[level-membership-for-neurosurgery-category]
Chapter 17 Medical Management of Hormone-Secreting Pituitary Tumors
The majority of pituitary tumors are hormone-secreting.1 With the exception of prolactinomas, surgery has historically been the mainstay of treatment for hormone-secreting as well as non-functioning pituitary tumors. In recent years, however, medical therapy has assumed an increasingly important role in the management of hormone-secreting pituitary adenomas. In addition to being the primary treatment for prolactinomas, medical therapy can serve an adjunctive role in the treatment of growth-hormone (GH) secreting tumors, refractory Cushing’s disease, and the rare thyroid-stimulating-hormone (TSH)-secreting adenomas. In a subset of patients with acromegaly, medical therapy can be the primary treatment approach. This chapter will review the pharmacological profile, therapeutic efficacy, safety, and side effects of the most common medications used in the treatment of pituitary tumors. In addition, the role of medical therapy in relation to neurosurgical management will be emphasized.
Prolactinomas
General Considerations
The decision of whether to initiate medical therapy in a patient with a prolactinoma should take into account the size of the tumor, fertility status of the patient, and the presence of symptoms related to hyperprolactinemia. In general, therapy is indicated for macroadenomas. A premenopausal woman with a microprolactinoma who does not have bothersome galactorrhea and does not wish to become pregnant may be reassured and treated with estrogen replacement. Likewise, a post-menopausal woman with a microprolactinoma may not require any treatment. This conservative approach is justified by the observation that approximately 90% of microprolactinomas will not grow in size over a 4- to 6-year period.2 In these patients, monitoring with periodic prolactin measurements and MRI’s is appropriate. While it is unlikely for a prolactinoma to grow without a concurrent rise in serum prolactin levels, this occurrence has been reported, and therefore some clinicians will monitor with periodic MRI’s even if prolactin levels are stable. Significant increases in serum prolactin levels or evidence of growth of a microprolactinoma are indications for treatment, given the possibility that such a tumor represents one of the small minority that will grow to become a macroadenoma. Alternatively, given the high surgical cure rate of microprolactinomas (up to 75%), surgery may be considered upfront in lieu of medical treatment in those patients who are interested in permanent and immediate cure.3
In the evaluation of hyperprolactinemia, it is important to recognize the phenomenon of macroprolactinemia, a laboratory finding that does not require any treatment. Patients with elevated prolactin levels who have either mild or no symptoms of hyperprolactinemia may, in fact, have large circulating complexes of the prolactin protein (more than 150 kDa), called macroprolactin. This larger prolactin protein, which can spuriously elevate serum prolactin levels, can be recognized by a simple laboratory method using PEG precipitation.4 As many as 10% to 26% of patients with idiopathic hyperprolactinemia have macroprolactinemia, and this entity should be suspected particularly in patients with discordant laboratory findings and clinical symptoms.4
Dopamine Agonists
In the majority of cases, dopamine receptor agonists are the mainstay of treatment for prolactinomas. These drugs inhibit prolactin secretion by binding to the D2 receptor of tumoral lactotrophs, which leads to decreased formation of prolactin secretory granules and thus reduced tumor volume. There may also be a cytocidal effect of dopamine agonist treatment on tumor cells resulting in tumor shrinkage and fibrosis.5 Among these drugs, cabergoline and bromocriptine are the most commonly used in practice today, as their use is substantiated by the greatest amount of clinical experience and evidence. The other dopamine agonists include quinagolide, a non-ergot derivative not available in the United States, and pergolide, an ergot derivative previously used to treat Parkinson’s disease that was removed from the U.S. market due to an increased risk of cardiac valvular disease.
Pharmacologic Aspects
There are currently three dopamine agonists available in the treatment of prolactinomas: cabergoline, bromocriptine, and quinagolide (Table 17-1).
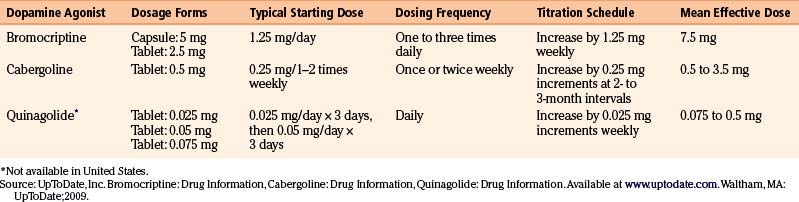
The semisynthetic ergot derivative, bromocriptine, a D2 agonist and weak D1 antagonist, was the first available medical treatment for prolactinomas. Administered orally, bromocriptine has a relatively short half-life (7 hours), often necessitating two to three daily doses, although once daily dosing may occasionally be effective. The standard dosage is 2.5 to 15 mg per day, and most patients are successfully treated with 7.5 mg or less.6 Since gastrointestinal side effects are common, it is usually recommended to start with very low doses (1.25 mg/day) and gradually titrate upward by 1.25 mg increments weekly until a dose of 7.5 mg is reached.6 In resistant patients, doses as high as 20 to 30 mg per day may be necessary.
Cabergoline, a preferential D2-receptor agonist, has largely supplanted bromocriptine as the first-line treatment of prolactinomas due to its superior efficacy, longer half-life, and greater tolerability. Its half-life of 2.5 to 4.5 days allows for once or twice weekly administration.7 The typical starting dose of cabergoline is 0.25 mg orally once or twice weekly, with escalation of dosing by 0.25-mg increments at 2- to 3-month intervals if plateau effect is reached, until prolactin levels normalize. In most patients, the therapeutic dose range is 0.5 to 3.5 mg weekly. A recent study found that more than 95% of patients are able to achieve normal prolactin levels with high-dose treatment (from 6 to 11 mg/wk).8
Therapeutic Efficacy
For microprolactinomas, bromocriptine is 80% to 90% effective at normalizing prolactin levels and shrinking tumor mass, whereas the success rate is around 70% for macroprolactinomas.2 The improvement in headache and visual field defects is rapid and dramatic in most patients, occurring within a few days after the first dose. These changes often precede radiographic evidence of tumor shrinkage. Likewise, gonadal and sexual function may improve even before complete normalization of serum prolactin levels, although sometimes normalization of testosterone may require several months of normal prolactin levels2. The degree of tumor shrinkage seen with bromocriptine does not necessarily correlate with the nadir prolactin level or the extent of decline in prolactin levels.2 Although most prolactinomas remain responsive to bromocriptine over time, the drug is generally not curative, as hyperprolactinemia and tumor growth will often recur after withdrawal of therapy. Prolonged use of bromocriptine has been associated with perivascular fibrosis and increased tumor consistency in prolactinomas that ultimately are surgically removed; the extent of fibrosis seems to correlate with the duration of pre-surgical treatment with bromocriptine and may impact the technical ease of surgical resection.9
Early studies found cabergoline to be 80% to 95% effective at normalizing prolactin levels, with varying degrees of tumor reduction seen in 70% to 90% of patients.10 The efficacy of cabergoline in normalizing prolactin levels in micro- and macro-prolactinomas is shown in Tables 17-2 and 17-3, respectively. Although there is considerable variability among studies with respect to tumor shrinkage, a recent large study showed a significant (>50%) tumor shrinkage in 80% of patients, and complete disappearance of the tumor mass in 57% of patients.11 Cabergoline is able to induce tumor shrinkage more effectively in dopamine agonist naïve patients compared with those who have received prior dopamine agonist treatment (Fig. 17-1). Furthermore, cabergoline offers the potential for cure after several years of treatment, allowing for withdrawal of therapy.12, 13
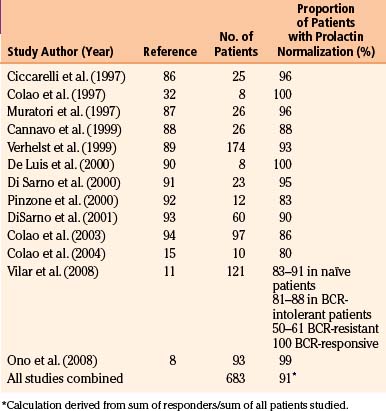
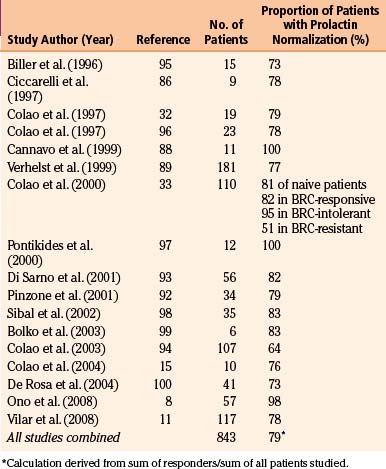
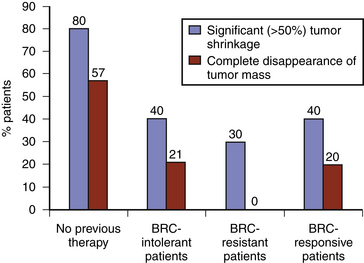
FIGURE 17-1 Efficacy of cabergoline on tumor shrinkage in patients with macroprolactinomas. BRC, bromocriptine.
(From Vilar L, Freitas MC, Naves LA, et al. Diagnosis and management of hyperprolactinemia: results of a Brazilian multicenter study with 1234 patients. J Endocrinol Invest. 2008;31:436-444. Editrice Kurtis.)
The beneficial effects of cabergoline on gonadal function are well documented in both sexes. Compared with bromocriptine, cabergoline is more effective at normalizing prolactin levels and restoring ovulation and is associated with fewer, milder, and shorter-lived side effects.14 Serum testosterone levels have been shown to normalize in the majority of patients, with concurrent improvement in sperm count and parameters.15
Quinagolide normalized prolactin levels in 73% of patients with microadenomas and 67% of patients with macroadenomas.16 Tumor reduction was seen in 55% of patients with microadenomas and 75% of patients with macroadenomas.16 Although cabergoline has been found to be more effective at normalizing prolactin levels and reducing tumor size compared with quinagolide, both drugs show similar efficacy on reproductive function.
Pergolide, a semisynthetic ergoline derivative, was initially approved in the United States only for the treatment of Parkinson’s disease. Typical doses used in the treatment of hyperprolactinemia range from 0.05 mg to 0.5 mg/day, whereas higher doses (>3 mg/day) have been used in Parkinson’s patients.17 Although pergolide has been found to be effective at normalizing prolactin levels and reducing the size of macroprolactinomas, this drug was withdrawn from the U.S. market in 2007 due to the increased risk of valvular heart disease in Parkinson’s patients receiving high daily doses.
Tolerability and Side Effects
The most common side effects of dopamine agonists include gastrointestinal, cardiovascular, and neurologic symptoms (Table 17-4). In general, if a patient cannot tolerate the first dopamine agonist administered, a trial of a second drug should be given.
| Common Side Effects of Dopamine Agonists |
|---|
| Gastrointestinal |
| Nausea |
| Vomiting |
| Cardiovascular |
| Postural hypotension |
| Dizziness |
| Syncope (rare) |
| Neurologic |
| Headache |
| Drowsiness |
| Exacerbation of pre-existing psychosis (rare) |
| Dyskinesias (high-dose treatment) |
Among the dopamine agonists, bromocriptine is the least well tolerated, with up to 12% of patients being unable to tolerate therapeutic doses.2 Up to one third of patients will experience nausea and vomiting, an effect that can be minimized by initiating a low dose and titrating very slowly.2 Taking the medication with a small snack may also reduce these symptoms.2 A gradual dose titration and taking the medication at bedtime may also help reduce postural hypotension and dizziness experienced by as many as 25% of patients.2 Syncope is rare but has been reported even with small initial doses.18 Tolerance to postural hypotension usually develops rapidly.18 Drowsiness, headache, and nasal congestion are common complaints. The safety of bromocriptine in psychiatric patients has not yet been established; there are case reports of onset or exacerbation of preexisting psychosis. Other psychiatric symptoms associated with high doses of bromocriptine include anxiety, depression, insomnia, paranoia, and hyperactivity.18 At higher doses used in the treatment of Parkinson’s disease, reversible pleuro-pulmonary changes and retroperitoneal fibrosis have been reported, but these changes are unlikely to occur at the doses used to treat prolactinomas.2
While cabergoline and bromocriptine have similar side effect profiles, cabergoline has been shown to be better tolerated in several large comparative studies.11, 19 Compared with bromocriptine, the side effects of cabergoline are generally less frequent, milder, and of shorter duration.2 Nausea and vomiting are most commonly observed, followed by headache and dizziness.2 In a multi-center randomized trial of hyperprolactinemic women, 3% could not tolerate cabergoline compared with 12% who had to stop taking bromocriptine.14 It has been proposed that fluctuations in concentrations of dopamine agonists are the main cause of side effects; cabergoline would be better tolerated due to its long half-life and relatively steady plasma concentration.18
Quinagolide has also shown better tolerability compared with bromocriptine. In a double-blind comparative study of 47 hyperprolactinemic patients, quinagolide was tolerated by 90% of patients versus 75% of patients treated with bromocriptine.20 As with the other dopamine agonists, the most frequent side effects include nausea, vomiting, headache, and dizziness. These effects are transient and occur within the first few days of starting therapy or during dose adjustments.18
Dopamine Agonists and Valvular Heart Disease
Recently, the safety of dopamine agonists has been brought into question after two large population-based studies showed an increased risk of cardiac valvular disease in Parkinson’s disease patients being treated with high doses of these drugs, and particularly cabergoline and pergolide.21, 22 It is now recognized that one of the “off target” effects of dopamine agonists is activation of the serotonin (5-HT) receptor, of which there are 7 distinct subtypes.23 High concentrations of the subtype 5-HT2B receptor are found on cardiac valves and the pulmonary arteries.23 Both pergolide and cabergoline have been implicated in the development of cardiac valvular fibrosis because of their agonist effects at the 5-HT2B receptor.23 Bromocriptine, which is only a partial agonist at the 5-HT2BR, had until recently not been thought to pose an increased risk of cardiac valvular disease, as there had only been a single case report suggesting an association.23 However, a recent prospective study suggests that bromocriptine may, in fact, be equally associated with the development of cardiac valvulopathy at high cumulative doses.24
Several studies have recently examined the effect of cabergoline on cardiac valvular disease in patients with prolactinomas.25 These patients typically receive much lower doses (10–20 times less) than in Parkinson’s disease. Only one of seven studies available to date showed an association between cabergoline use and valvular regurgitation. In this study, there was an increased risk of moderate tricuspid regurgitation in patients who had received cumulative cabergoline doses greater than 280 mg.26 However, in another study, patients receiving cabergoline (0.25–4 mg/wk) for 3 to 4 years (mean cumulative dose of 311 mg), had no increased prevalence of clinically significant valvular heart disease.16 Even in patients treated with cabergoline for 8 years who received cumulative doses as high as 1728 mg, there was no increased risk of clinically relevant heart disease.28
Therefore, conventional doses of cabergoline used to treat prolactinoma patients do not currently appear to pose an increased risk of cardiac valvular disease. However, until larger prospective studies clarify the issue, it may be prudent to monitor patients requiring higher doses of cabergoline (>2 or 3 mg/wk) with serial echocardiograms. If valvulopathy develops, it may be reasonable to switch to bromocriptine. Alternatively, surgical resection may be appropriate. This issue underscores the importance of using the smallest effective dose and attempting withdrawal from cabergoline treatment in patients who achieve normalization of prolactin levels and significant tumor shrinkage to minimize their cumulative lifetime exposure to the drug.25
Dopamine Agonists and Pregnancy
Two principal considerations arise in the management of women with prolactinomas who either desire fertility or who become pregnant: (1) the risk of tumor growth due to physiologic stimulation of tumoral lactotrophs induced by pregnancy, and (2) the effects of dopamine agonists on fetal development. These two considerations should be weighed against each other on an individualized basis, taking into account the size of the patient’s initial tumor, response to treatment, and the patient’s ability to tolerate side effects of treatment. It has been estimated that the risk for significant tumor enlargement during pregnancy is 1.6% to 5.5% in microprolactinomas and 15.6% to 35.7% in macroprolactinomas.18
For patients with macroadenomas, especially those in close proximity to the optic chiasm or cavernous sinuses, contraception should be encouraged while attempting to reduce the size of the tumor prior to pregnancy. If a patient with a macroprolactinoma becomes pregnant, there are several different therapeutic approaches that can be offered: (1) dopamine agonists may be stopped after conception with neuro-ophthalmological exams performed throughout pregnancy, (2) dopamine agonists may be continued throughout pregnancy, (3) trans-sphenoidal surgery may be performed in later stages of pregnancy if the enlarged tumor does not respond to resumption of dopamine agonists, or (4) delivery if tumor growth cannot be controlled and the pregnancy is advanced enough.2
Bromocriptine has conventionally been the drug of choice in women with prolactinomas who desire fertility or who become pregnant. There is substantial evidence showing that exposure to bromocriptine at the time of conception does not cause birth defects or increase the risk of spontaneous abortions, ectopic pregnancies, trophoblastic disease, or multiple pregnancies.29 However, evidence is also accumulating to show that cabergoline may have an equally good safety record in terms of pregnancy outcomes.30 A recent 12-year prospective study of cabergoline use in 329 pregnancies showed no increased risk of miscarriage or fetal malformation.30
Dopamine Agonist Resistance
A subset of patients with prolactin-secreting pituitary tumors demonstrate “biochemical” resistance (failure to normalize prolactin levels) or “mass” resistance (absence of tumor shrinkage) to dopamine agonists.2 Resistance to dopamine agonists occurs in both micro and macro-adenomas and is believed to be mediated by the downregulation of pituitary D2 receptors.31 Biochemical resistance has been estimated to occur in approximately 10% to 20% of patients on dopamine agonists. Mass resistance has been estimated to occur in approximately 30% to 40% and 15% of patients to bromocriptine and cabergoline, respectively.2, 31 The prevalence of resistance to quinagolide is unclear given the lack of data regarding its use in dopamine-agonist naïve patients.6
Most tumors resistant to bromocriptine will respond to cabergoline. When switched to cabergoline, 85% of patients resistant to bromocriptine and quinagolide achieved normal prolactin levels, and 70% had some change in tumor size.32 A recent large study showed that cabergoline normalized prolactin levels in 61% and 50% of bromocriptine-resistant microprolactinomas and macroprolactinomas, respectively.11
Therapeutic options in the management of dopamine-resistant prolactinomas include (1) increasing the dose of the dopamine agonist, (2) switching to an alternative dopamine agonist, (3) trans-sphenoidal surgery, and (4) radiation therapy. In most cases, the resistance to dopamine agonist is partial, and a response can be achieved by progressively increasing the dose of the drug. A recent study showed that in more than 95% of patients resistant to bromocriptine prolactin normalized with individualized high-dose cabergoline treatment (mean dose 5.2 mg/wk).8 Bromocriptine-resistant patients generally require higher doses of cabergoline and have less tumor shrinkage compared to treatment-naïve patients.33 In general, switching from cabergoline to bromocriptine is unlikely to be efficacious.
While trans-sphenoidal surgery is never a first-line treatment for macroprolactinomas, it remains an important option for patients who cannot tolerate dopamine agonists or when medical therapy is ineffective at controlling symptoms or restoring reproductive function. There is little long-term outcome data regarding patients with macroprolactinomas treated with dopamine agonists who subsequently proceed to surgery. In a recent study of 72 patients, 35% of patients required trans-sphenoidal surgery due to resistance and/or intolerance of dopamine agonists.34 This study, which an outlier given the disproportionately high number of dopamine agonist resistant patients, showed that additional tumor shrinkage was achieved in 57% of operated patients, while only 22% were able to attain normoprolactinemia without dopamine agonists following surgery.34 Surgery was associated with a high incidence of hypopituitarism regardless of whether patients received subsequent radiotherapy.35 The remission rate for surgery when used as second-line treatment for prolactinomas is quite low (around 22%). As opposed to its curative role in the other secretory macroadenomas, surgery plays only a “debulking” role in macroprolactinomas, and therefore should be reserved for patients who, despite maximal medical therapy, have progressive tumor enlargement and are at risk of visual compromise or neurological deficits.35
Conventional fractionated radiation therapy should be considered a last-line option in the management of dopamine-resistant prolactinomas given the delayed treatment effects and the high rate of panhypopituitarism. On the other hand, radiosurgery may be an effective treatment in secretory pituitary adenomas, with a lower risk of long-term hypopituitarism.36
Dopamine Agonist Withdrawal
The optimal duration of therapy for patients with prolactinomas is uncertain. Until recently, dopamine agonist treatment had been considered a “lifelong” requirement. However, after a landmark study by Colao et al. demonstrated disease remission in a considerable proportion of patients following withdrawal from cabergoline, attention shifted towards defining selection criteria for withdrawal and identifying predictors of long-term remission.12, 37 Defining the optimal timing and appropriate candidates for withdrawal is difficult because of the heterogeneity of studies that have examined this issue, which differ with respect to the causes of hyperprolactinemia (idiopathic vs. micro/macroprolactinoma), the type and duration of dopamine agonist treatment, and the treatment prior to the start of dopamine agonist therapy.38 Despite the inherent difficulty in extrapolating from these disparate studies, the Pituitary Society has proposed certain criteria for withdrawal, including (1) normoprolactinemia and (2) tumor absence or markedly reduced tumor volume after a minimum of 1 to 3 years of dopamine agonist treatment.39 A recent study tested the applicability of these criteria and found a recurrence rate of 54% after withdrawal of long-term cabergoline treatment.13 In this study, most patients had recurrence of disease within 1 year of discontinuation, and a similar recurrence rate (52% vs. 55%) was seen in patients with microprolactinomas and macroprolactinomas.13 The size of the tumor remnant prior to withdrawal appears to be predictive of recurrence risk.13
A recent meta-analysis showed that long-term remission occurs in only 21% of patients following dopamine agonist withdrawal.38 The best chance for a long-term remission is seen in patients treated with cabergoline for more than 2 years, with a mean remission rate around 54%, whereas remission is seen in only 20% of patients withdrawn from bromocriptine.38 Considering the financial burden, occasional problems with tolerability, and potential risk of valvulopathy with long-term dopamine agonist treatment, it is reasonable to attempt withdrawal in patients who have been on a dopamine agonist (particularly if cabergoline) for 2 years or more.
Acromegaly
General Considerations
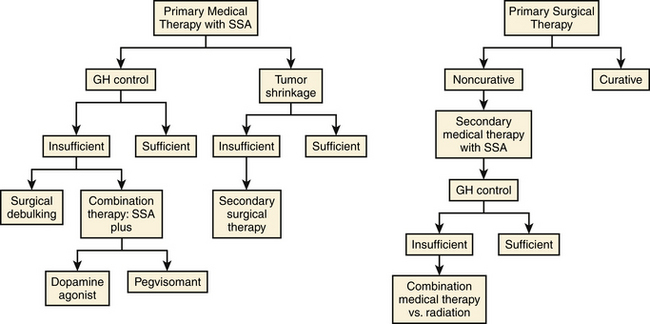
Trans-sphenoidal surgery is currently the first treatment approach in acromegaly when a definitive cure can be expected, as is the case with microadenomas, where cure rates are as high as 80% in the hands of an experienced neurosurgeon.38 Surgery is also indicated for decompressive purposes in the cases of large tumors that cause chiasmatic compression. Since the biochemical cure rate of macroadenomas following surgery is less than 50% (particularly if extrasellar extension of the tumor is present), many patients will require some form of adjuvant treatment following surgical resection.40 In addition, medical therapy should be considered as primary approach for patients without vision compromise who either have higher than normal surgical risk or whose tumor is deemed not to be surgically curable.41 Finally, two recent studies have suggested an increase in surgical cure rate in macroadenomas when they are pre-treated for 4 to 6 months with a long-acting somatostatin analog. If confirmed, this may expand the role of these drugs to all macroadenomas before surgical attempt.42, 43 The estimated benefit of postsurgical radiotherapy is often outweighed by the delayed onset of effects and the risk of panhypopituitarism.
1. Somatostatin analogues, octreotide and lanreotide, inhibit somatotroph cell proliferation and GH secretion by binding to the somatostatin receptors on tumoral cells.
2. Dopamine agonists bind to dopamine D2 receptors expressed on both somatotroph and mammotroph cells, exerting negative control.
3. The GH receptor antagonist Pegvisomant blocks the effect of GH. Somatostatin analogues are often considered first-line treatment.
Somatostatin Analogues: Pharmacologic Aspects
The presently commercially available somatostatin analogues act on two of the five somatostatin receptors existing in nature. The dosing frequencies of the somatostatin analogues are summarized in Table 17-5.
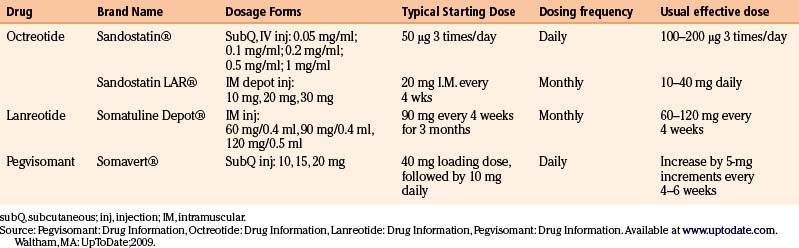
Octreotide was the first available somatostatin analogue. It has a relatively short half-life of 2 hours after subcutaneous injection, which requires a three times daily dosing to maintain therapeutic concentrations. The starting dose is usually 100 to 250 μg three times daily up to a maximum of 1500 μg daily.40 Nowadays, this formulation has been substituted by octreotide LAR (long-acting release), a long-acting depot formulation administered as an intramuscular injection every 4 weeks. The starting dose is 20 mg increasing or decreasing by 10 mg increments (maximal dose 40 mg) based on clinical and biochemical response.40
Lanreotide SR (sustained release) is a biodegradable polymer microparticle that allows a slow release following subcutaneous injection every 7 to 14 days.40 Lanreotide autogel is a longer-acting depot formulation that allows for injections every 28 days.40 In the United States, only the latter formulation is available.
Somatostatin Analogues: Therapeutic Efficacy
Somatostatin analogues are 40% to 60% effective at achieving biochemical control. Improvements in acromegaly symptoms generally correlate with the degree of IGF-1 reduction. In terms of tumor shrinkage, these drugs are 50% to 75% effective at reducing tumor size when used as primary treatment, whereas the effectiveness decreases to around 20% when used as secondary treatment.40, 44 The degree of tumor shrinkage is by no means as dramatic as the one seen with dopaminergic agents in prolactinomas. The primary treatment efficacy results are comparable to those observed in patients with macroadenomas previously treated by trans-sphenoidal surgery or pituitary irradiation,45, 46 validating the use of somatostatin analogues as a first-line modality in certain patients.
Biochemical Control
Within the class of somatostatin analogues, prospective studies have shown octreotide LAR to be modestly superior to lanreotide SR both with respect to biochemical normalization and tumor shrinkage, although the reliable interpretation of some of these studies may be compromised by a selection bias as patients were pre-selected according to sensitivity to somatostatin analogues (Fig. 17-2).47 In patients who had not been selected for somatostatin analogue responsiveness before study entry, octreotide LAR has been found to be 54% and 63% effective at normalizing GH and IGF-1 levels, respectively, whereas the efficacy rate for lanreotide SR is 48% and 42%, respectively.48 Higher-dose lanreotide SR, at 60 mg every 21 to 28 days, appears to be as effective as octreotide LAR at achieving biochemical normalization when pre-selection bias is not taken into account (Table 17-6). The depot formulation lanreotide autogel also appears to be as effective as octreotide LAR at lowering GH and IGF-1 levels, even though there is only a single randomized trial comparing the two drugs.49–55 Recent studies suggest that lanreotide autogel may be effective at tumor shrinkage in as many as 75% of patients (Table 17-6). The lanreotide autogel preparation has the advantage of being self- or partner-administered at home avoiding the need of monthly traveling to the doctor’s office.56 The relative efficacies of the somatostatin analogues are shown in Fig. 17-3.
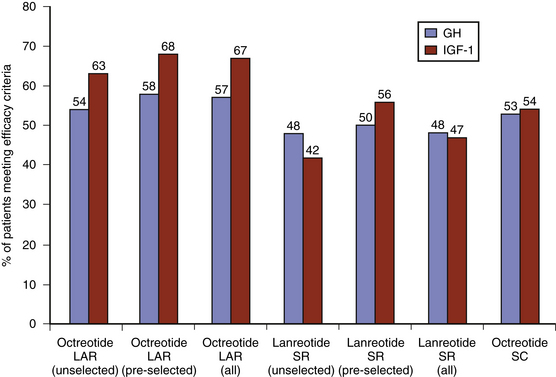
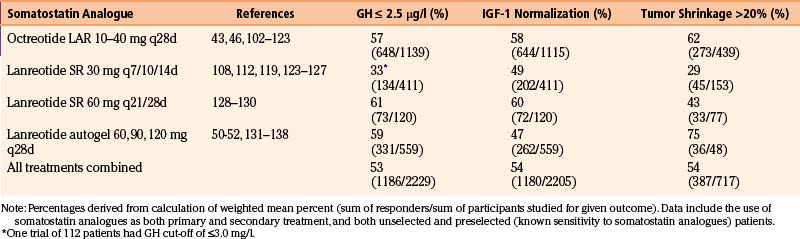
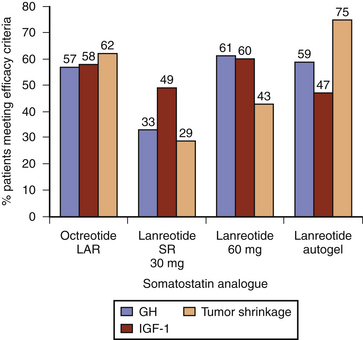
FIGURE 17-3 Efficacy of long-acting somatostatin analogues in acromegaly (data derived from Table 17-6). Studies include both preselected (sensitive to somatostatin therapy) and unselected (somatostatin analogue naive) patients, and combined primary (without surgical intervention) and secondary (following surgery or radiation) treatment outcome data. Efficacy criteria include GH ≤ 2.5 mg/l, IGF-1 normalization, and tumor volume reduction greater than 20%. LAR, long-acting release; SR, sustained release.
Tumor Shrinkage
The effects on tumor size have been shown to be slightly higher in naïve patients.47 A recent meta-analysis including treatment naïve and previously operated patients showed that 33% of patients treated with lanreotide SR or autogel experienced a variable tumor shrinkage (ranging from 10% to 77%).44 In three studies that included groups of treatment naïve patients, Lanreotide Autogel induced tumor shrinkage in 71% to 77% of patients.44 Thus far, the only predictor of response to tumor shrinkage appears to be the timing of somatostatin analogue therapy (primary vs. secondary), and data are conflicting with respect to other variables such as tumor size (micro. vs. macroadenoma), biochemical responsiveness, or dose of somatostatin analogues.
Somatostatin Analogues: Primary versus Secondary Therapy
Weighing the relative merits of surgical and medical treatment in acromegaly has proven to be difficult. The first randomized trial of medical treatment versus surgery showed a comparable success rate between octreotide LAR and trans-sphenoidal surgery, with both groups showing significant (>20%) shrinkage and similar biochemical response.57 Although this study was not powered to show superiority of medical vs. surgical treatment, it suggests that medical therapy may be used safely in patients who are not surgical candidates. Another argument for primary somatostatin treatment is the potential beneficial effect of pre-surgical treatment on surgical cure. Two recent prospective studies suggest that pre-operative treatment with somatostatin analogues may improve the chance of surgical cure in patients with macroadenomas.42, 43 Finally, the potential for reduced perioperative cardiovascular complications due to GH excess may be another reason to consider pre-operative somatostatin treatment.58 A general approach to the treatment of acromegaly is shown in Fig. 17-4.
Somatostatin Analogues: Tolerability and Side Effects
Somatostatin analogues are generally well tolerated. The most common side effects are gastrointestinal symptoms, such as abdominal cramps, flatulence, diarrhea or constipation, and nausea (Table 17-7).59 Biliary tract abnormalities, such as gallstones, sediment and sludge, and dilatation, are fairly common, occurring in as many as 50% of patients during the first 2 years of treatment, although they tend not to progress thereafter.59 Local skin irritation and pain at the injection site is mild and usually dose-dependent. Rarely, patients who require frequent injections or who are treated for long duration (i.e., metastatic carcinoid) may develop subcutaneous nodules in the gluteal area, believed to be a granulomatous reaction to the drug.60 Sinus bradycardia and conduction abnormalities have been described in 10% to 25% of patients, and medications that prolong the QT interval should not be used with somatostatin analogues.59 Impairment of pancreatic insulin secretion by somatostatin analogues may result in mild hyperglycemia, particularly in patients who have pre-existing glucose intolerance, although a clear diabetogenic effect of these medications has not been found.61 Data on the safety of somatostatin analogues during pregnancy are scarce, but there are case reports of octreotide LAR use in pregnant acromegalic women resulting in uneventful pregnancies and healthy babies.62
| Gastrointestinal (common) |
| Nausea |
| Flatulence |
| Abdominal cramps |
| Diarrhea |
| Constipation |
| Nausea |
| Gallbladder abnormalities (sediment, sludge, microlithiasis, gallstones) |
| Dermatologic |
| Local injection site irritation and pain |
| Cardiovascular |
| Asymptomatic sinus bradycardia |
| Conduction abnormalities |
| Endocrinologic |
| Impaired glucose tolerance |
| Hyperglycemia |
| Hypoglycemia (rarely) |
Dopamine Agonists in Acromegaly: Therapeutic Efficacy
As monotherapy, dopamine agonists have a limited role in acromegaly given their inferior efficacy compared with somatostatin analogues. Bromocriptine is only 15% effective at normalizing GH levels when used in high doses in acromegalic patients.40 Initial studies with cabergoline as primary treatment in acromegaly showed biochemical efficacy of 27% to 39% and suggested a higher likelihood of success in pituitary tumors that co-secrete GH and prolactin.63, 64 Interestingly, however, recent studies show that neither basal prolactin concentrations nor co-immunostaining of GH and prolactin predicts a more favorable response.65, 66 Instead, prior radiotherapy appears to be associated with an enhanced efficacy of GH response to dopamine agonists compared with somatostatin analogues.65 In general, dopamine agonists are reserved as adjuvant treatment to optimize medical therapy in partially somatostatin-resistant acromegalic patients; in these patients, the addition of cabergoline to a somatostatin analogue normalized IGF-1 levels in 42% of cases.66
Pegvisomant: Pharmacologic Aspects
Pegvisomant, a genetically engineered GH analogue, is a competitive inhibitor acting at the receptor level that prevents that action of native GH. The drug is administered as a subcutaneous injection, with an initial loading dose of 40 mg, followed by an initial dose of 10 mg daily (Table 17-5). Since pegvisomant typically causes elevations in serum GH levels, only serum IGF-1 levels can be used as markers of effective dose response. Doses are adjusted by 5 mg increments at 4- to 6-week intervals according to IGF-1 levels.
Pegvisomant: Therapeutic Efficacy
Pegvisomant was found in initial trails to be highly effective at normalizing IGF-1 levels (90%–97% patients) and controlling acromegalic symptoms; however, because of its mechanism of action at the peripheral level, this drug does not reduce tumor size or GH levels.67 A large post-marketing study has placed it efficacy in IGF-1 normalization at 70%, possibly in part due to failure of dose titration.68
Pegvisomant: Tolerability and Side Effects
Pegvisomant is usually well tolerated, with the most common side effects listed in Table 17-8. The main concern is the risk of liver damage, with up to 9% of patients reported to have elevated transaminases, sometimes as high as 8-10 times the upper limit of normal.40 Liver function tests should be assessed before treatment and monthly for the first 6 months of treatment, quarterly for the next 6 months, and biannually the following year.69
| Gastrointestinal |
| Hepatic impairment |
| Diarrhea |
| Nausea |
| Dermatologic |
| Injection site pain or skin irritation |
| Infection |
| Cardiovascular |
| Hypertension |
| Chest pain |
| Peripheral edema |
| Neurologic |
| Dizziness |
| Risk of progressive pituitary tumor growth |
Whether pegvisomant poses an increased risk of tumor growth via blockage of negative feedback by IGF-1 on the pituitary gland is uncertain. A 5-year observational study found a risk of tumor growth in 5% of patients treated with pegvisomant, which is higher than the expected 2.2% risk of continuous tumor progression during somatostatin analogue treatment.70 Another 5-year observational study found a 2.8% incidence of increased tumor growth, which appears reassuring; however this study had a lower biochemical normalization rate raising the possibility that the lower rate of tumor growth may be influenced by failure to titrate to therapeutic dose ranges.68 Given the uncertainty about tumor growth related to pegvisomant, it is recommended that patients on this drug be followed with serial MRI studies every 6 months.40
In acromegalic patients who are partially resistant to treatment with somatostatin analogues alone, the addition of once weekly subcutaneous pegvisomant injections normalized IGF-1 concentrations in 90% to 95% of cases.71 This reduced frequency dosing offers a financial advantage in patients who might otherwise require high-dose pegvisomant monotherapy. Combination therapy with pegvisomant is currently reserved for those patients who achieve insufficient control after somatostatin therapy and surgical treatment.
Emerging Therapies in Acromegaly
Pasireotide (SOM230), a novel somatostatin analogue with affinity for multiple somatostatin receptor subtypes is a promising emerging therapy for acromegaly. Initial studies have shown this drug to be as effective as the currently available somatostatin analogues.72 The effect on IGF-1 levels by pasireotide appears to be more sustained compared with octreotide, suggesting a longer half-life.72 Further long-term studies examining the effect on tumor shrinkage and sustained biochemical response are presently under way to determine whether pasireotide is a reasonable chronic treatment option in acromegaly. In addition, a newer chimeric molecule, BIM-23A387, directed towards both somatostatin and dopamine receptors, may offer improved efficacy in acromegaly due to its proposed synergistic receptor activation.73
Cushing’s Disease
General Considerations
Trans-sphenoidal surgery is the definitive treatment in Cushing’s disease; in the hands of an experienced neurosurgeon, the cure rate of ACTH-secreting pituitary microadenomas is high.74 However, approximately 10% to 30% of patients will not be cured and will require some form of adjuvant treatment, including conventional radiotherapy, stereotactic radiosurgery, medical therapy, and bilateral adrenalectomy.75 This subset of patients with refractory or residual Cushing’s disease poses a significant management challenge, as each of these secondary therapeutic options carries adverse risks. These patients are best managed in a multidisciplinary setting, such as in dedicated pituitary centers, where there is close collaboration between experienced neurosurgeons, endocrinologists, and radiation therapists.
Criteria for Cure and Remission
A step-wise approach should be taken in the postoperative evaluation of Cushing’s disease patients to assess their response to surgery.75 An expert consensus statement published in 2008 recommends assessment of remission by measurement of morning serum cortisol during the first postoperative week, either by withholding exogenous glucocorticoids or by using low dose dexamethasone testing.76 Given the likelihood of central adrenal insufficiency in patients who are surgically cured, close monitoring for signs of hypoadrenalism is necessary if glucocorticoid treatment is not empirically initiated following surgery. Persistent postoperative morning serum cortisol levels of less than 2 μg/dl are associated with remission, and a 10-year recurrence rate of approximately 10%.76 A persistent serum cortisol level above 5 μg/dl at 6 weeks following surgery requires evaluation for persistent hypercortisolemia. Biochemical evidence of pathologic ACTH hypersecretion following surgery may be indicated by repeatedly elevated urine free cortisol levels, abnormal diurnal variation patterns in serum or salivary cortisol levels, or an abnormal cortisol response to dexamethasone suppression.
Persistent Disease after Surgery
Once the diagnosis of residual (or recurrent) Cushing’s disease is confirmed, treatment options should be considered in the context of the individual patient. Repeating trans-sphenoidal surgery should be considered in most patients, especially those with accessible tumor visible on MRI. Cure rates for repeated surgery range from approximately 40% to 70%.75 The main risk associated with repeated surgery is the development of hypopituitarism. Conventional radiotherapy may not be effective until months to years following treatment, which is often not acceptable in a patient with persistent, severe hypercortisolemia. Single-treatment stereotactic radiosurgery offers the advantage of minimized radiation to normal pituitary or adjacent structures, and has been reported to have a remission rate of 50%.36 A recent study using stereotactic radiosurgery as either primary or adjuvant therapy in the treatment of secretory pituitary adenomas (acromegaly, Cushing’s disease, prolactinomas) found a 23% incidence of hypopituitarism at 4- to 8-year follow-up.36
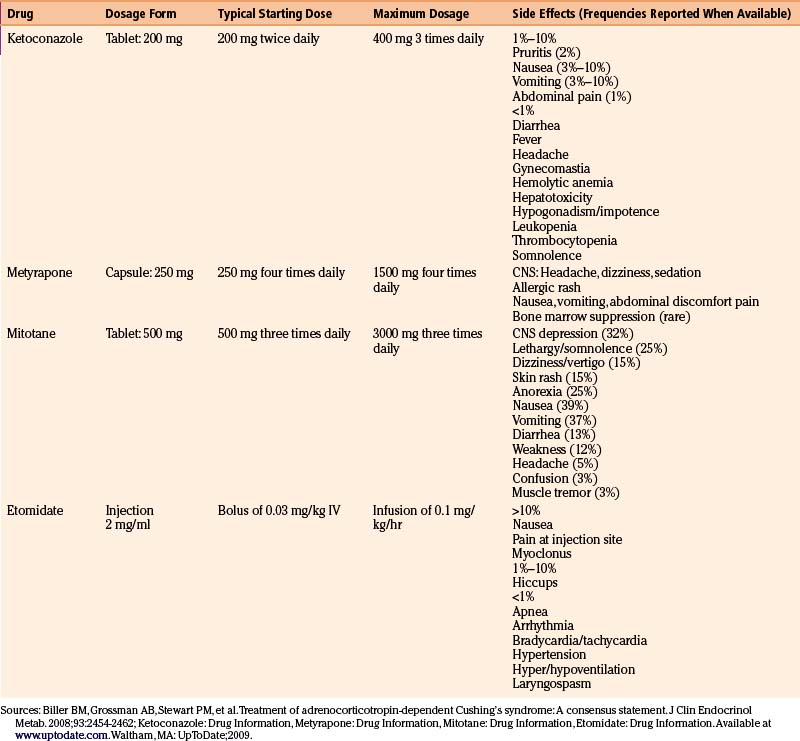
Adrenal Inhibitors: Pharmacologic Aspects, Therapeutic Efficacy, and Side Effects
Ketoconazole is the first-line drug used to lower cortisol levels in Cushing’s disease patients. This oral antifungal agent, at higher doses, blocks cortisol production by inhibiting two enzymes in the glucocorticoid synthesis pathway, 17 alpha-hydroxylase and 17,20 lyase.67 Typical starting doses are 200 mg twice daily, titrated up to a maximal dose of 400 mg four times daily based on serum cortisol levels and 24-hour urine free cortisol levels.67 Although remission rates on ketoconazole have been reported in 25% to 76% of patients, the medication is generally poorly tolerated, and carries a high risk of hepatitis at the chronically high doses often required to treat hypercortisolemia.67 Hepatitis occurs in as many as 12% of patients and therefore close monitoring of liver function tests is required. The increased risk of hypogonadism in men may be a reason to consider metyrapone as an alternative in this patient population. In order to be absorbed, ketoconazole requires low gastric pH. Absorption is therefore reduced in patients with achlorhydria or on proton pump inhibitors or H2 blockers. The use of acidic drinks such as coca cola improves absorption of the drug.77
Although no longer commercially available in the United States, metyrapone can be provided on a compassionate basis by contacting the manufacturer (Novartis) directly.76 This drug, which works by blocking the final step in cortisol synthesis, is 80% effective at controlling hypercortisolemia.78 Cortisol suppression can be maintained chronically with doses of 500 to 2000 mg/day.79 Nausea, vomiting, and dizziness may occur with metyrapone, possibly due to sudden cortisol withdrawal and relative adrenal insufficiency.79 Another adverse side effect is acne and hirsutism (in women), which results from shunting of steroidogenic precursors towards the androgen pathway. This side effect may be a reason to favor ketoconazole in female patients.
Mitotane, a cytotoxic drug primarily used in the treatment of patients with adrenocortical carcinoma, can be used at lower doses to control hypercortisolemia. The mechanism of the adrenolytic effect is postulated to involve covalent binding to target proteins within adrenal cells resulting in oxidative damage and adrenal cortical atrophy.79 Typical therapeutic doses range from 2 to 4 g, with the maximum daily dose of 9 g.76 The main drawbacks to this medication are its slow onset of action (weeks to months) and poor tolerability due to gastrointestinal and central nervous system side effects. Careful monitoring of drug and cortisol levels is required.76
Intravenous etomidate therapy is reserved for situations where rapid control of cortisol levels is required and there are barriers to oral therapy.76 The drug lowers cortisol and aldosterone levels by blocking 11-β-hydroxylase. Short-term continuous infusions of etomidate reduce serum cortisol in 11 to 24 hours.79
The pharmacologic aspects and side effects of the adrenal inhibitors76,139 are summarized in Table 17-9.
Experimental Treatments in Cushing’s Disease
There are currently no medical therapies that are effective at shrinking ACTH-secreting pituitary tumors, and many tumor-targeted drugs are being investigated. The dopamine receptors, D2 and D4, are expressed in about 75% of ACTH-pituitary adenomas, making dopamine agonists appealing experimental agents. Cabergoline may be partially effective in controlling the hypercortisolemia in patients with Cushing’s disease, but the long-term efficacy and the effects on tumor shrinkage remain to be seen.80 The newer somatostatin analogue, pasireotide (SOM230), has been shown to reduce ACTH secretion in vitro; early clinical studies show promising results in patients with de novo, persistent, or recurrent Cushing’s disease.81 Studies of patients with ectopic ACTH syndrome suggest that the combination of somatostatin analogues and dopamine agonists may offer additional therapeutic benefit in patients with pituitary-dependent Cushing’s disease.10
TSH-Secreting Pituitary Adenomas
TSH-secreting pituitary adenomas are exceedingly rare. The goals of therapy include tumor removal and restoration of the euthyroid state. While trans-sphenoidal surgery is the first approach, because most patients are diagnosed when adenomas are macros, the reported cure rate is only around 38%.82 For those patients who do not achieve surgical remission, adjuvant medical therapy offers promising results. Medical treatment relies mainly on the use of somatostatin analogues, which inhibit thyrotroph cell growth by binding to the somatostatin receptor found on both normal pituitary and tumoral cells. Octreotide is 90% effective at normalizing thyroid hormone levels and 50% effective at reducing tumor size.10 As with growth-hormone secreting tumors, there is evidence that pre-surgical treatment with somatostatin analogues may impact the ease of surgical removal.83 Typically, the doses of somatostatin analogues required to normalize TSH levels are lower than the doses used in the treatment of GH-secreting tumors: octreotide SC (300 mg/day), lanreotide SR (30 mg every 14 days), and octreotide LAR (10 mg/day).10 However, due to the rarity of this disease, it is difficult to make generalizations about dose requirements, so each patient’s response and tolerance to somatostatin therapy should be considered individually.
Clinically Nonfunctioning Pituitary Tumors
Nonfunctioning pituitary tumors are usually not amenable to medical treatment. However, because the absence of hormonal hypersecretion frequently leads to a delayed diagnosis, complete surgical cure may not always be possible due to the large size of these tumors at presentation. Radiotherapy may be offered to prevent tumor regrowth. Recently, medical treatment with dopamine agonists, somatostatin analogues, or a combination of the two has been explored as adjuvant treatment in nonfunctioning tumors. Treatment with these drugs relies on the fact that many nonfunctioning adenomas express dopamine and somatostatin receptors. Improvements in visual field were seen in 20% and 32% of tumors treated with dopamine agonists and somatostatin analogues, respectively; tumor shrinkage was seen in 28% and 5%, respectively.84 Larger controlled trials are still needed to determine the effectiveness of medical therapy in this tumor type.
Beck-Peccoz P., Persani L. Thyrotropinomas. Endocrinol Metab Clin North Am. 2008;37:123-134. viii-ix
Ben-Shlomo A., Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2008;37:101-122. viii
Blevins L.S.Jr., Sanai N., Kunwar S., Devin J.K. An approach to the management of patients with residual Cushing’s disease. J Neurooncol. 2009 Sep;94(3):313-319.
Brue T. ACROSTUDY: Status update on 469 patients. Horm Res. 2009 Jan;71(Suppl 1):34-38.
Carlsen S.M., Lund-Johansen M., Schreiner T., et al. Preoperative octreotide treatment in newly diagnosed acromegalic patients with macroadenomas increases cure short-term postoperative rates: a prospective, randomized trial. J Clin Endocrinol Metab. 2008 Aug;93(8):2984-2990.
Casanueva F.F., Molitch M.E., Schlechte J.A., et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin Endocrinol (Oxf). 2006 Aug;65(2):265-273.
Chanson P., Boerlin V., Ajzenberg C., et al. Comparison of octreotide acetate LAR and lanreotide SR in patients with acromegaly. Clin Endocrinol (Oxf). 2000 Nov;53(5):577-586.
Colao A., Pivonello R., Di Somma C., et al. Medical therapy of pituitary adenomas: effects on tumor shrinkage. Rev Endocr Metab Disord. 2009 Jun;10(2):111-123.
Dekkers O.M., Lagro J., Burman P., et al. Recurrence of hyperprolactinemia after withdrawal of dopamine agonists: systematic review and meta-analysis. J Clin Endocrinol Metab. 2010;95:43-51.
Freda P.U., Katznelson L., van der Lely A.J., et al. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005 Aug;90(8):4465-4473.
Kharlip J., Salvatori R., Yenokyan G., Wand G.S. Recurrence of hyperprolactinemia after withdrawal of long-term cabergoline therapy. J Clin Endocrinol Metab. 2009 Jul;94(7):2428-2436.
Maiza J.C., Vezzosi D., Matta M., et al. Long-term (up to 18 years) effects on GH/IGF-1 hypersecretion and tumour size of primary somatostatin analogue (SSTa) therapy in patients with GH-secreting pituitary adenoma responsive to SSTa. Clin Endocrinol (Oxf). 2007 Aug;67(2):282-289.
Mancini T., Casanueva F.F., Giustina A. Hyperprolactinemia and prolactinomas. Endocrinol Metab Clin North Am. 2008 Mar;37(1):67-99. viii
Mao Z.G., Zhu Y.H., Tang H.L., et al. Preoperative lanreotide treatment in acromegalic patients with macroadenomas increases short-term postoperative cure rates: a prospective, randomised trial. Eur J Endocrinol. 2010 Apr;162(4):661-666.
Mazziotti G., Giustina A. Effects of lanreotide SR and Autogel on tumor mass in patients with acromegaly: a systematic review. Pituitary. 2010;13(1):60-67.
Molitch M.E. Pituitary disorders during pregnancy. Endocrinol Metab Clin North Am. 2006 Mar;35(1):99-116. vi
Ono M., Miki N., Kawamata T., et al. Prospective study of high-dose cabergoline treatment of prolactinomas in 150 patients. J Clin Endocrinol Metab. 2008 Dec;93(12):4721-4727.
Pascal-Vigneron V., Weryha G., Bosc M., Leclere J. [Hyperprolactinemic amenorrhea: treatment with cabergoline versus bromocriptine. Results of a national multicenter randomized double-blind study]. Presse Med. 1995 Apr 29;24(16):753-757.
Schteingart D.E. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs. 2009 Dec;14(4):661-671.
Valassi E., Klibanski A., Biller B.M. Clinical review: potential cardiac valve effects of dopamine agonists in hyperprolactinemia. J Clin Endocrinol Metab. 2010 Mar;95(3):1025-1033.
Vilar L., Freitas M.C., Naves L.A., et al. Diagnosis and management of hyperprolactinemia: results of a Brazilian multicenter study with 1234 patients. J Endocrinol Invest. 2008 May;31(5):436-444.
Webster J., Piscitelli G., Polli A., et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group. N Engl J Med. 1994 Oct 6;331(14):904-909.
1. Osamura R.Y., Kajiya H., Takei M., et al. Pathology of the human pituitary adenomas. Histochem Cell Biol. 2008 Sep;130(3):495-507.
2. Mancini T., Casanueva F.F., Giustina A. Hyperprolactinemia and prolactinomas. Endocrinol Metab Clin North Am. 2008 Mar;37(1):67-99. viii
3. Gillam M.P., Middler S., Freed D.J., Molitch M.E. The novel use of very high doses of cabergoline and a combination of testosterone and an aromatase inhibitor in the treatment of a giant prolactinoma. J Clin Endocrinol Metab. 2002 Oct;87(10):4447-4451.
4. Hattori N. Macroprolactinemia: a new cause of hyperprolactinemia. J Pharmacol Sci. 2003 Jul;92(3):171-177.
5. Mori H., Maeda T., Saitoh Y., Onishi T. Changes in prolactinomas and somatotropinomas in humans treated with bromocriptine. Pathol Res Pract. 1988 Sep;183(5):580-583.
6. Gillam M.P., Molitch M.E., Lombardi G., Colao A. Advances in the treatment of prolactinomas. Endocr Rev. 2006 Aug;27(5):485-534.
7. Del Dotto P., Bonuccelli U. Clinical pharmacokinetics of cabergoline. Clin Pharmacokinet. 2003;42(7):633-645.
8. Ono M., Miki N., Kawamata T., Makino R., Amano K., Seki T., et al. Prospective study of high-dose cabergoline treatment of prolactinomas in 150 patients. J Clin Endocrinol Metab. 2008;93:4721-4727.
9. Esiri M.M., Bevan J.S., Burke C.W., Adams C.B. Effect of bromocriptine treatment on the fibrous tissue content of prolactin-secreting and nonfunctioning macroadenomas of the pituitary gland. J Clin Endocrinol Metab. 1986 Aug;63(2):383-388.
10. Colao A., Pivonello R., Di Somma C., et al. Medical therapy of pituitary adenomas: effects on tumor shrinkage. Rev Endocr Metab Disord. 2009 Jun;10(2):111-123.
11. Vilar L., Freitas M.C., Naves L.A., et al. Diagnosis and management of hyperprolactinemia: results of a Brazilian multicenter study with 1234 patients. J Endocrinol Invest. 2008 May;31(5):436-444.
12. Colao A., Di Sarno A., Guerra E., et al. Predictors of remission of hyperprolactinaemia after long-term withdrawal of cabergoline therapy. Clin Endocrinol (Oxf). 2007 Sep;67(3):426-433.
13. Kharlip J., Salvatori R., Yenokyan G., Wand G.S. Recurrence of hyperprolactinemia after withdrawal of long-term cabergoline therapy. J Clin Endocrinol Metab. 2009 Jul;94(7):2428-2436.
14. Webster J., Piscitelli G., Polli A., et al. A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group. N Engl J Med. 1994 Oct 6;331(14):904-909.
15. Colao A., Vitale G., Cappabianca P., et al. Outcome of cabergoline treatment in men with prolactinoma: effects of a 24-month treatment on prolactin levels, tumor mass, recovery of pituitary function, and semen analysis. J Clin Endocrinol Metab. 2004 Apr;89(4):1704-1711.
16. Schultz P.N., Ginsberg L., McCutcheon I.E., et al. Quinagolide in the management of prolactinoma. Pituitary. 2000 Dec;3(4):239-249.
17. Orrego J.J., Chandler W.F., Barkan A.L. Pergolide as primary therapy for macroprolactinomas. Pituitary. 2000 Dec;3(4):251-256.
18. Colao A., di Sarno A., Pivonello R., et al. Dopamine receptor agonists for treating prolactinomas. Expert Opin Investig Drugs. 2002 Jun;11(6):787-800.
19. Pascal-Vigneron V., Weryha G., Bosc M., Leclere J. [Hyperprolactinemic amenorrhea: treatment with cabergoline versus bromocriptine. Results of a national multicenter randomized double-blind study]. Presse Med. 1995 Apr 29;24(16):753-757.
20. van der Heijden P.F., de Wit W., Brownell J., et al. 205-502, a new dopamine agonist, versus bromocriptine in the treatment of hyperprolactinaemia. Eur J Obstet Gynecol Reprod Biol. 1991 Jul 1;40(2):111-118.
21. Zanettini R., Antonini A., Gatto G. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med. 2007 Jan 4;356(1):39-46.
22. Schade R., Andersohn F., Suissa S., et al. Dopamine agonists and the risk of cardiac-valve regurgitation. N Engl J Med. 2007 Jan 4;356(1):29-38.
23. Cheung D., Heaney A. Dopamine agonists and valvular heart disease. Curr Opin Endocrinol Diabetes Obes. 2009 Aug;16(4):316-320.
24. Tan L.C., Ng K.K., Au W.L., Lee R.K., Chan Y.H., Tan N.C. Bromocriptine use and the risk of valvular heart disease. Mov Disord. 2009 Feb 15;24(3):344-349.
25. Valassi E., Klibanski A., Biller B.M. Clinical review: potential cardiac valve effects of dopamine agonists in hyperprolactinemia. J Clin Endocrinol Metab. 2010 Mar;95(3):1025-1033.
26. Colao A., Galderisi M., Di Sarno A., et al. Increased prevalence of tricuspid regurgitation in patients with prolactinomas chronically treated with cabergoline. J Clin Endocrinol Metab. 2008 Oct;93(10):3777-3784.
27. Wakil A., Rigby A.S., Clark A.L., et al. Low dose cabergoline for hyperprolactinaemia is not associated with clinically significant valvular heart disease. Eur J Endocrinol. 2008 Oct;159(4):R11-R14.
28. Kars M., Delgado V., Holman E.R., et al. Aortic valve calcification and mild tricuspid regurgitation but no clinical heart disease after 8 years of dopamine agonist therapy for prolactinoma. J Clin Endocrinol Metab. 2008 Sep;93(9):3348-3356.
29. Molitch M.E. Pituitary disorders during pregnancy. Endocrinol Metab Clin North Am. 2006 Mar;35(1):99-116. vi
30. Colao A., Abs R., Barcena D.G., et al. Pregnancy outcomes following cabergoline treatment: extended results from a 12-year observational study. Clin Endocrinol (Oxf). 2008 Jan;68(1):66-71.
31. Olafsdottir A., Schlechte J. Management of resistant prolactinomas. Nat Clin Pract Endocrinol Metab. 2006 Oct;2(10):552-561.
32. Colao A., Di Sarno A., Sarnacchiaro F., et al. Prolactinomas resistant to standard dopamine agonists respond to chronic cabergoline treatment. J Clin Endocrinol Metab. 1997 Mar;82(3):876-883.
33. Colao A., Di Sarno A., Landi M.L., et al. Macroprolactinoma shrinkage during cabergoline treatment is greater in naive patients than in patients pretreated with other dopamine agonists: a prospective study in 110 patients. J Clin Endocrinol Metab. 2000 Jun;85(6):2247-2252.
34. Grings F., Salvia M., Karszenbaum H., et al. Exploring the capacity of radar remote sensing to estimate wetland marshes water storage. J Environ Manage. 2009 May;90(7):2189-2198.
35. Kars M., Pereira A.M., Smit J.W., Romijn J.A. Long-term outcome of patients with macroprolactinomas initially treated with dopamine agonists. Eur J Intern Med. 2009 Jul;20(4):387-393.
36. Castinetti F., Nagai M., Morange I., et al. Long-term results of stereotactic radiosurgery in secretory pituitary adenomas. J Clin Endocrinol Metab. 2009 Sep;94(9):3400-3407.
37. Colao A., Di Sarno A., Cappabianca P., et al. Withdrawal of long-term cabergoline therapy for tumoral and nontumoral hyperprolactinemia. N Engl J Med. 2003 Nov 20;349(21):2023-2033.
38. Dekkers O.M., Lagro J., Burman P., et al. Recurrence of hyperprolactinemia after withdrawal of dopamine agonists: systematic review and meta-analysis. J Clin Endocrinol Metab. 2009 Oct 30.
39. Casanueva F.F., Molitch M.E., Schlechte J.A., et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin Endocrinol (Oxf). 2006 Aug;65(2):265-273.
40. Ben-Shlomo A., Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2008 Mar;37(1):101-122. viii
41. Bush Z.M., Vance M.L. Management of acromegaly: is there a role for primary medical therapy? Rev Endocr Metab Disord. 2008 Mar;9(1):83-94.
42. Mao Z.G., Zhu Y.H., Tang H.L., et al. Preoperative lanreotide treatment in acromegalic patients with macroadenomas increases short-term postoperative cure rates: a prospective, randomised trial. Eur J Endocrinol. 2010 Apr;162(4):661-666.
43. Carlsen S.M., Lund-Johansen M., Schreiner T., et al. Preoperative octreotide treatment in newly diagnosed acromegalic patients with macroadenomas increases cure short-term postoperative rates: a prospective, randomized trial. J Clin Endocrinol Metab. 2008 Aug;93(8):2984-2990.
44. Mazziotti G., Giustina A. Effects of lanreotide SR and Autogel on tumor mass in patients with acromegaly: a systematic review. Pituitary. 2010;13(1):60-67.
45. Maiza J.C., Vezzosi D., Matta M., et al. Long-term (up to 18 years) effects on GH/IGF-1 hypersecretion and tumour size of primary somatostatin analogue (SSTa) therapy in patients with GH-secreting pituitary adenoma responsive to SSTa. Clin Endocrinol (Oxf). 2007 Aug;67(2):282-289.
46. Mercado M., Borges F., Bouterfa H., et al. A prospective, multicentre study to investigate the efficacy, safety and tolerability of octreotide LAR (long-acting repeatable octreotide) in the primary therapy of patients with acromegaly. Clin Endocrinol (Oxf). 2007 Jun;66(6):859-868.
47. Feelders R.A., Hofland L.J., van Aken M.O., et al. Medical therapy of acromegaly: efficacy and safety of somatostatin analogues. Drugs. 2009;69(16):2207-2226.
48. Freda P.U., Katznelson L., van der Lely A.J., et al. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005 Aug;90(8):4465-4473.
49. Caron P., Beckers A., Cullen D.R., et al. Efficacy of the new long-acting formulation of lanreotide (lanreotide Autogel) in the management of acromegaly. J Clin Endocrinol Metab. 2002 Jan;87(1):99-104.
50. Ashwell S.G., Bevan J.S., Edwards O.M., et al. The efficacy and safety of lanreotide Autogel in patients with acromegaly previously treated with octreotide LAR. Eur J Endocrinol. 2004 Apr;150(4):473-480.
51. Alexopoulou O., Abrams P., Verhelst J., et al. Efficacy and tolerability of lanreotide Autogel therapy in acromegalic patients previously treated with octreotide LAR. Eur J Endocrinol. 2004 Sep;151(3):317-324.
52. Ronchi C.L., Boschetti M., Degli Uberti E.C., et al. Efficacy of a slow-release formulation of lanreotide (Autogel 120 mg) in patients with acromegaly previously treated with octreotide long acting release (LAR): an open, multicentre longitudinal study. Clin Endocrinol (Oxf). 2007 Oct;67(4):512-519.
53. Andries M., Glintborg D., Kvistborg A., et al. 12-month randomized crossover study on the effects of lanreotide Autogel and octreotide long-acting repeatable on GH and IGF-l in patients with acromegaly. Clin Endocrinol (Oxf). 2008 Mar;68(3):473-480.
54. Kelly P., Maher K., Chew S.L., et al. A single-center open-label study to investigate the efficacy and safety of repeat subcutaneous injections of lanreotide Autogel (ATG; Somatuline Depot) in patients with acromegaly previously treated with octreotide. Endocr Pract. 2009 Oct 15:1-27.
55. Melmed S., Cook D., Schopohl J., et al. Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel((R)) therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension. Pituitary. 2010;13:18-28.
56. Salvatori R., Nachtigall L.B., Cook D.M., et al. Effectiveness of self- or partner-administration of an extended-release aqueous-gel formulation of lanreotide in lanreotide-naive patients with acromegaly. Pituitary. 2010;13:115-122.
57. Colao A., Cappabianca P., Caron P., et al. Octreotide LAR vs. surgery in newly diagnosed patients with acromegaly: a randomized, open-label, multicentre study. Clin Endocrinol (Oxf). 2009 May;70(5):757-768.
58. Nemergut E.C., Dumont A.S., Barry U.T., Laws E.R. Perioperative management of patients undergoing transsphenoidal pituitary surgery. Anesth Analg. 2005 Oct;101(4):1170-1181.
59. Ben-Shlomo A., Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2001 Sep;30(3):565-583. vi
60. Debono M., Hon L.Q., Bax N., et al. Gluteal nodules in patients with metastatic midgut carcinoid disease treated with depot somatostatin analogs. J Clin Endocrinol Metab. 2008 May;93(5):1860-1864.
61. Colao A., Auriemma R.S., Savastano S., et al. Glucose tolerance and somatostatin analog treatment in acromegaly: a 12-month study. J Clin Endocrinol Metab. 2009 Aug;94(8):2907-2914.
62. Maffei P., Tamagno G., Nardelli G.B., et al. Effects of octreotide exposure during pregnancy in acromegaly. Clin Endocrinol (Oxf). 2010;72:668-677.
63. Abs R., Verhelst J., Maiter D., et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998 Feb;83(2):374-378.
64. Cozzi R., Attanasio R., Barausse M., et al. Cabergoline in acromegaly: a renewed role for dopamine agonist treatment? Eur J Endocrinol. 1998 Nov;139(5):516-521.
65. Sherlock M., Fernandez-Rodriguez E., Alonso A.A., et al. Medical therapy in patients with acromegaly: predictors of response and comparison of efficacy of dopamine agonists and somatostatin analogues. J Clin Endocrinol Metab. 2009 Apr;94(4):1255-1263.
66. Cozzi R., Attanasio R., Lodrini S., Lasio G. Cabergoline addition to depot somatostatin analogues in resistant acromegalic patients: efficacy and lack of predictive value of prolactin status. Clin Endocrinol (Oxf). 2004 Aug;61(2):209-215.
67. Patil C.G., Hayden M., Katznelson L., Chang S.D. Non-surgical management of hormone-secreting pituitary tumors. J Clin Neurosci. 2009 Aug;16(8):985-993.
68. Trainer P.J. ACROSTUDY: The first 5 years. Eur J Endocrinol. 2009 Nov;161(suppl 1):S19-S24.
69. Pegvisomant. Drug Information, Available at www.uptodate.com, Waltham, MA, UpToDate, 2009.
70. Brue T. ACROSTUDY: Status update on 469 patients. Horm Res. 2009 Jan;71(suppl 1):34-38.
71. Feenstra J., de Herder W.W., ten Have S.M., et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005 May 7-13;365(9471):1644-1646.
72. van der Hoek J., van der Lelij A.J., Feelders R.A., et al. The somatostatin analogue SOM230, compared with octreotide, induces differential effects in several metabolic pathways in acromegalic patients. Clin Endocrinol (Oxf). 2005 Aug;63(2):176-184.
73. Jaquet P., Gunz G., Saveanu A., et al. Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. Eur J Endocrinol. 2005 Jul;153(1):135-141.
74. Porterfield J.R., Thompson G.B., Young W.F.Jr., et al. Surgery for Cushing’s syndrome: an historical review and recent ten-year experience. World J Surg. 2008 May;32(5):659-677.
75. Blevins L.S.Jr., Sanai N., Kunwar S., Devin J.K. An approach to the management of patients with residual Cushing’s disease. J Neurooncol. 2009 Sep;94(3):313-319.
76. Biller B.M., Grossman A.B., Stewart P.M., et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008 Jul;93(7):2454-2462.
77. Chin T.W., Loeb M., Fong I.W. Effects of an acidic beverage (Coca-Cola) on absorption of ketoconazole. Antimicrob Agents Chemother. 1995 Aug;39(8):1671-1675.
78. Shalet S., Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes. 2008 Jun;15(3):234-238.
79. Schteingart D.E. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs. 2009 Dec;14(4):661-671.
80. Pivonello R., De Martino M.C., Cappabianca P., et al. The medical treatment of Cushing’s disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab. 2009 Jan;94(1):223-230.
81. Boscaro M., Ludlam W.H., Atkinson B., et al. Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. J Clin Endocrinol Metab. 2009 Jan;94(1):115-122.
82. Beck-Peccoz P., Persani L. Thyrotropinomas. Endocrinol Metab Clin North Am. 2008 Mar;37(1):123-134. viii-ix
83. Beck-Peccoz P., Brucker-Davis F., Persani L., Smallridge R.C., Weintraub B.D. Thyrotropin-secreting pituitary tumors. Endocr Rev. 1996 Dec;17(6):610-638.
84. Colao A., Di Somma C., Pivonello R., et al. Medical therapy for clinically non-functioning pituitary adenomas. Endocr Relat Cancer. 2008 Dec;15(4):905-915.
85. . UpToDate, Inc. Bromocriptine: Drug Information, Cabergoline: Drug Information, Quinagolide: Drug Information, Available at www.uptodate.com, Waltham, MA, UpToDate, 2009.
86. Ciccarelli E., Grottoli S., Razzore P., et al. Long-term treatment with cabergoline, a new long-lasting ergoline derivate, in idiopathic or tumorous hyperprolactinaemia and outcome of drug-induced pregnancy. J Endocrinol Invest. 1997 Oct;20(9):547-551.
87. Muratori M., Arosio M., Gambino G., et al. Use of cabergoline in the long-term treatment of hyperprolactinemic and acromegalic patients. J Endocrinol Invest. 1997 Oct;20(9):537-546.
88. Cannavo S., Curto L., Squadrito S., et al. Cabergoline: a first-choice treatment in patients with previously untreated prolactin-secreting pituitary adenoma. J Endocrinol Invest. 1999 May;22(5):354-359.
89. Verhelst J., Abs R., Maiter D., et al. Cabergoline in the treatment of hyperprolactinemia: a study in 455 patients. J Clin Endocrinol Metab. 1999 Jul;84(7):2518-2522.
90. De Luis D.A., Becerra A., Lahera M., et al. A randomized cross-over study comparing cabergoline and quinagolide in the treatment of hyperprolactinemic patients. J Endocrinol Invest. 2000 Jul-Aug;23(7):428-434.
91. Di Sarno A., Landi M.L., Marzullo P., et al. The effect of quinagolide and cabergoline, two selective dopamine receptor type 2 agonists, in the treatment of prolactinomas. Clin Endocrinol (Oxf). 2000 Jul;53(1):53-60.
92. Pinzone J.J., Katznelson L., Danila D.C., et al. Primary medical therapy of micro- and macroprolactinomas in men. J Clin Endocrinol Metab. 2000 Sep;85(9):3053-3057.
93. Di Sarno A., Landi M.L., Cappabianca P., et al. Resistance to cabergoline as compared with bromocriptine in hyperprolactinemia: prevalence, clinical definition, and therapeutic strategy. J Clin Endocrinol Metab. 2001 Nov;86(11):5256-5261.
94. Colao A., Sarno A.D., Cappabianca P., et al. Gender differences in the prevalence, clinical features and response to cabergoline in hyperprolactinemia. Eur J Endocrinol. 2003 Mar;148(3):325-331.
95. Biller B.M., Molitch M.E., Vance M.L., et al. Treatment of prolactin-secreting macroadenomas with the once-weekly dopamine agonist cabergoline. J Clin Endocrinol Metab. 1996 Jun;81(6):2338-2343.
96. Colao A., Di Sarno A., Landi M.L., et al. Long-term and low-dose treatment with cabergoline induces macroprolactinoma shrinkage. J Clin Endocrinol Metab. 1997 Nov;82(11):3574-3579.
97. Pontikides N., Krassas G.E., Nikopoulou E., Kaltsas T. Cabergoline as a first-line treatment in newly diagnosed macroprolactinomas. Pituitary. 2000 May;2(4):277-281.
98. Sibal L., Ugwu P., Kendall-Taylor P., et al. Medical therapy of macroprolactinomas in males: I. Prevalence of hypopituitarism at diagnosis. II. Proportion of cases exhibiting recovery of pituitary function. Pituitary. 2002;5(4):243-246.
99. Bolko P., Jaskula M., Wasko R., Wolun M., Sowinski J. The assessment of cabergoline efficacy and tolerability in patients with pituitary prolactinoma type. Pol Arch Med Wewn. 2003 May;109(5):489-495.
100. De Rosa M., Zarrilli S., Vitale G., et al. Six months of treatment with cabergoline restores sexual potency in hyperprolactinemic males: an open longitudinal study monitoring nocturnal penile tumescence. J Clin Endocrinol Metab. 2004 Feb;89(2):621-625.
101. Octreotide. Drug Information, Lanreotide: Drug Information, Pegvisomant: Drug Information, www.uptodate.com, Available at, Waltham, MA, UpToDate, 2009.
102. Baldys-Waligorska A., Golkowski F., Krzentowska A., et al. [Evaluation of the efficacy of octreotide LAR in the treatment of acromegaly—a yearly observation]. Przegl Lek. 2009;66(5):218-221.
103. Colao A., Pivonello R., Rosato F., et al. First-line octreotide-LAR therapy induces tumour shrinkage and controls hormone excess in patients with acromegaly: Results from an open, prospective, multicentre trial. Clin Endocrinol (Oxf). 2006 Mar;64(3):342-351.
104. Cozzi R., Montini M., Attanasio R., et al. Primary treatment of acromegaly with octreotide LAR: a long-term (up to nine years) prospective study of its efficacy in the control of disease activity and tumor shrinkage. J Clin Endocrinol Metab. 2006 Apr;91(4):1397-1403.
105. Trepp R., Stettler C., Zwahlen M., et al. Treatment outcomes and mortality of 94 patients with acromegaly. Acta Neurochir (Wien). 2005 Mar;147(3):243-251. discussion 50–51
106. Mangupli R., Lisette A., Ivett C., et al. Improvement of acromegaly after octreotide LAR treatment. Pituitary. 2003;6(1):29-34.
107. Gilbert J., Ketchen M., Kane P., et al. The treatment of de novo acromegalic patients with octreotide-LAR: Efficacy, tolerability and cardiovascular effects. Pituitary. 2003;6(1):11-18.
108. Cozzi R., Dallabonzana D., Attanasio R., et al. A comparison between octreotide-LAR and lanreotide-SR in the chronic treatment of acromegaly. Eur J Endocrinol. 1999 Sep;141(3):267-271.
109. Grottoli S., Celleno R., Gasco V., et al. Efficacy and safety of 48 weeks of treatment with octreotide LAR in newly diagnosed acromegalic patients with macroadenomas: an open-label, multicenter, non-comparative study. J Endocrinol Invest. 2005 Dec;28(11):978-983.
110. Ayuk J., Stewart S.E., Stewart P.M., Sheppard M.C. Long-term safety and efficacy of depot long-acting somatostatin analogs for the treatment of acromegaly. J Clin Endocrinol Metab. 2002 Sep;87(9):4142-4146.
111. Ayuk J., Stewart S.E., Stewart P.M., Sheppard M.C. Efficacy of Sandostatin LAR (long-acting somatostatin analogue) is similar in patients with untreated acromegaly and in those previously treated with surgery and/or radiotherapy. Clin Endocrinol (Oxf). 2004 Mar;60(3):375-381.
112. Chanson P., Boerlin V., Ajzenberg C., et al. Comparison of octreotide acetate LAR and lanreotide SR in patients with acromegaly. Clin Endocrinol (Oxf). 2000 Nov;53(5):577-586.
113. Colao A., Ferone D., Marzullo P., et al. Long-term effects of depot long-acting somatostatin analog octreotide on hormone levels and tumor mass in acromegaly. J Clin Endocrinol Metab. 2001 Jun;86(6):2779-2786.
114. Cozzi R., Attanasio R., Montini M., et al. Four-year treatment with octreotide-long-acting repeatable in 110 acromegalic patients: predictive value of short-term results? J Clin Endocrinol Metab. 2003 Jul;88(7):3090-3098.
115. Flogstad A.K., Halse J., Bakke S., et al. Sandostatin LAR in acromegalic patients: long-term treatment. J Clin Endocrinol Metab. 1997 Jan;82(1):23-28.
116. Jallad R.S., Musolino N.R., Salgado L.R., Bronstein M.D. Treatment of acromegaly with octreotide-LAR: extensive experience in a Brazilian institution. Clin Endocrinol (Oxf). 2005 Aug;63(2):168-175.
117. Lancranjan I., Atkinson A.B. Results of a European multicentre study with Sandostatin LAR in acromegalic patients. Sandostatin LAR Group. Pituitary. 1999;1(2):105-114.
118. Lancranjan I., Bruns C., Grass P., et al. Sandostatin LAR: a promising therapeutic tool in the management of acromegalic patients. Metabolism. 1996 Aug;45(8 suppl 1):67-71.
119. Amato G., Mazziotti G., Rotondi M., et al. Long-term effects of lanreotide SR and octreotide LAR on tumour shrinkage and GH hypersecretion in patients with previously untreated acromegaly. Clin Endocrinol (Oxf). 2002 Jan;56(1):65-71.
120. Plockinger U., Quabbe H.J. Presurgical octreotide treatment in acromegaly: no improvement of final growth hormone (GH) concentration and pituitary function. A long-term case–control study. Acta Neurochir (Wien). 2005 May;147(5):485-493. discussion 93
121. Bevan J.S., Atkin S.L., Atkinson A.B., et al. Primary medical therapy for acromegaly: an open, prospective, multicenter study of the effects of subcutaneous and intramuscular slow-release octreotide on growth hormone, insulin-like growth factor-I, and tumor size. J Clin Endocrinol Metab. 2002 Oct;87(10):4554-4563.
122. Davies P.H., Stewart S.E., Lancranjan L., et al. Long-term therapy with long-acting octreotide (Sandostatin-LAR) for the management of acromegaly. Clin Endocrinol (Oxf). 1998 Mar;48(3):311-316.
123. Turner H.E., Vadivale A., Keenan J., Wass J.A. A comparison of lanreotide and octreotide LAR for treatment of acromegaly. Clin Endocrinol (Oxf). 1999 Sep;51(3):275-280.
124. Attanasio R., Barausse M., Cozzi R. GH/IGF-I normalization and tumor shrinkage during long-term treatment of acromegaly by lanreotide. J Endocrinol Invest. 2001 Apr;24(4):209-216.
125. Verhelst J.A., Pedroncelli A.M., Abs R., et al. Slow-release lanreotide in the treatment of acromegaly: a study in 66 patients. Eur J Endocrinol. 2000 Nov;143(5):577-584.
126. Lucas T., Astorga R., Catala M. Preoperative lanreotide treatment for GH-secreting pituitary adenomas: effect on tumour volume and predictive factors of significant tumour shrinkage. Clin Endocrinol (Oxf). 2003 Apr;58(4):471-481.
127. al-Maskari M., Gebbie J., Kendall-Taylor P. The effect of a new slow-release, long-acting somatostatin analogue, lanreotide, in acromegaly. Clin Endocrinol (Oxf). 1996 Oct;45(4):415-421.
128. Attanasio R., Baldelli R., Pivonello R., et al. Lanreotide 60 mg, a new long-acting formulation: effectiveness in the chronic treatment of acromegaly. J Clin Endocrinol Metab. 2003 Nov;88(11):5258-5265.
129. Ambrosio M.R., Franceschetti P., Bondanelli M., et al. Efficacy and safety of the new 60-mg formulation of the long-acting somatostatin analog lanreotide in the treatment of acromegaly. Metabolism. 2002 Mar;51(3):387-393.
130. Cozzi R., Barausse M., Sberna M., et al. Lanreotide 60 mg, a longer-acting somatostatin analog: tumor shrinkage and hormonal normalization in acromegaly. Pituitary. 2000 Dec;3(4):231-238.
131. Lombardi G., Minuto F., Tamburrano G., et al. Efficacy of the new long-acting formulation of lanreotide (lanreotide Autogel) in somatostatin analogue-naive patients with acromegaly. J Endocrinol Invest. 2009 Mar;32(3):202-209.
132. van Thiel S.W., Romijn J.A., Biermasz N.R., et al. Octreotide long-acting repeatable and lanreotide Autogel are equally effective in controlling growth hormone secretion in acromegalic patients. Eur J Endocrinol. 2004 Apr;150(4):489-495.
133. Attanasio R., Lanzi R., Losa M., et al. Effects of lanreotide Autogel on growth hormone, insulinlike growth factor 1, and tumor size in acromegaly: a 1-year prospective multicenter study. Endocr Pract. 2008 Oct;14(7):846-855.
134. Chanson P., Borson-Chazot F., Kuhn J.M., et al. Control of IGF-I levels with titrated dosing of lanreotide Autogel over 48 weeks in patients with acromegaly. Clin Endocrinol (Oxf). 2008 Aug;69(2):299-305.
135. Caron P., Bex M., Cullen D.R., et al. One-year follow-up of patients with acromegaly treated with fixed or titrated doses of lanreotide Autogel. Clin Endocrinol (Oxf). 2004 Jun;60(6):734-740.
136. Caron P. [Somatuline(R) Autogel(R), a new formulation of lanreotide for the treatment of acromegalic patients]. Ann Endocrinol (Paris). 63(2 Pt 3), 2002 Apr. 2S19-24
137. Caron P., Cogne M., Raingeard I., Bex-Bachellerie V., Kuhn J.M. Effectiveness and tolerability of 3-year lanreotide Autogel treatment in patients with acromegaly. Clin Endocrinol (Oxf). 2006 Feb;64(2):209-214.
138. Lucas T., Astorga R. Efficacy of lanreotide Autogel administered every 4–8 weeks in patients with acromegaly previously responsive to lanreotide microparticles 30 mg: a phase III trial. Clin Endocrinol (Oxf). 2006 Sep;65(3):320-326.
139. Ketoconazole. Drug Information, Metyrapone: Drug Information, Mitotane: Drug Information, Etomidate: Drug Information, www.uptodate.com, Available at, Waltham, MA, UpToDate, 2009.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 17 Medical Management of Hormone-Secreting Pituitary Tumors
The majority of pituitary tumors are hormone-secreting.1 With the exception of prolactinomas, surgery has historically been the mainstay of treatment for hormone-secreting as well as non-functioning pituitary tumors. In recent years, however, medical therapy has assumed an increasingly important role in the management of hormone-secreting pituitary adenomas. In addition to being the primary treatment for prolactinomas, medical therapy can serve an adjunctive role in the treatment of growth-hormone (GH) secreting tumors, refractory Cushing’s disease, and the rare thyroid-stimulating-hormone (TSH)-secreting adenomas. In a subset of patients with acromegaly, medical therapy can be the primary treatment approach. This chapter will review the pharmacological profile, therapeutic efficacy, safety, and side effects of the most common medications used in the treatment of pituitary tumors. In addition, the role of medical therapy in relation to neurosurgical management will be emphasized.
Prolactinomas
General Considerations
The decision of whether to initiate medical therapy in a patient with a prolactinoma should take into account the size of the tumor, fertility status of the patient, and the presence of symptoms related to hyperprolactinemia. In general, therapy is indicated for macroadenomas. A premenopausal woman with a microprolactinoma who does not have bothersome galactorrhea and does not wish to become pregnant may be reassured and treated with estrogen replacement. Likewise, a post-menopausal woman with a microprolactinoma may not require any treatment. This conservative approach is justified by the observation that approximately 90% of microprolactinomas will not grow in size over a 4- to 6-year period.2 In these patients, monitoring with periodic prolactin measurements and MRI’s is appropriate. While it is unlikely for a prolactinoma to grow without a concurrent rise in serum prolactin levels, this occurrence has been reported, and therefore some clinicians will monitor with periodic MRI’s even if prolactin levels are stable. Significant increases in serum prolactin levels or evidence of growth of a microprolactinoma are indications for treatment, given the possibility that such a tumor represents one of the small minority that will grow to become a macroadenoma. Alternatively, given the high surgical cure rate of microprolactinomas (up to 75%), surgery may be considered upfront in lieu of medical treatment in those patients who are interested in permanent and immediate cure.3
In the evaluation of hyperprolactinemia, it is important to recognize the phenomenon of macroprolactinemia, a laboratory finding that does not require any treatment. Patients with elevated prolactin levels who have either mild or no symptoms of hyperprolactinemia may, in fact, have large circulating complexes of the prolactin protein (more than 150 kDa), called macroprolactin. This larger prolactin protein, which can spuriously elevate serum prolactin levels, can be recognized by a simple laboratory method using PEG precipitation.4 As many as 10% to 26% of patients with idiopathic hyperprolactinemia have macroprolactinemia, and this entity should be suspected particularly in patients with discordant laboratory findings and clinical symptoms.4
Dopamine Agonists
In the majority of cases, dopamine receptor agonists are the mainstay of treatment for prolactinomas. These drugs inhibit prolactin secretion by binding to the D2 receptor of tumoral lactotrophs, which leads to decreased formation of prolactin secretory granules and thus reduced tumor volume. There may also be a cytocidal effect of dopamine agonist treatment on tumor cells resulting in tumor shrinkage and fibrosis.5 Among these drugs, cabergoline and bromocriptine are the most commonly used in practice today, as their use is substantiated by the greatest amount of clinical experience and evidence. The other dopamine agonists include quinagolide, a non-ergot derivative not available in the United States, and pergolide, an ergot derivative previously used to treat Parkinson’s disease that was removed from the U.S. market due to an increased risk of cardiac valvular disease.
Pharmacologic Aspects
There are currently three dopamine agonists available in the treatment of prolactinomas: cabergoline, bromocriptine, and quinagolide (Table 17-1).
The semisynthetic ergot derivative, bromocriptine, a D2 agonist and weak D1 antagonist, was the first available medical treatment for prolactinomas. Administered orally, bromocriptine has a relatively short half-life (7 hours), often necessitating two to three daily doses, although once daily dosing may occasionally be effective. The standard dosage is 2.5 to 15 mg per day, and most patients are successfully treated with 7.5 mg or less.6 Since gastrointestinal side effects are common, it is usually recommended to start with very low doses (1.25 mg/day) and gradually titrate upward by 1.25 mg increments weekly until a dose of 7.5 mg is reached.6 In resistant patients, doses as high as 20 to 30 mg per day may be necessary.
Cabergoline, a preferential D2-receptor agonist, has largely supplanted bromocriptine as the first-line treatment of prolactinomas due to its superior efficacy, longer half-life, and greater tolerability. Its half-life of 2.5 to 4.5 days allows for once or twice weekly administration.7 The typical starting dose of cabergoline is 0.25 mg orally once or twice weekly, with escalation of dosing by 0.25-mg increments at 2- to 3-month intervals if plateau effect is reached, until prolactin levels normalize. In most patients, the therapeutic dose range is 0.5 to 3.5 mg weekly. A recent study found that more than 95% of patients are able to achieve normal prolactin levels with high-dose treatment (from 6 to 11 mg/wk).8
Therapeutic Efficacy
For microprolactinomas, bromocriptine is 80% to 90% effective at normalizing prolactin levels and shrinking tumor mass, whereas the success rate is around 70% for macroprolactinomas.2 The improvement in headache and visual field defects is rapid and dramatic in most patients, occurring within a few days after the first dose. These changes often precede radiographic evidence of tumor shrinkage. Likewise, gonadal and sexual function may improve even before complete normalization of serum prolactin levels, although sometimes normalization of testosterone may require several months of normal prolactin levels2. The degree of tumor shrinkage seen with bromocriptine does not necessarily correlate with the nadir prolactin level or the extent of decline in prolactin levels.2 Although most prolactinomas remain responsive to bromocriptine over time, the drug is generally not curative, as hyperprolactinemia and tumor growth will often recur after withdrawal of therapy. Prolonged use of bromocriptine has been associated with perivascular fibrosis and increased tumor consistency in prolactinomas that ultimately are surgically removed; the extent of fibrosis seems to correlate with the duration of pre-surgical treatment with bromocriptine and may impact the technical ease of surgical resection.9
Early studies found cabergoline to be 80% to 95% effective at normalizing prolactin levels, with varying degrees of tumor reduction seen in 70% to 90% of patients.10 The efficacy of cabergoline in normalizing prolactin levels in micro- and macro-prolactinomas is shown in Tables 17-2 and 17-3, respectively. Although there is considerable variability among studies with respect to tumor shrinkage, a recent large study showed a significant (>50%) tumor shrinkage in 80% of patients, and complete disappearance of the tumor mass in 57% of patients.11 Cabergoline is able to induce tumor shrinkage more effectively in dopamine agonist naïve patients compared with those who have received prior dopamine agonist treatment (Fig. 17-1). Furthermore, cabergoline offers the potential for cure after several years of treatment, allowing for withdrawal of therapy.12, 13
FIGURE 17-1 Efficacy of cabergoline on tumor shrinkage in patients with macroprolactinomas. BRC, bromocriptine.
(From Vilar L, Freitas MC, Naves LA, et al. Diagnosis and management of hyperprolactinemia: results of a Brazilian multicenter study with 1234 patients. J Endocrinol Invest. 2008;31:436-444. Editrice Kurtis.)
The beneficial effects of cabergoline on gonadal function are well documented in both sexes. Compared with bromocriptine, cabergoline is more effective at normalizing prolactin levels and restoring ovulation and is associated with fewer, milder, and shorter-lived side effects.14 Serum testosterone levels have been shown to normalize in the majority of patients, with concurrent improvement in sperm count and parameters.15
Quinagolide normalized prolactin levels in 73% of patients with microadenomas and 67% of patients with macroadenomas.16 Tumor reduction was seen in 55% of patients with microadenomas and 75% of patients with macroadenomas.16 Although cabergoline has been found to be more effective at normalizing prolactin levels and reducing tumor size compared with quinagolide, both drugs show similar efficacy on reproductive function.
Pergolide, a semisynthetic ergoline derivative, was initially approved in the United States only for the treatment of Parkinson’s disease. Typical doses used in the treatment of hyperprolactinemia range from 0.05 mg to 0.5 mg/day, whereas higher doses (>3 mg/day) have been used in Parkinson’s patients.17 Although pergolide has been found to be effective at normalizing prolactin levels and reducing the size of macroprolactinomas, this drug was withdrawn from the U.S. market in 2007 due to the increased risk of valvular heart disease in Parkinson’s patients receiving high daily doses.
Tolerability and Side Effects
The most common side effects of dopamine agonists include gastrointestinal, cardiovascular, and neurologic symptoms (Table 17-4). In general, if a patient cannot tolerate the first dopamine agonist administered, a trial of a second drug should be given.
| Common Side Effects of Dopamine Agonists |
|---|
| Gastrointestinal |
| Nausea |
| Vomiting |
| Cardiovascular |
| Postural hypotension |
| Dizziness |
| Syncope (rare) |
| Neurologic |
| Headache |
| Drowsiness |
| Exacerbation of pre-existing psychosis (rare) |
| Dyskinesias (high-dose treatment) |
Among the dopamine agonists, bromocriptine is the least well tolerated, with up to 12% of patients being unable to tolerate therapeutic doses.2 Up to one third of patients will experience nausea and vomiting, an effect that can be minimized by initiating a low dose and titrating very slowly.2 Taking the medication with a small snack may also reduce these symptoms.2 A gradual dose titration and taking the medication at bedtime may also help reduce postural hypotension and dizziness experienced by as many as 25% of patients.2 Syncope is rare but has been reported even with small initial doses.18 Tolerance to postural hypotension usually develops rapidly.18 Drowsiness, headache, and nasal congestion are common complaints. The safety of bromocriptine in psychiatric patients has not yet been established; there are case reports of onset or exacerbation of preexisting psychosis. Other psychiatric symptoms associated with high doses of bromocriptine include anxiety, depression, insomnia, paranoia, and hyperactivity.18 At higher doses used in the treatment of Parkinson’s disease, reversible pleuro-pulmonary changes and retroperitoneal fibrosis have been reported, but these changes are unlikely to occur at the doses used to treat prolactinomas.2
While cabergoline and bromocriptine have similar side effect profiles, cabergoline has been shown to be better tolerated in several large comparative studies.11, 19 Compared with bromocriptine, the side effects of cabergoline are generally less frequent, milder, and of shorter duration.2 Nausea and vomiting are most commonly observed, followed by headache and dizziness.2 In a multi-center randomized trial of hyperprolactinemic women, 3% could not tolerate cabergoline compared with 12% who had to stop taking bromocriptine.14 It has been proposed that fluctuations in concentrations of dopamine agonists are the main cause of side effects; cabergoline would be better tolerated due to its long half-life and relatively steady plasma concentration.18
Quinagolide has also shown better tolerability compared with bromocriptine. In a double-blind comparative study of 47 hyperprolactinemic patients, quinagolide was tolerated by 90% of patients versus 75% of patients treated with bromocriptine.20 As with the other dopamine agonists, the most frequent side effects include nausea, vomiting, headache, and dizziness. These effects are transient and occur within the first few days of starting therapy or during dose adjustments.18
Dopamine Agonists and Valvular Heart Disease
Recently, the safety of dopamine agonists has been brought into question after two large population-based studies showed an increased risk of cardiac valvular disease in Parkinson’s disease patients being treated with high doses of these drugs, and particularly cabergoline and pergolide.21, 22 It is now recognized that one of the “off target” effects of dopamine agonists is activation of the serotonin (5-HT) receptor, of which there are 7 distinct subtypes.23 High concentrations of the subtype 5-HT2B receptor are found on cardiac valves and the pulmonary arteries.23 Both pergolide and cabergoline have been implicated in the development of cardiac valvular fibrosis because of their agonist effects at the 5-HT2B receptor.23 Bromocriptine, which is only a partial agonist at the 5-HT2BR, had until recently not been thought to pose an increased risk of cardiac valvular disease, as there had only been a single case report suggesting an association.23 However, a recent prospective study suggests that bromocriptine may, in fact, be equally associated with the development of cardiac valvulopathy at high cumulative doses.24
Several studies have recently examined the effect of cabergoline on cardiac valvular disease in patients with prolactinomas.25 These patients typically receive much lower doses (10–20 times less) than in Parkinson’s disease. Only one of seven studies available to date showed an association between cabergoline use and valvular regurgitation. In this study, there was an increased risk of moderate tricuspid regurgitation in patients who had received cumulative cabergoline doses greater than 280 mg.26 However, in another study, patients receiving cabergoline (0.25–4 mg/wk) for 3 to 4 years (mean cumulative dose of 311 mg), had no increased prevalence of clinically significant valvular heart disease.16 Even in patients treated with cabergoline for 8 years who received cumulative doses as high as 1728 mg, there was no increased risk of clinically relevant heart disease.28
Therefore, conventional doses of cabergoline used to treat prolactinoma patients do not currently appear to pose an increased risk of cardiac valvular disease. However, until larger prospective studies clarify the issue, it may be prudent to monitor patients requiring higher doses of cabergoline (>2 or 3 mg/wk) with serial echocardiograms. If valvulopathy develops, it may be reasonable to switch to bromocriptine. Alternatively, surgical resection may be appropriate. This issue underscores the importance of using the smallest effective dose and attempting withdrawal from cabergoline treatment in patients who achieve normalization of prolactin levels and significant tumor shrinkage to minimize their cumulative lifetime exposure to the drug.25
Dopamine Agonists and Pregnancy
Two principal considerations arise in the management of women with prolactinomas who either desire fertility or who become pregnant: (1) the risk of tumor growth due to physiologic stimulation of tumoral lactotrophs induced by pregnancy, and (2) the effects of dopamine agonists on fetal development. These two considerations should be weighed against each other on an individualized basis, taking into account the size of the patient’s initial tumor, response to treatment, and the patient’s ability to tolerate side effects of treatment. It has been estimated that the risk for significant tumor enlargement during pregnancy is 1.6% to 5.5% in microprolactinomas and 15.6% to 35.7% in macroprolactinomas.18
For patients with macroadenomas, especially those in close proximity to the optic chiasm or cavernous sinuses, contraception should be encouraged while attempting to reduce the size of the tumor prior to pregnancy. If a patient with a macroprolactinoma becomes pregnant, there are several different therapeutic approaches that can be offered: (1) dopamine agonists may be stopped after conception with neuro-ophthalmological exams performed throughout pregnancy, (2) dopamine agonists may be continued throughout pregnancy, (3) trans-sphenoidal surgery may be performed in later stages of pregnancy if the enlarged tumor does not respond to resumption of dopamine agonists, or (4) delivery if tumor growth cannot be controlled and the pregnancy is advanced enough.2
Bromocriptine has conventionally been the drug of choice in women with prolactinomas who desire fertility or who become pregnant. There is substantial evidence showing that exposure to bromocriptine at the time of conception does not cause birth defects or increase the risk of spontaneous abortions, ectopic pregnancies, trophoblastic disease, or multiple pregnancies.29 However, evidence is also accumulating to show that cabergoline may have an equally good safety record in terms of pregnancy outcomes.30 A recent 12-year prospective study of cabergoline use in 329 pregnancies showed no increased risk of miscarriage or fetal malformation.30
Dopamine Agonist Resistance
A subset of patients with prolactin-secreting pituitary tumors demonstrate “biochemical” resistance (failure to normalize prolactin levels) or “mass” resistance (absence of tumor shrinkage) to dopamine agonists.2 Resistance to dopamine agonists occurs in both micro and macro-adenomas and is believed to be mediated by the downregulation of pituitary D2 receptors.31 Biochemical resistance has been estimated to occur in approximately 10% to 20% of patients on dopamine agonists. Mass resistance has been estimated to occur in approximately 30% to 40% and 15% of patients to bromocriptine and cabergoline, respectively.2, 31 The prevalence of resistance to quinagolide is unclear given the lack of data regarding its use in dopamine-agonist naïve patients.6
Most tumors resistant to bromocriptine will respond to cabergoline. When switched to cabergoline, 85% of patients resistant to bromocriptine and quinagolide achieved normal prolactin levels, and 70% had some change in tumor size.32 A recent large study showed that cabergoline normalized prolactin levels in 61% and 50% of bromocriptine-resistant microprolactinomas and macroprolactinomas, respectively.11
Therapeutic options in the management of dopamine-resistant prolactinomas include (1) increasing the dose of the dopamine agonist, (2) switching to an alternative dopamine agonist, (3) trans-sphenoidal surgery, and (4) radiation therapy. In most cases, the resistance to dopamine agonist is partial, and a response can be achieved by progressively increasing the dose of the drug. A recent study showed that in more than 95% of patients resistant to bromocriptine prolactin normalized with individualized high-dose cabergoline treatment (mean dose 5.2 mg/wk).8 Bromocriptine-resistant patients generally require higher doses of cabergoline and have less tumor shrinkage compared to treatment-naïve patients.33 In general, switching from cabergoline to bromocriptine is unlikely to be efficacious.
While trans-sphenoidal surgery is never a first-line treatment for macroprolactinomas, it remains an important option for patients who cannot tolerate dopamine agonists or when medical therapy is ineffective at controlling symptoms or restoring reproductive function. There is little long-term outcome data regarding patients with macroprolactinomas treated with dopamine agonists who subsequently proceed to surgery. In a recent study of 72 patients, 35% of patients required trans-sphenoidal surgery due to resistance and/or intolerance of dopamine agonists.34 This study, which an outlier given the disproportionately high number of dopamine agonist resistant patients, showed that additional tumor shrinkage was achieved in 57% of operated patients, while only 22% were able to attain normoprolactinemia without dopamine agonists following surgery.34 Surgery was associated with a high incidence of hypopituitarism regardless of whether patients received subsequent radiotherapy.35 The remission rate for surgery when used as second-line treatment for prolactinomas is quite low (around 22%). As opposed to its curative role in the other secretory macroadenomas, surgery plays only a “debulking” role in macroprolactinomas, and therefore should be reserved for patients who, despite maximal medical therapy, have progressive tumor enlargement and are at risk of visual compromise or neurological deficits.35
Conventional fractionated radiation therapy should be considered a last-line option in the management of dopamine-resistant prolactinomas given the delayed treatment effects and the high rate of panhypopituitarism. On the other hand, radiosurgery may be an effective treatment in secretory pituitary adenomas, with a lower risk of long-term hypopituitarism.36
Dopamine Agonist Withdrawal
The optimal duration of therapy for patients with prolactinomas is uncertain. Until recently, dopamine agonist treatment had been considered a “lifelong” requirement. However, after a landmark study by Colao et al. demonstrated disease remission in a considerable proportion of patients following withdrawal from cabergoline, attention shifted towards defining selection criteria for withdrawal and identifying predictors of long-term remission.12, 37 Defining the optimal timing and appropriate candidates for withdrawal is difficult because of the heterogeneity of studies that have examined this issue, which differ with respect to the causes of hyperprolactinemia (idiopathic vs. micro/macroprolactinoma), the type and duration of dopamine agonist treatment, and the treatment prior to the start of dopamine agonist therapy.38 Despite the inherent difficulty in extrapolating from these disparate studies, the Pituitary Society has proposed certain criteria for withdrawal, including (1) normoprolactinemia and (2) tumor absence or markedly reduced tumor volume after a minimum of 1 to 3 years of dopamine agonist treatment.39 A recent study tested the applicability of these criteria and found a recurrence rate of 54% after withdrawal of long-term cabergoline treatment.13 In this study, most patients had recurrence of disease within 1 year of discontinuation, and a similar recurrence rate (52% vs. 55%) was seen in patients with microprolactinomas and macroprolactinomas.13 The size of the tumor remnant prior to withdrawal appears to be predictive of recurrence risk.13
A recent meta-analysis showed that long-term remission occurs in only 21% of patients following dopamine agonist withdrawal.38 The best chance for a long-term remission is seen in patients treated with cabergoline for more than 2 years, with a mean remission rate around 54%, whereas remission is seen in only 20% of patients withdrawn from bromocriptine.38 Considering the financial burden, occasional problems with tolerability, and potential risk of valvulopathy with long-term dopamine agonist treatment, it is reasonable to attempt withdrawal in patients who have been on a dopamine agonist (particularly if cabergoline) for 2 years or more.
Acromegaly
General Considerations
Trans-sphenoidal surgery is currently the first treatment approach in acromegaly when a definitive cure can be expected, as is the case with microadenomas, where cure rates are as high as 80% in the hands of an experienced neurosurgeon.38 Surgery is also indicated for decompressive purposes in the cases of large tumors that cause chiasmatic compression. Since the biochemical cure rate of macroadenomas following surgery is less than 50% (particularly if extrasellar extension of the tumor is present), many patients will require some form of adjuvant treatment following surgical resection.40 In addition, medical therapy should be considered as primary approach for patients without vision compromise who either have higher than normal surgical risk or whose tumor is deemed not to be surgically curable.41 Finally, two recent studies have suggested an increase in surgical cure rate in macroadenomas when they are pre-treated for 4 to 6 months with a long-acting somatostatin analog. If confirmed, this may expand the role of these drugs to all macroadenomas before surgical attempt.42, 43 The estimated benefit of postsurgical radiotherapy is often outweighed by the delayed onset of effects and the risk of panhypopituitarism.
1. Somatostatin analogues, octreotide and lanreotide, inhibit somatotroph cell proliferation and GH secretion by binding to the somatostatin receptors on tumoral cells.
2. Dopamine agonists bind to dopamine D2 receptors expressed on both somatotroph and mammotroph cells, exerting negative control.
3. The GH receptor antagonist Pegvisomant blocks the effect of GH. Somatostatin analogues are often considered first-line treatment.
Somatostatin Analogues: Pharmacologic Aspects
The presently commercially available somatostatin analogues act on two of the five somatostatin receptors existing in nature. The dosing frequencies of the somatostatin analogues are summarized in Table 17-5.
Octreotide was the first available somatostatin analogue. It has a relatively short half-life of 2 hours after subcutaneous injection, which requires a three times daily dosing to maintain therapeutic concentrations. The starting dose is usually 100 to 250 μg three times daily up to a maximum of 1500 μg daily.40 Nowadays, this formulation has been substituted by octreotide LAR (long-acting release), a long-acting depot formulation administered as an intramuscular injection every 4 weeks. The starting dose is 20 mg increasing or decreasing by 10 mg increments (maximal dose 40 mg) based on clinical and biochemical response.40
Lanreotide SR (sustained release) is a biodegradable polymer microparticle that allows a slow release following subcutaneous injection every 7 to 14 days.40 Lanreotide autogel is a longer-acting depot formulation that allows for injections every 28 days.40 In the United States, only the latter formulation is available.
