mutation analysis became available. It is possible for RET mutation-negative members of MEN2 families to have elevated calcitonin responses to pentagastrin stimulation.45
In the early literature, some of these positive PG tests led to thyroidectomy, and C-cell hyperplasia was found in some patients but not in others.46 This is probably because CCH can be a normal variant.34 PG testing can also be used to detect hereditary MTC carriers in families where no RET mutation is identified, although these families are now rare. Adverse effects from the PG test include retrosternal discomfort, nausea, vomiting, metallic taste, abdominal cramps, esophageal spasm, and tachycardia.
Genetic Screening
RET mutations are clustered tightly, and the analysis can be confined to specific exons. This compares favorably with genetic analysis in MEN1, where the mutations are scattered. Once a mutation is identified, other family members are screened with a straightforward analysis for that specific mutation. Various techniques have been used, and currently, direct polymerase chain reaction (PCR)-based sequencing is performed in most laboratories. Other methods include restriction enzyme digestion of PCR products47 with subsequent sequencing,48 single-strand conformation polymorphism, and denaturing gradient gel electrophoresis.49 Denaturing high-performance liquid chromatography can also be used to identify mutations, provided that appropriate positive controls are available, since there are multiple polymorphisms in the RET gene in normal individuals.50 Linkage analysis is not commonly performed, although it may rarely have a role if no RET mutation can be identified with other techniques.51 Testing should be performed on two separate occasions on two different blood samples to avoid the risk of sample mix-ups. RET mutation-negative members of known MEN2 families (i.e., where RET mutation is identified in the family) can be reassured that their risk of developing MTC is no greater than for the general population.
Genetic screening must therefore be comprehensive and include sequencing of exons 10, 11, 13, 14, 15, and 16.52
Management Implications of Genetic Results
Timing of Prophylactic Thyroidectomy
The advent of genetic screening has allowed prophylactic thyroidectomy to be performed earlier than it would be by traditional biochemical screening. Logically, this would be expected to lead to an improvement in the prognosis of RET-mutation carriers. Long-term data is slowly accumulating to support the practice of prophylactic thyroidectomy based on genetic screening rather than on clinical or biochemical screening. Consensus was reached at the MEN ’97 Workshop that the decision to perform thyroidectomy in MEN2 should be based predominantly on the result of RET-mutation testing rather than on calcitonin testing.53
The specific mutated RET codon may be correlated with the degree of aggressiveness of the MTC. A three-level risk stratification system has been adopted since its inception at the Seventh International Workshop on Multiple Endocrine Neoplasia in 1999.52 Category 1, the highest risk group, includes all MEN2B-mutation carriers, because mutations in RET codon 883 and 918 are associated with the most aggressive MTC. Such patients should have thyroidectomy within the first 6 months of life because metastases within the first year of life have been described. Those with RET codon 611, 618, 620, or 634 are classified as category 2, having a high risk of MTC, and thyroidectomy before 5 years of age is recommended. MTC with the codon 634 mutation has been reported as early as age 2 years, although cases of MTC are generally rare before the age of 5 years in MEN2A and FMTC. Thyroidectomy at this age is technically possible without significant morbidity. Category 3 includes those with RET codon 609, 768, 790, 791, 804, and 891 mutations, having the lowest risk among the stratification categories. It should be noted that a single case of metastatic MTC in a 6-year-old was reported in a family with the codon 804 RET mutation.54 Some have recommended thyroidectomy by age 5 years, whereas others have suggested it by age 10 years, for gene-positive members of category 3–risk families. If baseline calcitonin is elevated, then thyroidectomy should be performed immediately, regardless of the RET mutation found. Histology in this setting is likely to show MTC rather than C-cell hyperplasia, with the associated risk of lymph node spread.55 Fig. 19-2 describes the earliest age of reported MTC for each of the known RET mutations. Early prophylactic surgery also alleviates the need for ongoing unpleasant PG stimulation testing being performed in a child.
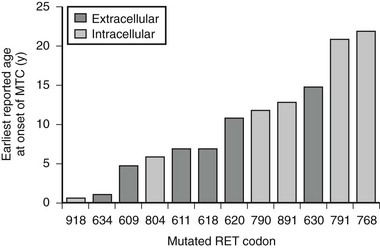
The Role of Somatic RET Mutations
Somatic mutations of RET are observed in a proportion of sporadic MTC cases. The commonest somatic RET mutation is the codon 918 ATG-to-ACG (M918T) mutation. Its prevalence varies widely (from 28% to 86%) in different series.56 This may reflect either geographic differences in population studies or perhaps methodological differences. Other somatic mutations reported in sporadic MTC include codon 883 in exon 15 and codon 634 in exon 11.57 Although not a universal finding, smaller series had suggested that the somatic RET codon 918 mutation was associated with a poorer prognosis in sporadic MTC, correlating the mutation with distant metastases and tumor recurrence.58,59,60 A recent large study of 100 sporadic MTC patients with a 10-year follow-up period supported this concept.57 This study reported increased lymph node metastases at diagnosis, increased likelihood of persistent disease, and lower overall survival in those subjects with a somatic RET mutation.57
Somatic Mutations in Hereditary MTC
Somatic RET mutations have also been described in hereditary MTC. The somatic codon 918 RET mutation has been reported, albeit rarely, in association with germline RET mutations in exon 10 or 11.61 This important observation negated the theory that the codon 918 somatic RET mutation could be used as a specific marker of sporadic disease. Others have observed the somatic codon 918 RET mutation occurring in patients with a germline codon 768 RET mutation.62 Microdissection techniques have uncovered the fact that somatic RET mutations may be found in some regions of an MTC tumor but not in others.63 It has been suggested that the presence of a somatic mutation in addition to the germline RET mutation provides a “second hit” that may explain the more aggressive behavior of some familial cases of MTC.
Routine Germline Screening of Sporadic MTCs
It is now recommended that all new cases of sporadic MTC should have germline screening for RET mutations. Unsuspected familial cases are uncovered in approximately 6% of such patients despite an apparently negative family history.64 This was confirmed in a recent series of apparently sporadic MTC, in which 7.3% of subjects were found to possess a germline mutation. Lower-risk RET mutations in the non-cysteine codons were most commonly discovered, including codons 804, 891, and 768, although cysteine codon mutations were also found. The majority of these unsuspected familial cases were diagnosed as FMTC, based on the absence of other endocrine neoplasia at follow up.65 Of note, genetic screening of first-degree relatives should be undertaken if a germline RET mutation is identified in a subject with MTC.
Given that more than 97% of MEN2 families have identifiable RET mutations, a negative-germline RET-mutation test provides a high degree of certainty that a particular individual has sporadic MTC. This is assuming the genetic analysis is comprehensive. It is important to include exons 13, 14, and 15 because mutations in these exons are most likely to manifest late, with a lower prevalence of pheochromocytoma, and are more likely to escape detection as familial.66
Natural Progression and Behavior of MTC
Hereditary Versus Sporadic MTC
Hereditary MTC is characterized by the early onset of bilateral and multifocal disease. Sporadic MTC may present as a palpable thyroid mass or a nodule on ultrasound, and lymph node involvement is present in 30% to 60% of cases at diagnosis.67 Local lymph-node spread occurs early in the course of the disease in both hereditary and sporadic forms.
A study by Raue and colleagues evaluating prognostic factors in MTC found that the stage of disease at diagnosis, age, sex, and type of disease (sporadic versus hereditary) were relevant prognostic factors in a univariate analysis, with better prognosis for young female patients with familial disease diagnosed at an early stage. The difference in the survival rate of patients with hereditary disease versus those with the sporadic form disappeared in multivariate proportional hazards analysis, but the prognostic information provided by age and sex was still significant. The overall adjusted survival rate was 86.7% at 5 years and 64.2% at 10 years. These rates are comparable to those quoted by other studies.68–70 A more recent study found only the stage of disease and presence of extrathyroidal growth at presentation with MTC to be independent predictors of clinical behavior and life expectancy. In this study, increased age at diagnosis was found to be associated with a reduction in overall survival; however, when adjusted for the baseline mortality rate of the general population on multivariate analysis, this effect was lost.70
Management
Current therapy for MTC is restricted to surgical removal of all neoplastic tissue, and alternative forms of treatment using radiotherapy or chemotherapy provide little benefit. Overall, 50% of MTC patients will have lymph node involvement at presentation.71 Patients with MEN2B and sporadic MTC have a higher rate of extrathyroidal spread than do those with MEN2A.72
The primary operation should include total thyroidectomy and the removal of all lymph nodes in the central neck compartment bounded by the hyoid bone, sternal notch, and internal jugular veins. Lymph nodes in the upper mediastinum and lateral neck regions should also be examined and removed if macroscopically abnormal. Scollo and colleagues have advocated the routine performance of central and bilateral neck dissection for all sporadic and hereditary MTC cases. They showed that the ipsilateral, contralateral, and central compartments were involved with the same frequency in patients with either sporadic or hereditary MTC, even in the case of unilateral thyroid tumors. It was suggested that contralateral lymph node dissection could be avoided only in those with unilateral tumor involvement and no central or ipsilateral lymph node involvement.73 It should be mentioned that whenever a patient is assessed for thyroid surgery, urinary catecholamines and metanephrines should be measured prior to undergoing general anesthesia to exclude unrecognized pheochromocytoma. Serum calcium and parathyroid hormone levels should also be measured preoperatively.
Localization of Recurrent MTC
Localization of recurrence is often very difficult, particularly when calcitonin levels are only modestly elevated. One commonly faces the problem of failure of detection of metastatic disease even when its presence is suggested by persistently elevated serum calcitonin or CEA. The biological behavior of MTC varies between subjects. As well as being a tumor marker, calcitonin doubling time has been reported to be a prognostic factor for poor survival.74,75 One recent study reported both calcitonin and CEA doubling times to be associated with progressive disease in most patients,76 suggesting these could be used to guide the frequency and intensity of serial imaging. Of note, calcitonin and CEA markers can fluctuate significantly in the short term, so caution in assessing trends is required.77
Ultrasonography/Computed Tomography/Magnetic Resonance Imaging
There is a paucity of systematic studies assessing various radiographic imaging protocols. One recent study compared ultrasonography, spiral computed tomography (CT) scan, magnetic resonance imaging (MRI) of the liver and whole body, as well as 18-fluoro-2-deoxy-d-glucose (FDG) positron emission tomography (PET) in a group of 55 patients with MTC and persistently elevated calcitonin.78 They found neck ultrasound to be the most sensitive tool in detecting lymph node metastases. Chest CT was found to be superior in detecting lung lesions, and liver MRI the most sensitive in diagnosing hepatic metastases. Bone scan complemented MRI in the detection of axial skeletal metastases but was superior in assessing peripheral skeletal lesions. As discussed below, 18FDG PET was disappointing.
Nuclear Imaging
A variety of nuclear imaging methods have been used in an attempt to take advantage of the specific expression of hormonal receptors or transporters by endocrine tumors to allow disease localization. The radionuclide agents used in trials include 201T1-chloride, 131I-meta-iodobenzylguanidine (MIBG), 99mTc pentavalent-dimercapto-succinic acid (DMSA), 131I anti-CEA or anticalcitonin antibodies or fragments, labeled somatostatin receptors, and technetium-99m-sestamibi (99mTc-sestamibi).79 PET scanning has also been investigated. Unfortunately, no single method is adequately sensitive or specific for definitive diagnosis.
Imaging with 131I MIBG and 131I anti-CEA is not sensitive but is specific. These tracers are therefore inappropriate for primary use in the early diagnosis or exclusion of MTC. Once MTC has been diagnosed, it is worthwhile establishing whether these tracers accumulate in the tumor to evaluate their potential therapeutic use. 201T1 Chloride and 99mTc DMSA have slightly better sensitivity, with the advantage of no competitive tracer uptake by normal thyroid tissue, but neither is specific for MTC.80
The use of 99mTc-sestamibi can image differentiated thyroid carcinomas, parathyroid tumors, and cardiac muscles because of its avid mitochondrial uptake. The use of 99mTc-sestamibi in MTC has been limited. It was found to be useful in detecting MTC recurrence in the soft tissues of the neck and chest in patients with calcitonin values in excess of 6000 ng/L81, and it complements other modalities such as CT in the overall assessment of patients with very high calcitonin levels. The drawback is that this radiopharmaceutical is excreted via the hepatic route, thereby confounding the identification of liver metastasis.
6-(18F)-Fluorodopamine is a positron-emitting analogue of dopamine used as a tracer for PET scanning. It has shown particular promise in imaging the sympathetic nervous system (e.g., in pheochromocytomas). In a case report, metastatic histologically proven MTC was unequivocally detected by 6-(18F)-fluorodopamine PET scanning.82 However, the broader use of this agent remains to be evaluated, and its availability is limited. Results with 18-FDG PET have also been disappointing, with one recent study reporting a sensitivity of only 58% in imaging metastatic MTC. This study found a low standardized uptake value of FDG in the metastatic MTCs, possibly due to slow growth, small size, or the sclerotic, necrotic, or calcified nature of the tumors.78
Selective Venous Catheterization
Selective venous catheterization (SVC) has been regarded as the most sensitive tool for the localization of residual disease and for the early detection of distant metastases in MTC.83 In this study, neck and/or mediastinal tumor foci were found in 12 out of 18 patients with calcitonin gradients in the neck and/or the mediastinum. Distant metastases emerged clinically in all five patients with significant gradients.
The procedure of SVC involves the insertion of catheters via the femoral vein into the main neck veins, a hepatic vein, and a peripheral vein. Sequential samples of calcitonin values are obtained at these sites and are expressed as a ratio with the mean peripheral value. A local/peripheral calcitonin gradient of 2.5, yielding a sensitivity of 100%, has been reported.84 The invasive nature of this technique and need for angiographic expertise limits its widespread applicability.
Reoperation
Elevated levels of calcitonin persist after primary operation in many patients with MTC. Failure of normalization of calcitonin is more likely in subjects with a higher preoperative calcitonin level and in those in whom nodal metastases were confirmed at surgery.85 Because of the lack of a highly sensitive and specific diagnostic tool to detect micrometastases postoperatively, one is often confronted with the unique situation of whether to reoperate without definite localization and, if so, to what extent. Gimm and Dralle found that lymph node metastases were found in 94% of those patients with elevated calcitonin levels after primary therapy. Four-compartment lymphadenectomy gives a chance of cure in 35% of patients without distant metastases.86
Reoperation is recommended if local disease in the neck and/or upper mediastinum is found without evidence of distant metastases. Exploration of the mediastinum is controversial because of the greater morbidity and the lower chance of cure. If distant metastases are found, there is no indication for surgical intervention unless the patient develops diarrhea, for which tumor debulking may be beneficial.53
Other Modalities of Treatment
Therapy with different formulations of octreotide and lanreotide does not seem to modify serum concentrations of calcitonin and CEA in patients with recurrent MTC. No significant decrease in the size of metastases has been found with the use of somatostatin analogues in patients with advanced MTC.87 However, such agents do have a role in symptomatic control of diarrhea, flushing, weight loss, or bone pain.88 Sandostatin has been used in practice, with some partial relief of symptoms.
Chemotherapy
Chemotherapy has been of limited value. Doxorubicin has been the most widely used single chemotherapeutic agent in the treatment of MTC and therefore is the one associated with most reported responses. Dacarbazine is the most widely used agent in combination therapy. Petursson reported a patient with metastatic MTC who demonstrated a complete response with combination chemotherapy of dacarbazine and 5-fluorouracil.89 Combined chemotherapy has been tried in patients with advanced MTC using cyclophosphamide, vincristine, and dacarbazine; this yielded only moderate antitumor activity.90 Trials of other combination therapy are ongoing. Small studies have investigated selective arterial chemoembolization to treat hepatic metastases, with some tumor response and symptom palliation.91,92
Radiotherapy
There is no demonstrable survival benefit of adjuvant external beam radiotherapy. A retrospective study by Brierley and colleagues in 73 patients demonstrated no overall difference in local or regional relapse-free rate between those who received external irradiation and those who did not.93 Also of concern is the potential for radiation treatment to cause tissue damage, thus increasing the potential for complications from future surgery. Radiotherapy does have a role in controlling local disease and in alleviating symptoms such as bone pain.94 If reoperation is deemed unfeasible, then the effect on local tumor shrinkage may be significant, particularly if there is concern about mass obstruction or extension of tumor into an important structure such as the trachea, esophagus, or carotid artery.95
Recent Advances in Treatment of Medullary Thyroid Carcinoma
Radionuclide Treatment
Radioactive iodine (131I) has not demonstrated efficacy in treatment of MTC.96 This is attributed to the fact that MTC arises from parafollicular C cells of the thyroid, which do not concentrate iodine. The therapeutic response of 131I-MIBG in treatment has been poor in a limited experience.95 Other attempts to deliver targeted radioactivity are under ongoing investigation. One approach has been to administer monoclonal antibodies coupled to radioisotopes. A recent non-randomized study reported disease stabilization and prolonged survival with two-part radioimmunotherapy against CEA in MTC patients with a high calcitonin doubling time.97 Other groups have used yttrium-90-labeled somatostatin analogues for targeted radiotherapy, with some promise in early reports.98 As yet these studies are preliminary.
Immunotherapy
Immunotherapy for MTC has been studied with the rationale that stimulation of immune responses against tumor antigens may result in a clinical response. One study, by Schott and colleagues, explored immunization with calcitonin and/or CEA peptide-pulsed dendritic cells to induce T-cellular antigen-specific immune responses in patients with MTC to achieve a clinical response.99 One of their seven patients had a regression of detectable liver metastases and pulmonary lesions. Other approaches have included genetic immunization using transfer of cytokines into tumor tissues to mount an immune response against tumor antigens. The cytokines investigated include interleukin 2 (IL-2) and 12 (IL-12).100 IL-2, also known as T-cell growth factor, may exhibit antineoplastic capacity through activation of cytotoxic T lymphocytes, CD4+ and CD8+ lymphocytes, and natural killer cells. This was illustrated in a murine MTC study by Zhang and colleagues, who demonstrated some tumor response by in vivo delivery of adenoviral vectors expressing murine IL-2.101 A follow-up study showed that the injection of adenovirus expressing murine IL-2 directly into rat MTC had antitumor effect.102 IL-12 has also been studied by Zhang and DeGroot. They studied a rat MTC model to investigate murine IL-12 therapy using an adenoviral vector system, demonstrating tumor regression in 86% of treated animals and evidence of long-term immunity to tumor cells.103 Studies of immunomodulation in MTC are ongoing.
Gene Therapy
Targeting gene therapy to the inhibition of oncogenic “activated RET” is another attractive treatment modality for MTC, and several methods have been investigated.104 The practical considerations in endocrine tumor gene therapy were well explored in a review by Messina and colleagues.105 Adenoviral vectors expressing a dominant-negative RET mutant provide one potential mechanism by which endogenous RET signaling may be blocked.104 Another approach has been the use of small interfering ribonucleic acid (siRNA) to silence RET gene expression.17 Suicide gene therapy has also been investigated. This involves the gene transfer of prodrug activating enzymes, along with the prodrugs, to result in targeted antitumor effects.106 Unfortunately, early promise in gene therapy has not yet been translated into clinical results.
Molecular Targets
MTC expresses the constitutively active RET receptor, tyrosine kinase. This forms the basis of studies using tyrosine kinase inhibitors as potential therapeutic tools. Several small molecule inhibitors have been assessed in small, phase 2 clinical trials, and other agents are in preclinical development. These compounds have been designed to block the RET signaling cascade at varying levels.17,18 Tyrosine kinase inhibitors such as imatinib block autophosphorylation. Despite success in treating chronic myeloid leukemia and gastrointestinal stromal tumors, imatinib appears disappointing in the treatment of MTC.107,108 Another tyrosine kinase inhibitor, vandetanib (ZD6474), has shown promise in phase 2 studies,109 with results of both the open-labeled study in hereditary MTC and the double-blind, randomized, phase 3 study in sporadic MTC patients awaited. Other potential molecular targets include inhibition of adaptor protein recruitment or of downstream pathways such as Ras/Raf/ERK, P13K/Akt, PLCγ/PKC and c-Jun/JNK.17,18 Sorafenib (Bay43-9006) is an orally available kinase inhibitor that was initially developed to inhibit BRAF. It has subsequently been found to inhibit multiple other pathways including RET, and phase 2 clinical trials in MTC are ongoing.18 Of note, the trial compounds to date also inhibit the vascular endothelial growth factor receptor (VEGFR), thereby potentially inhibiting tumor angiogenesis in addition to oncogenic RET signaling.17,18 Indeed, no agent in clinical trial demonstrates specificity for the RET tyrosine kinase, creating the potential for multiple side effects. The toxicity with these agents is not insignificant, and photosensitive skin rashes, hypertension, fatigue, and diarrhea have been observed.18,109 Also of interest is the potential for RET mutations at distinct codons to confer selective resistance to these compounds. In vitro work has suggested that the codon 804 mutation results in resistance to vandetinib.110 Study into these compounds is ongoing, and with our increasing understanding of molecular oncology, these targeted biological agents will remain a prime area of investigation for medullary thyroid carcinoma therapy.
References
1. Marsh, DJ, Learoyd, DL, Robinson, BG. Medullary thyroid carcinoma: recent advances and management update. Thyroid. 1995;5:407–424.
2. Kidd, KK, Simpson, NE. Search for the gene for multiple endocrine neoplasia type 2A. Recent Prog Horm Res. 1990;46:305–343.
3. Komminoth, P. The RET proto-oncogene in medullary and papillary thyroid carcinoma. Molecular features, pathophysiology and clinical implications. Virchows Arch. 1997;431:1–9.
4. Easton, DF, Ponder, MA, Cummings, T, et al. The clinical and screening age-at-onset distribution for the MEN-2 syndrome. Am J Hum Genet. 1989;44:208–215.
5. Chandrasekharappa, SC, Guru, SC, Manickam, P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407.
6. Donis-Keller, H, Dou, S, Chi, D, et al. Mutations in the RET proto-oncogene are associated with MEN2A and FMTC. Hum Mol Genet. 1993;2:851–856.
7. Mulligan, LM, Kwok, JB, Healey, CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–460.
8. Pasini, B, Hofstra, RM, Yin, L, et al. The physical map of the human RET proto-oncogene. Oncogene. 1995;11:1737–1743.
9. Hofstra, RM, Landsvater, RM, Ceccherini, I, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367:375–376.
10. Eng, C, Clayton, D, Schuffenecker, I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276:1575–1579.
11. Machens, A, Niccoli-Sire, P, Hoegel, J, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349:1517–1525.
12. Cote, GJ, Gagel, RF. Lessons learned from the management of a rare genetic cancer. N Engl J Med. 2003;349:1566–1568.
13. Takahashi, M, Buma, Y, Iwamoto, T, et al. Cloning and expression of the RET proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene. 1988;3:571–578.
14. Ullrich, A, Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212.
15. Takahashi, M, Asai, N, Iwashita, T, et al. Characterization of the RET proto-oncogene products expressed in mouse L cells. Oncogene. 1993;8:2925–2929.
16. Salvatore, D, Barone, MV, Salvatore, G, et al. Tyrosines 1015 and 1062 are in vivo autophosphorylation sites in RET and RET-derived oncoproteins. J Clin Endocrinol Metab. 2000;85:3898–3907.
17. de Groot, JW, Links, TP, Plukker, JTM, et al. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocrine Reviews. 2006;27:535–560.
18. Schlumberger, M, Carlomagno, F, Baudin, E, et al. New therapeutic approaches to treat medullary thyroid carcinoma. Nat Clin Pract Endocrinol Metab. 2008;4:22–32.
19. Santoro, M, Carlomagno, F. Drug insight: small-molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nat Clin Pract Endocrinol Metab. 2006;2:42–51.
20. Mulligan, LM, Eng, C, Attie, T, et al. Diverse phenotypes associated with exon 10 mutations of the RET proto-oncogene. Hum Mol Genet. 1994;3:2163–2167.
21. Donovan, DT, Levy, ML, Furst, EJ, et al. Familial cutaneous lichen amyloidosis in association with multiple endocrine neoplasia type 2A: a new variant. Henry Ford Hosp Med J. 1989;37:147–150.
22. Jing, S, Wen, D, Yu, Y, et al. GDNF-induced activation of the RET protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 1996;85:1113–1124.
23. Klein, RD, Sherman, D, Ho, WH, et al. A GPI-linked protein that interacts with Ret to form a candidate neurturin receptor. Nature. 1997;387:717–721.
24. Baloh, RH, Tansey, MG, Lampe, PA, et al. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron. 1998;21:1291–1302.
25. Milbrandt, J, de Sauvage, FJ, Fahrner, TJ, et al. Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron. 1998;20:245–253.
26. Schuchardt, A, D’Agati, V, Larsson-Blomberg, L, et al. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor RET. Nature. 1994;367:380–383.
27. Treanor, JJ, Goodman, L, de Sauvage, F, et al. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83.
28. Woodward, ER, Eng, C, McMahon, R, et al. Genetic predisposition to phaeochromocytoma: Analysis of candidate genes GDNF, RET and VHL. Hum Mol Genet. 1997;6:1051–1056.
29. Asai, N, Iwashita, T, Matsuyama, M, et al. Mechanism of activation of the ret proto-oncogene by multiple endocrine neoplasia 2A mutations. Mol Cell Biol. 1995;15:1613–1619.
30. Santoro, M, Carlomagno, F, Romano, A, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN-2A and MEN-2B. Science. 1995;267:381–383.
31. Pasini, A, Geneste, O, Legrand, P, et al. Oncogenic activation of RET by two distinct FMTC mutations affecting the tyrosine kinase domain. Oncogene. 1997;15:393–402.
32. Russo, AF, Clark, MS, Durham, PL. Thyroid parafollicular cells. An accessible model for the study of serotonergic neurons. Mol Neurobiol. 1996;13:257–276.
33. Rosai, J, Carcangiu, ML, De Lellis, RA. Tumors of the thyroid gland. In: Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology; 1995.
34. Guyetant, S, Rousselet, MC, Durigon, M, et al. Sex-related C cell hyperplasia in the normal human thyroid: a quantitative autopsy study. J Clin Endocrinol Metab. 1997;82:42–47.
35. Papotti, M, Botto Micca, F, Favero, A, et al. Poorly differentiated thyroid carcinomas with primordial cell component. A group of aggressive lesions sharing insular, trabecular, and solid patterns. Am J Surg Pathol. 1993;17:291–301.
36. Berndt, I, Reuter, M, Saller, B, et al. A new hot spot for mutations in the ret protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab. 1998;83:770–774.
37. Carlson, KM, Dou, S, Chi, D, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci U S A. 1994;91:1579–1583.
38. Carlson, KM, Bracamontes, J, Jackson, CE, et al. Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet. 1994;55:1076–1082.
39. Gimm, O, Marsh, DJ, Andrew, SD, et al. Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J Clin Endocrinol Metab. 1997;82:3902–3904.
40. Punales, MK, Graf, H, Gross, JL, et al. RET codon 634 mutations in multiple endocrine neoplasia type 2: variable clinical features and clinical outcome. J Clin Endocrinol Metab. 2003;88:2644–2649.
41. Nilson, O, Tissell, L, Janson, S, et al. Adrenal and extra-adrenal pheochromocytomas in a family with germline RET V804L mutation. JAMA. 1999;281:1587.
42. Learoyd, DL, Gosnell, J, Elston, MS, et al. Experience of prophylactic thyroidectomy in multiple endocrine neoplasia type 2A kindreds with RE codon 804 mutations. Clin Endocrinol. 2005;63:636–641.
43. Mulligan, LM, Eng, C, Attie, T, et al. Diverse phenotypes associated with exon 10 mutations of the RET proto-oncogene. Hum Mol Genet. 1994;3:2163–2167.
44. Donovan, DT, Levy, ML, Furst, EJ, et al. Familial cutaneous lichen amyloidosis in association with multiple endocrine neoplasia type 2A: a new variant. Henry Ford Hosp Med J. 1989;37:147–150.
45. Lips, CJ, Landsvater, RM, Hoppener, JW, et al. Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Engl J Med. 1994;331:828–835.
46. Marsh, DJ, McDowall, D, Hyland, VJ, et al. The identification of false positive responses to the pentagastrin stimulation test in RET mutation negative members of MEN2A families. Clin Endocrinol (Oxf). 1996;44:213–220.
47. Marsh, DJ, Robinson, BG, Andrew, S, et al. A rapid screening method for the detection of mutations in the RET proto-oncogene in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma families. Genomics. 1994;23:477–479.
48. Frilling, A, Dralle, H, Eng, C, et al. Presymptomatic DNA screening in families with multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Surgery. 1995;118:1099–1103.
49. Decker, RA, Peacock, ML, Borst, MJ, et al. Progress in genetic screening of multiple endocrine neoplasia type 2A: is calcitonin testing obsolete? Surgery. 1995;118:257–263.
50. Marsh, DJ, Theodosopoulos, G, Howell, V, et al. Rapid mutation scanning of genes associated with familial cancer syndromes using denaturing high-performance liquid chromatography. Neoplasia. 2001;3:236–244.
51. Tsai, MS, Ledger, GA, Khosla, S, et al. Identification of multiple endocrine neoplasia, type 2 gene carriers using linkage analysis and analysis of the RET proto-oncogene. J Clin Endocrinol Metab. 1994;78:1261–1264.
52. Brandi, ML, Gagel, RF, Angeli, A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–5671.
53. Lips, CJ. Clinical management of the multiple endocrine neoplasia syndromes: results of a computerized opinion poll at the Sixth International Workshop on Multiple Endocrine Neoplasia and von Hippel-Lindau disease. J Intern Med. 1998;243:589–594.
54. Fronhauer, MK, Decker, RA. Update on MEN2A c804 RET mutation: is prophylactic thyroidectomy indicated? Surgery. 2000;128:1052–1058.
55. Learoyd, DL, Robinson, BG. Do all patients with RET mutations associated with multiple endocrine neoplasia type 2 require surgery? Nat Clin Pract Endocrinol Metab. 2005;1:60–61.
56. Learoyd, DL, Capes, AG, Robinson, BG. Multiple endocrine neoplasia type 2 and glial cell line-derived neurotrophic factor. Curr Opin Endocrinol Diabetes. 1997;4:130–137.
57. Elisei, R, Cosci, B, Romei, C, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93:682–687.
58. Zedenius, J, Larsson, C, Bergholm, U, et al. Mutations of codon 918 in the RET proto-oncogene correlate to poor prognosis in sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab. 1995;80:3088–3090.
59. Romei, C, Elisei, R, Pinchera, A, et al. Somatic mutations of the RET protooncogene in sporadic medullary thyroid carcinoma are not restricted to exon 16 and are associated with tumor recurrence. J Clin Endocrinol Metab. 1996;81:1619–1622.
60. Marsh, DJ, Learoyd, DL, Andrew, SD, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf). 1996;44:249–257.
61. Marsh, DJ, Andrew, SD, Eng, C, et al. Germline and somatic mutations in an oncogene: RET mutations in inherited medullary thyroid carcinoma. Cancer Res. 1996;56:1241–1243.
62. Miyauchi, A, Egawa, S, Futami, H, et al. A novel somatic mutation in the RET proto-oncogene in familial medullary thyroid carcinoma with a germline codon 768 mutation. Jpn J Cancer Res. 1997;88:527–531.
63. Eng, C, Mulligan, LM, Healey, CS, et al. Heterogeneous mutation of the RET proto-oncogene in subpopulations of medullary thyroid carcinoma. Cancer Res. 1996;56:2167–2170.
64. Wohllk, N, Cote, GJ, Bugalho, MM, et al. Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab. 1996;81:3740–3745.
65. Elisei, R, Romei, C, Cosci, B, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92:4725–4729.
66. Eng, C, Mulligan, LM, Smith, DP, et al. Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf). 1995;43:123–127.
67. Rougier, P, Parmentier, C, Laplanche, A, et al. Medullary thyroid carcinoma: prognostic factors and treatment. Int J Radiat Oncol Biol Phys. 1983;9:161–169.
68. Saad, MF, Ordonez, NG, Rashid, RK, et al. Medullary carcinoma of the thyroid. A study of the clinical features and prognostic factors in 161 patients. Medicine (Baltimore). 1984;63:319–342.
69. Raue, F, Kotzerke, J, Reinwein, D, et al. Prognostic factors in medullary thyroid carcinoma: evaluation of 741 patients from the German Medullary Thyroid Carcinoma Register. Clin Investig. 1993;71:7–12.
70. de Groot, JWB, Plukker, JTM, Wolffenbuttel, BHR, et al. Determinants of life expectancy in medullary thyroid cancer: age does not matter. Clin Endocrinol. 2006;65:729–736.
71. Wells, SA, Jr., Baylin, SB, Gann, DS, et al. Medullary thyroid carcinoma: relationship of method of diagnosis to pathologic staging. Ann Surg. 1978;188:377–383.
72. Russell, CF, Van Heerden, JA, Sizemore, GW, et al. The surgical management of medullary thyroid carcinoma. Ann Surg. 1983;197:42–48.
73. Scollo, C, Baudin, E, Travagli, JP, et al. Rationale for central and bilateral lymph node dissection in sporadic and hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 2003;88:2070–2075.
74. Miyauchi, A, Onishi, T, Morimoto, S, et al. Relation of doubling time of plasma calcitonin levels to prognosis and recurrence of medullary thyroid carcinoma. Ann Surg. 1984;1999:461–466.
75. Barbet, J, Campion, L, Kraeber-Bodere, L, et al. Prognostic impact of serum calcitonin and carcinoembryonic antigen doubling-times in patients with medullary thyroid carcinoma. J Clin Endocrinol Metab. 2005;90:6077–6084.
76. Giraudet, AL, Ghulzan, AA, Auperin, A, et al. Progression of medullary thyroid carcinoma: assessment with calcitonin and carcinoembryonic antigen doubling times. Eur J Endocrinol. 2008;158:239–248.
77. de Groot, JW, Kema, IP, Breukelman, H, et al. Biochemical markers in the follow-up of medullary thyroid cancer. Thyroid. 2006;16:1163–1170.
78. Giraudet, AL, Vanel, D, Leboulleux, S, et al. Imaging medullary thyroid carcinoma with persistent elevated calcitonin levels. J Clin Endocrinol Metab. 2007;93:4185–4190.
79. Lebouthillier, G, Morais, J, Picard, M, et al. Tc-99m sestamibi and other agents in the detection of metastatic medullary carcinoma of the thyroid. Clin Nucl Med. 1993;18:657–661.
80. Hoefnagel, CA, Delprat, CC, Zanin, D, et al. New radionuclide tracers for the diagnosis and therapy of medullary thyroid carcinoma. Clin Nucl Med. 1988;13:159–165.
81. Learoyd, DL, Roach, PJ, Briggs, GM, et al. Technetium-99m-sestamibi scanning in recurrent medullary thyroid carcinoma. J Nucl Med. 1997;38:227–230.
82. Gourgiotis, L, Sarlis, NJ, Reynolds, JC, et al. Localization of medullary thyroid carcinoma metastasis in a multiple endocrine neoplasia type 2A patient by 6-[18F]-fluorodopamine positron emission tomography. J Clin Endocrinol Metab. 2003;88:637–641.
83. Abdelmoumene, N, Schlumberger, M, Gardet, P, et al. Selective venous sampling catheterisation for localization of persisting medullary thyroid carcinoma. Br J Cancer. 1994;69:1141–1144.
84. Ben Mrad, MD, Gardet, P, Roche, A, et al. Value of venous catheterization and calcitonin studies in the treatment and management of clinically inapparent medullary thyroid carcinoma. Cancer. 1989;63:133–138.
85. Machens, A, Schneyer, U, Holzhausen, H-J, et al. Prospects of remission in medullary thyroid carcinoma according to basal calcitonin level. J Clin Endocrinol Metab. 2005;90:2029–2034.
86. Gimm, O, Dralle, H. Reoperation in metastasizing medullary thyroid carcinoma: is a tumor stage–oriented approach justified? Surgery. 1997;122:1124–1130.
87. Diez, JJ, Iglesias, P. Somatostatin analogs in the treatment of medullary thyroid carcinoma. J Endocrinol Invest. 2002;25:773–778.
88. Kebebew, E, Clark, OH. Medullary thyroid cancer. Curr Treat Options Oncol. 2000;1:359–367.
89. Petursson, SR. Metastatic medullary thyroid carcinoma. Complete response to combination chemotherapy with dacarbazine and 5-fluorouracil. Cancer. 1988;62:1899–1903.
90. Wu, LT, Averbuch, SD, Ball, DW, et al. Treatment of advanced medullary thyroid carcinoma with a combination of cyclophosphamide, vincristine, and dacarbazine. Cancer. 1994;73:432–436.
91. Lorenz, K, Brauckhoff, M, Behrmann, C, et al. Selective arterial chemoembolization for hepatic metastases from medullary thyroid carcinoma. Surgery. 2005;138:986–993.
92. Fromigue, J, De Baere, T, Baudin, E, et al. Chemoembolization for liver metastases from medullary thyroid carcinoma. J Clin Endocrinol Metab. 2006;91:2496–2499.
93. Brierley, J, Tsang, R, Simpson, WJ, et al. Medullary thyroid cancer: analyses of survival and prognostic factors and the role of radiation therapy in local control. Thyroid. 1996;6:305–310.
94. Steinfield, AD. The role of radiation therapy in medullary carcinoma of the thyroid. Radiology. 1977;123:745–746.
95. Gagel, RF, Robinson, MF, Donovan, DT, et al. Clinical review 44: medullary thyroid carcinoma: recent progress. J Clin Endocrinol Metab. 1993;76:809–814.
96. Saad, MF, Guido, JJ, Samaan, NA. Radioactive iodine in the treatment of medullary carcinoma of the thyroid. J Clin Endocrinol Metab. 1983;57:124–128.
97. Chatal, J-F, Campion, L, Kraeber-Bodere, F, et al. Survival improvement in patients with medullary thyroid carcinoma who undergo pretargeted anti-carcinoembryonic-antigen radioimmunotherapy: a collaborative study with the French Endocrine Tumor Group. J Clin Oncol. 2006;24:1075–1711.
98. Iten, F, Muller, B, et al. Response to [90Yttrium-DOTA]-TOC treatment is associated with long-term survival benefit in metastasized medullary thyroid cancer: a phase II clinical trial. Clin Cancer Res. 2007;13:6696–6702.
99. Schott, M, Seissler, J, Lettmann, M, et al. Immunotherapy for medullary thyroid carcinoma by dendritic cell vaccination. J Clin Endocrinol Metab. 2001;86:4965–4969.
100. Drosten, M, Putzer, BM. Gene therapeutic approaches for medullary thyroid carcinoma treatment. J Mol Med. 2003;81:411–419.
101. Zhang, R, Baunoch, D, DeGroot, LJ. Genetic immunotherapy for medullary thyroid carcinoma: destruction of tumors in mice by in vivo delivery of adenoviral vector transducing the murine interleukin-2 gene. Thyroid. 1998;8:1137–1146.
102. Zhang, R, Straus, FH, DeGroot, LJ. Effective genetic therapy of established medullary thyroid carcinomas with murine interleukin-2: dissemination and cytotoxicity studies in a rat tumor model. Endocrinology. 1999;140:2152–2158.
103. Zhang, R, DeGroot, LJ. Genetic immunotherapy of established tumors with adenoviral vectors transducing murine interleukin 12 (mIL-12) subunits in a rat medullary thyroid carcinoma model. Clin Endocrinol (Oxf). 2000;52:687–694.
104. Drosten, M, Frilling, A, Stiewe, T, et al. A new therapeutic approach in medullary thyroid cancer treatment: inhibition of oncogenic RET signaling by adenoviral vector-mediated expression of a dominant-negative RET mutant. Surgery. 2002;132:991–997.
105. Messina, M, Learoyd, DL, Both, GW, et al. Gene therapy for endocrine tumors: strategies and progress. Curr Opin Endocrinol Diabetes. 2001;8:35–40.
106. Aghi, M, Hochberg, F, Breakefield, XO. Prodrug activation enzymes in cancer gene therapy. J Gene Med. 2000;2:148–164.
107. Frank-Raue, K, Fabel, M, Delorme, S, et al. Efficacy of imatinib mesylate in advanced medullary thyroid carcinoma. Eur J Endocrinol. 2007;157:215–220.
108. de Groot, JWB, Zonnenberg, BA, Ufford-Mannesse, PQ, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3466–3469.
109. Wells SW, Gosnell JE, Gagel RF, et al: Vandetanib in metastatic hereditary medullary thyroid cancer: follow-up results of an open-label phase II trial. Abstract 6018 presented at the 43rd Annual Meeting of the American Society of Clinical Oncology (ASCO 2007) ASCO Meeting Abstracts June 20, 2007.
110. Carlomagno, F, Guida, T, Anaganti, S, et al. Disease-associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene. 2004;23:6056–6063.