Chapter 22 Mechanisms of Oxidative Stress in Retinal Injury
Overview of oxidative stress in the retina
Reactive oxygen species (ROS) are a major source of retinal oxidative stress. These highly reactive particles include free radicals, peroxides, and singlet oxygen. Free radicals, such as the hydroxyl radical (OH•), hydroperoxyl radicals (HO2•), superoxide anion (O2–•), and lipid peroxyl radicals, are strong oxidizing agents with an unpaired electron in the outer shell. Peroxides (e.g., hydrogen peroxide (H2O2), lipid peroxides) and singlet oxygen (1O2) have a full complement of electrons in an unstable state.1
Under physiologic conditions, the body produces ROS through normal metabolic processes, such as glycolysis and the Krebs cycle. Aging and disease may disturb the balance between ROS generation and clearance, resulting in oxidative damage to macromolecules.2 The majority of endogenous ROS are produced by mitochondria through the electron transport chain, which converts 2–3% of all utilized oxygen into ROS.3 Stimuli such as aging, inflammation, irradiation, air pollutants, and cigarette smoke lead to increased ROS, and thus increased cellular oxidative injury.1,4,5
The body’s defense against increasing oxidative stress consists of molecules with antioxidant capacity, including vitamins C and E, carotenoids, and other free radical scavengers. Most ROS are eliminated immediately by antioxidant enzymes, such as superoxide dismutase (SOD), glutathione peroxidase, and catalase. For example, the superoxide anion, produced by the mitochondria during the electron transport stage of cellular respiration, is converted to the less noxious hydrogen peroxide molecule (H2O2) by SOD.1 Smaller antioxidant molecules (e.g., vitamin C (ascorbate), vitamin E (tocopherol), and carotenoids) act on free radicals directly, reducing ROS such as the hydroxyl radical.
As most free radical chain reactions are prevented by free radical-scavenging molecules, it appears that free radicals are often not the direct cause of oxidative damage but rather act to initiate further oxidative damage by nonradical oxidants. The redox hypothesis describes radical-free oxidative stress in which disrupted thiol redox circuits interfere with regulation of cellular redox status, affecting cell signaling and physiological regulation. Redox-sensitive thiols include the amino acid cysteine (Cys), the Cys-derived disulfide cystine (CySS), the Cys-containing tripeptide glutathione (GSH), and glutathione disulfide (GSSG). Sulfur redox couples act as “on/off” switches regulating gene expression and protein function. Because the Cys/CySS and GSH/GSSG couples are not in equilibrium, it is plausible that abnormal levels of nonradical oxidants could be sufficient to disrupt normal function.6
The retina is particularly vulnerable to oxidative stress because of its high oxygen consumption and high proportion of polyunsaturated fatty acids (PUFAs).1,2 High light exposure and phagocytosis in the RPE contribute to the oxidative burden of the retina. The turnover rate of photoreceptors is high, with these cells shedding about 10% of their outer-segment discs each day. The disc membranes, in particular the PUFAs, are subject to peroxidation, which is highly damaging to the RPE. Two carotenoids, lutein and zeaxanthin, comprise the macular pigment that protects against ROS in the retina. Lutein and zeaxanthin can quench the reactive singlet oxygen and form an optical filter that blocks highly damaging blue light from reaching the photoreceptors.1
Retinal diseases related to oxidative stress
Oxidative stress contributes to diseases of the retina, including AMD, diabetic retinopathy, and inherited retinal degenerations (Table 22.1). To set the stage for a detailed discussion of the underlying mechanisms of oxidative damage to the retina, we will discuss the pathology and evidence for a role of oxidative stress in each of these diseases.
Table 22.1 Oxidative stress in the pathophysiology of common retinal diseases
AMD, age-related macular degeneration; RPE, retinal pigment epithelium; ROS, reactive oxygen species; AGE, advanced glycation end-product; RAGE, receptor of AGEs; VEGF, vascular endothelial growth factor; t-BHP, tert-butyl hydroperoxide; NF-κB, nuclear factor-κB; GSH, glutathione; ERGs, electroretinograms; GST, glutathione-S-transferase; GSHPx, glutathione peroxidase.
Age-related macular degeneration
AMD, the leading cause of irreversible vision loss in older individuals in the western world, is a complex disease influenced by factors such as genetics, demographics, and environmental exposure. Approximately 1.5% of individuals in the USA over the age of 40 (about 1.75 million people) develop the sight-threatening advanced stages of the disease, and this number is projected to approach 3 million by 2020.7
AMD can be divided into an early, typically asymptomatic form, and a late form that often results in severe central vision loss. In the early stages of AMD, whitish-yellow waste deposits called drusen accumulate in the macula, usually localized between the RPE and Bruch’s membrane. These deposits may be small and discrete (hard drusen) or larger and more confluent (soft drusen). Drusen may also be present between the photoreceptors and RPE or within the photoreceptor cell layer (reticular drusen).8,9 Histologically, drusen consist of numerous proteins (e.g., complement, immunoglobulins, amyloid-β) and lipids (e.g., phospholipids, cholesterol, apolipoproteins).10,11 Early dry AMD may develop into advanced dry or advanced wet AMD. Advanced dry AMD (geographic atrophy) is marked by cell death in the choriocapillaris and overlying RPE. Advanced wet AMD is characterized by choroidal neovascularization, in which abnormal choroidal vessels extend into the subretinal space.
Risk factors for AMD include older age, high body mass index, and high light exposure.12,13 Smoking, the strongest environmental risk factor for AMD, has been linked to disease onset and progression in multiple large, epidemiologic studies.14–19 Additionally, high dietary intake of carotenoids and antioxidant supplementation have been linked with lower risk of AMD.20,21
Multiple lines of evidence point to the role of oxidative stress in the pathogenesis of AMD. Several AMD risk factors, particularly aging, smoking, and light exposure, have been linked to increased levels of oxidative stress.1 Additionally, proteome analysis of human donor eyes revealed oxidative modifications to proteins and DNA in Bruch’s membrane, drusen, and RPE.23 The higher prevalence of these oxidative changes in AMD patients versus controls23 suggests an increased oxidative state in the retinas of AMD patients.
Impairment of the retinal antioxidant defense system may also play a role in AMD, as some studies have reported decreased macular pigment in AMD patients.24–26 Significantly lower levels of macular pigment (lutein and zeaxanthin) were found by direct measurement in AMD versus control eyes at autopsy.24 Additionally, dietary supplementation with antioxidants (vitamin C, vitamin E, and β-carotene) and zinc was shown to decrease the risk of progression to advanced AMD in the Age-Related Eye Disease Study (AREDS).22 In past observational studies, high dietary intake of lutein/zeaxanthin and omega-3 fatty acids has corresponded to reduced prevalence and incidence of AMD.27 These compounds are now under investigation for the treatment of AMD in the AREDS2 clinical trial.
Diabetic retinopathy
Diabetic retinopathy, a common microvascular complication of both type 1 and type 2 diabetes mellitus (DM), is the leading cause of vision loss in working-age adults.28 In the USA, approximately 26 million people are living with diabetes, and the number of diabetic adults with retinopathy is projected to triple to 16 million by the year 2050.29 The majority of DM (90–95%) is represented by type 2 diabetes, which involves impaired insulin action at target tissues and impaired insulin release. In contrast, the primary cause of type 1 diabetes is the autoimmune destruction of pancreatic β cells.
Interestingly, retinal capillary cells in diabetics undergo accelerated apoptosis that precedes the detection of any of the histopathological changes characteristic of diabetic retinopathy.30 Although the exact mechanism for this accelerated apoptosis remains uncertain, studies have pointed to the involvement of oxidative stress-activated caspases and nuclear factor-κB (NF-κB) in retinal cell death.31,32 Additional experiments revealed that inhibition of superoxide accumulation in diabetes prevents this early retinal cell death.33,34
Oxidative stress appears to be a major risk factor for the onset and progression of diabetes. Many of the common DM risk factors, including obesity and age, foster an oxidative environment that may alter insulin sensitivity by increasing insulin resistance or impairing glucose tolerance. Hyperglycemia, a common result of both types of diabetes, contributes in turn to the progression and maintenance of an overall oxidative environment.35
Hyperglycemia may contribute to oxidative stress by generating ROS directly or disrupting the cellular redox balance. This hyperglycemia-induced oxidative stress can occur via several well-studied mechanisms, including increased polyol pathway flux, increased intracellular formation of advanced glycation end products (AGEs), activation of protein kinase C (PKC), and disturbance of the hexosamine biosynthesis pathway. The overproduction of superoxide by the mitochondrial transport chain has been proposed as a possible root cause for these metabolic changes.36
The polyol pathway leads to the reduction of glucose to sorbitol via aldose reductase in an nicotinamide adenine dinucleotide phosphate (NADPH)-dependent manner. Sorbitol is then oxidized to form fructose by sorbitol dehydrogenase. The polyol pathway consumes NADPH, an essential cofactor for regenerating the intracellular antioxidant GSH. By decreasing the amount of free GSH, the polyol pathway may increase the susceptibility of cells to oxidative injury.36
AGEs are modifications of proteins or lipids that become nonenzymatically glycated and oxidized after contact with aldose sugars. Early glycation and oxidation result in the formation of Schiff bases and Amadori products. Further glycation of proteins and lipids causes molecular rearrangements that lead to the generation of AGEs.37 AGEs appear to damage cells by modifying intracellular proteins, such as transcription factors, and altering extracellular proteins and extracellular matrix molecules. These modified proteins can then bind and activate receptor of AGEs (RAGE), resulting in the upregulation of the transcription factor NF-κB and the production of inflammatory cytokines and growth factors.36
The activation of PKC leads to multiple characteristics of diabetic retinopathy, including increased vascular permeability, endothelial cell proliferation and apoptosis, and neovascularization.38 Hyperglycemia-induced ROS inhibit the key glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH), leading to an increase in glycolytic pathway precursors. Increased levels of triose phosphate upregulate diacylglycerol, the primary activator of PKC.38,39 Activated PKC can induce expression of vascular endothelial growth factor (VEGF),40 a major effector of blood flow and vessel permeability and a primary stimulator of neovascularization. PKC also leads to the synthesis of NF-κB, a proinflammatory transcription factor, and transforming growth factor-β1 (TGF-β), which can contribute to the occlusion of capillaries seen in diabetic retinopathy.36,38
The hexosamine biosynthesis pathway may also mediate some of the toxic effects of hyperglycemia. The inhibition of GADPH by ROS causes diversion of glycolytic metabolites to the hexosamine pathway, producing uridine diphosphate N-acetylglucosamine. N-acetylglucosamine is added to serine and threonine residues of transcription factors, often resulting in pathological changes in gene expression. Examples include increased expression of TGF-β and plasminogen activator inhibitor-1.36
Hyperglycemia-induced alterations in each of these DM-associated pathways can be linked to oxidative stress. Toward that end, Brownlee has proposed a “unifying mechanism” that hypothesizes that all of these pathway changes are a result of mitochondrial superoxide overproduction in the endothelial cells of both large and small blood vessels.41 In this model, diabetic hyperglycemia causes increased production of ROS by mitochondria. Greater concentration of ROS in the body induces nuclear DNA strand breaks. This DNA damage activates the DNA repair enzyme poly(ADP-ribose) polymerase, which reduces GADPH activity by the addition of ADP ribose polymers. Decreased GAPDH activity results in a backup of the glycolytic pathway and activation of the polyol, AGE, PKC, and hexosamine pathways, discussed above.36
Inherited retinal degenerations
Inherited retinal degenerations are a group of disorders characterized by primary and progressive loss of photoreceptor cells, leading to irreversible vision loss. Over 170 genes and greater than 200 loci have been associated with inherited retinal degenerations (www.sph.uth.tmc.edu/retnet). The proteins encoded by these genes are diverse in function, including not only proteins required for phototransduction but also structural proteins, RNA splicing factors, intracellular trafficking molecules, and proteins involved in the phagocytosis and regulation of intracellular pH.42
The most common form of these diseases, retinitis pigmentosa (RP), which has an estimated prevalence of around 1 in 4000,43 involves a primary insult to rods that initiates rod cell death followed by cone cell death (rod–cone dystrophy). Other forms of inherited retinal degeneration include cone dystrophy, cone–rod dystrophy, and macular dystrophy. The most common inherited macular dystrophy is Stargardt macular dystrophy, an autosomal recessive disease with a carrier frequency of approximately 1 in 50.44
The unifying feature in animal models and human cases of RP seems to be the apoptotic cell death of rod photoreceptors, although the mechanisms that lead to photoreceptor cell death in RP remain poorly defined. While most mutations in RP specifically affect rods, both rods and cones die. Several mechanisms have been proposed for the dependence of cone survival on the presence of functioning rods. One theory is that rods produce a survival factor that is required by cones and that modulates the thioredoxin antioxidant defense system in the retina.45 Oxidative stress or alterations in the cellular redox state have also been proposed by several groups as a common mediator of photoreceptor cell death.46 Several lines of evidence support this hypothesis, including the induction of apoptosis by oxidants such as H2O2 and the inhibition of apoptosis by antioxidants.
Oxidative injury to the retina
Retinal pigment epithelium
Oxidative damage to the RPE can be induced by oxidizing agents or light. Under normal conditions, phagocytosis of photoreceptor outer segments by the RPE generates ROS via the NADPH oxidase system, leading to increased intracellular H2O2 and increased catalase activity.47 The phosphoinositide 3-kinase (PI3K)-Akt pathway may protect RPE cells from such oxidative stress. The oxidant H2O2 was demonstrated to induce PI3K-dependent Akt activation in cultured RPE cells. Akt activation can enhance RPE survival by phosphorylating and thus inactivating multiple proapoptotic factors.48 Cultured human RPE cells exhibited signs of senescence when treated with the oxidizing agents tert-butyl hydroperoxide (t-BHP)49 or H2O2,50 suggesting that oxidative stress may contribute to the RPE senescence seen in AMD. Oxidative stress induced by hypoxia/reoxygenation in cultured human RPE cells led to the accumulation of extracellular matrix and may be linked to the thickening of Bruch’s membrane seen in early AMD.51 Additionally, animal and laboratory studies have shown that blue light damages the RPE through ROS generation.52 RPE cells treated with subapoptotic levels of ultraviolet light showed increases in both ROS and AGEs.53
The AGE-induced activation of RAGE may play a key role in RPE apoptosis. In human donor eyes, RAGE were found to colocalize with AGE deposits and macular disease in AMD retinas, while normal retinas displayed little or no immunolabeling for either AGE or RAGE. This study also showed that AGEs stimulate RAGE-mediated activation in cultured RPE cells in a dose-dependent fashion.54 RAGE increased secretion of VEGF in cultured RPE cells, suggesting that AGE-mediated activation of the RAGE axis in RPE cells could contribute to neovascular macular disease.55
Further evidence supports the idea that oxidative stress in the RPE has a significant influence on VEGF regulation. Treatment of cultured human RPE cells with the oxidant dl-buthionine-(S,R)-sulfoximine caused a significant decrease in intracellular GSH and GSH/GSSG ratios. This change in thiol redox status was associated with increased VEGF-A secretion as well as significant induction of VEGF receptors VEGFR-1 and VEGFR-2.56 Both VEGF-A and VEGF-C were upregulated in cultured RPE cells treated with t-BHP, with secretion higher to the apical side than to the basal side,57 suggesting that VEGF may directly affect the photoreceptors. VEGF secretion in RPE cells is regulated, in part, by the mitogen-activated protein kinases, including c-Jun-activated kinase, p38, and Erk. Studies in cultured RPE cells demonstrated that constitutive VEGF secretion is regulated by p38, while oxidative stress-induced VEGF secretion is regulated by both p38 and Erk.58 Finally, it appears that autocrine VEGF-A can enhance RPE cell survival under oxidative stress, utilizing the autocrine VEGF-A/VEGFR-2/PI3K/Akt pathway.59
Retinal vasculature
Increased oxidative stress can lead to alterations in both the retinal and choroidal vasculature. In diabetes, hyperglycemia-induced ROS may link elevated glucose and multiple metabolic abnormalities associated with the development of retinopathy.41 Retinal capillary cell cultures containing endothelial cells and pericytes incubated in high-glucose medium exhibited increased oxidative stress markers. In these cells, increased caspase-3 and apoptosis were seen after 5 days in culture.32
Oxidative stress has been correlated with increased production of the angiogenic growth factor and vascular permeability factor VEGF and may be involved in the upregulation of endothelial cell VEGF expression seen in diabetes.60 It appears that increased VEGF expression in vascular endothelial cells may result from peroxynitrite, a highly reactive oxidant formed by the combination of superoxide anion with nitric oxide.61 Peroxynitrite mediates a variety of biological processes including inhibition of key metabolic enzymes, lipid peroxidation, nitration of protein tyrosine residues, and oxidation of thiol pools.62
A hyperglycemic environment may promote the damaging effects of VEGF observed in diabetes. Interestingly, VEGF has been shown to be a potent survival factor for endothelial cells in vitro and in vivo.63 Despite its neuroprotective properties, the increased levels of VEGF seen in diabetic retinas64 are associated with increased retinal endothelial cell death. Cultured endothelial cells subjected to serum withdrawal-induced apoptosis were protected by VEGF in normal glucose but not in high-glucose or peroxynitrite media.62 Further experiments determined that high-glucose treatment blocks the prosurvival effect of VEGF and causes accelerated endothelial cell apoptosis via the action of peroxynitrite.62
Oxidative stress can also affect retinal endothelial cells’ ability to transport glucose into the cell. Exposure of endothelial cells to sustained oxidative stress in the form of H2O2 resulted in decreased glucose transport activity due to increased internalization of GLUT1, the most common retinal glucose transporter.65 H2O2-induced oxidative stress was also found to increase the rate of GLUT1 internalization by a proteasome-dependent mechanism involving inactivation of Akt.
Oxidative stress may have direct and indirect effects on the choroidal endothelial cells (CEC), and thus the choroidal neovascularization seen in AMD. Treatment of cultured RPE and CEC with the oxidant t-BHP resulted in decreased viability and increased proliferation of both cell types.66 RPE cells exposed to t-BHP for 24 hours were found to release basic fibroblast growth factor, a prominent proangiogenic factor. This finding suggests that oxidative stress may stimulate choroidal neovascularization via RPE-mediated growth factor release. Interestingly, CECs exposed to oxidative stress-induced AGEs in culture demonstrated increased proliferation as well as upregulation of VEGF, suggesting that AGEs may also promote choroidal neovascularization.67
Photoreceptors
Photoreceptor cell loss or a reduction in energy-demanding activities like phototransduction can lead to elevated tissue oxygen concentrations because choroidal blood vessels are not autoregulated by local oxygen levels. Accordingly, increases in outer retinal oxygen concentrations have been confirmed in multiple animal models.68–70 The inner retinal arterioles, in contrast, do autoregulate, leading to their attenuation in the presence of high oxygen concentrations.71 This oxygen imbalance induced by photoreceptor dysfunction is manifest by the thinned retinal vessels seen clinically in RP.
Alterations in the cellular redox state appear to mediate photoreceptor cell death. An in vitro model of photoreceptor apoptosis demonstrated an early and sustained increase in intracellular ROS accompanied by a rapid depletion of intracellular GSH.46 Evidence suggests that multiple oxidative stress-related mechanisms underlie the programmed cell death of photoreceptors. Classically, caspase-dependent apoptotic pathways have been described.72,73 More recently, it has been shown that programmed cell death can also be accomplished through caspase-independent apoptosis,74–76 as well as via autophagy.77 In a photoreceptor cell line treated with the nitric oxide donor sodium nitroprusside, cytosolic calcium levels increased during photoreceptor apoptosis, leading to activation of caspases as well as the calcium-dependent proteases calpains.78 Inhibitors of both caspases78 and calpains,79 applied independently to the same photoreceptor cell line, failed to prevent apoptosis. These results indicate that, in addition to the classic caspase-dependent pathways of apoptosis, calpain activity may be crucial for apoptosis.
Calpains may lead to retinal oxidative damage by impairing DNA repair mechanisms. Studies in retinal degeneration (rd1) mice, which have a mutation in exon 7 of the beta subunit of the rod photoreceptor PDE6 gene, have demonstrated downregulation in defensive mechanisms against oxidative stress (e.g., glutathione-S-transferase and glutathione peroxidase), potentially allowing an increase in intracellular ROS.80,81 A major product of ROS reactions in the DNA is the highly mutagenic oxidated nucleotide 8-oxo-guanosine (8-oxoG). Mismatches of 8-oxoG with adenine or cytosine are corrected by the specific DNA repair enzyme 8-oxoG DNA glycosylase (OGG1).82 OGG1 is regulated by cAMP response element-binding protein (CREB), and OGG1 and CREB have both been found to be downregulated in the rd1 retina. Since OGG1 can be inactivated by calpains, an increase in calpain activity could severely compromise a cell’s ability to repair oxidative stress-induced DNA damage.
Cones appear to be sensitive to retinal oxygen concentration. In multiple animal models of RP, increased levels of oxygen in the retina result in progressive oxidative damage to cones.71,83,84 Treatment of rd1–/– mice with a cocktail of antioxidants downregulated markers of oxidative damage in cones, reduced cone cell loss, and preserved cone ERGs. These results suggest that oxidative damage is important in cone death regardless of the underlying mechanism that kills rods.85 Most recently, studies in rd1–/– mice showed that NADPH oxidase plays a critical role in generation of the oxidative stress that leads to cone death in RP.85
Initial experiments investigating the progressive death of cones after rod cell death showed that grafting of normal photoreceptors (97% rods) into the eye of the rodless rd1–/– mouse before the cones degenerated had a positive effect on the host cones, even at some distance from the transplant.86 Further studies identified a protein that allowed for cone survival, termed rod-derived cone viability factor (RdCVF).45 RdCVF, encoded by the exon 1 of the nucleoredoxin-like 1 (Nxnl1) gene, corresponds to a truncated thioredoxin (TRX)-like protein. Because TRX proteins are key to maintaining a reducing intracellular environment,87 disruption of TRXs can lead to conditions of increased oxidative stress. Nxnl1 knockout mice have been shown to be sensitive to oxidative stress, suggesting that RdCVF is part of an endogenous redox-based signaling pathway involved in retinal maintenance.87
Mitochondria
Cumulative oxidative damage to mitochondria appears to play a role in aging and retinal diseases.88 Mitochondria are especially susceptible to oxidative injury due to the high concentration of ROS produced by the electron transport chain and the limited capacity for mitochondrial DNA (mtDNA) repair.89 RPE cells treated with ultraviolet light demonstrated conformational changes in mitochondria,53 and oxidized photoreceptor outer segments in in vitro damaged RPE mtDNA.90 Additionally, variations in the mitochondrial genome may modulate ROS production and thus susceptibility to disease.
Accumulating evidence, reviewed in Table 22.2, suggests a role for mitochondrial damage in the pathophysiology of both AMD and diabetic retinopathy.88 Abnormalities in the number and structure of mitochondria in the RPE have been reported in studies of AMD donor eyes.91 Proteomic analyses have suggested that mitochondrial proteins92 and mtDNA93 are altered in AMD, and long-extension polymerase chain reaction (PCR) confirmed that mtDNA is increasingly damaged with AMD progression.94 Oxidative damage to mitochondria can lead to apoptosis in human RPE cells,95 and decreased levels of the mitochondrial antioxidant manganese SOD (MnSOD) led to the development of dry AMD-like lesions in mice.96
Table 22.2 Evidence of mitochondrial oxidative stress in age-related macular degeneration and diabetic retinopathy
AMD, age-related macular degeneration; RPE, retinal pigment epithelium; mtDNA, mitochondrial DNA; MnSOD, manganese superoxide dismutase.
In the case of diabetes, hyperglycemia causes excess production of superoxide by the mitochondrial electron transport chain, inhibiting GAPDH activity.36 As mentioned previously, Brownlee has proposed that this excess of mitochondrial superoxide in endothelial cells of both large and small blood vessels leads to an array of complications seen in diabetes, including increased activity of the polyol pathway and increased production of AGEs. Specifically, hyperglycemia causes increased oxidation of glucose, leading to an elevation in voltage gradient across the mitochondrial membrane. Once a critical threshold in voltage gradient is reached, electron transfer inside complex III of the electron transport chain is blocked. The electrons accumulate at coenzyme Q, which donates them to molecular oxygen, creating large amounts of superoxide. Ultimately, the increased number of ROS in diabetes can alter the mitochondrial membrane potential, leading to cell death.36
Animal studies have demonstrated the effects of hyperglycemia-induced impairment of the mitochondrial membrane. Increased levels of cytochrome c, a key component of the electron transport chain and the precursor to capase-9 in the apoptotic cascade, were detected in rats after 8 months of diabetes. These rats also had increased levels of the proapoptotic Bcl-2-associated X protein (BAX) in the mitochondria. In retinal endothelial cells and pericytes, high concentrations of glucose enhanced the permeability of the mitochondrial membrane, increasing levels of cytochrome c in the cytosol and BAX in the mitochondria.97 These glucose-mediated responses were inhibited by the antioxidant MnSOD. A subsequent study by the same group showed that increased expression of MnSOD can prevent the reduction of GSH levels and total antioxidant capacity caused by diabetes.98 In nontransgenic diabetic mice, upregulation of MnSOD protected against diabetes-related changes in the mitochondria and reduced vascular pathology.33
Genetic association studies have indicated that polymorphisms in mtDNA influence the risk of developing AMD and diabetic retinopathy. In the Blue Mountain Eye Study, mitochondrial haplogroup H was associated with a reduced prevalence of any AMD (odds ratio (OR) 0.75, 95% confidence interval (CI) 0.58–0.97) and early AMD (OR 0.75, 95% CI 0.57–0.98), whereas haplogroups J and U were associated with early AMD only.99 A US study determined that individuals with the T2 haplogroup were 2.5 times more likely to have advanced AMD than those without the haplogroup (OR 2.54, 95% CI 1.36–4.80).100 This finding was supported by studies showing that the mtDNA 4917G polymorphism associated with haplogroup T independently predicted the presence of AMD (OR 2.16, 95% CI 1.21–3.91).101 Subsequently, PCR analysis of mtDNA from AMD and control donor retinas found that single-nucleotide polymorphisms T16126C and A73G, associated with haplogroups J and T, were more frequent in AMD retinas than in normal retinas.102 The authors then replicated these associations in blood DNA from 99 cases and 92 controls.102 The mtDNA haplogroup T was associated with risk of retinopathy among type 2 diabetics.103 As this haplogroup has been associated with multifactorial age-related diseases such as coronary artery disease103 and AMD,101 it seems plausible that carriers of haplogroup T may be exposed to greater oxidative stress or have greater susceptibility to oxidative damage than individuals with other haplogroups.
The A69S polymorphism in the ARMS2 gene, which has been linked to mitochondrial function in some studies, is strongly associated with AMD.104,105 ARMS2 mRNA encoding a 12-kDa protein has been detected in human retina. Cell culture experiments have localized this protein to the mitochondrial outer membrane106 and specifically to mitochondria in the ellipsoid region of the photoreceptors.107 These findings suggest a functional role for ARMS2 in mitochondrial homeostasis and have led to an attractive hypothesis linking ARMS2 and oxidative stress. Such a connection fits well with previous reports of an interaction between ARMS2 A69S and cigarette smoking.108,109 The A69S variant plus smoking was shown to confer a higher risk of AMD than either factor alone, suggesting a link between the ARMS2 protein and smoking-induced oxidative stress. The localization of ARMS2 to the mitochondria has been called into question, however, by a recent immunohistologic study that found no colocalization of ARMS2 antibodies and mitochondrial markers, instead reporting localization of the ARMS2 protein to the cytosol in cultured RPE cells.110 Due to potential inconsistencies in antibodies and visualization, further investigation is necessary to confirm or refute the relationship between ARMS2 and the mitochondria.
Oxidative stress and inflammation
Growing evidence from both cell culture and animal studies points to a mechanistic link between oxidative stress and inflammation (Table 22.3). Decreased SOD2 and increased superoxide production were associated with mononuclear phagocyte-induced apoptosis of the RPE in sod2+/– mice.111 In mice with oxygen-induced retinopathy, injections of the serine proteinase inhibitor SERPINA3K reduced expression of proinflammatory factors (e.g., VEGF, tumor necrosis factor-α, and intercellular adhesion molecule-1), decreased ROS production, and increased SOD and GSH levels.112 The anti-inflammatory effect of SERPINA3K was also demonstrated in cultured retinal cells exposed to hypoxia.112
Table 22.3 Inflammation and its relationship with oxidative stress in age-related macular degeneration (AMD) and diabetic retinopathy
IL, interleukin; PDR, proliferative diabetic retinopathy; COX-2, cyclooxygenase-2; PGE2, prostaglandin E2; VEGF, vascular endothelial growth factor; RPE, retinal pigment epithelium; ROS, reactive oxygen species; NF-κB, nuclear factor-κB; CEP, carboxyethylpyrrole; SOD2, superoxide dismutase 2.
Immunologic and inflammatory processes appear to play a major role in AMD, including drusen formation, complement activation, recruitment of tissue-destructive macrophages, and microglial activation and accumulation.113 The association of multiple complement gene polymorphisms with AMD,114–128 the presence of complement proteins in drusen,10,11 and the link between systemic complement activation and AMD129–131 suggest that innate immunity plays a critical role in AMD pathogenesis. These discoveries have led to experiments designed to define the role of oxidative stress in complement activation in the RPE.
Cell culture studies have linked oxidative stress to reduced expression of the complement factor H (CFH) gene in RPE cells. Investigation of constitutive CFH production in a stable RPE cell line demonstrated that long-term treatment of RPE cells with oxidized photoreceptor outer segments (POS), but not normal POS, markedly downregulated CFH mRNA expression. Further, phagocytosis of both oxidized and normal POS reduced CFH protein expression.132 These results suggest that the phagocytosis of POS, particularly if oxidized, impairs synthesis and secretion of CFH, resulting in reduced regulatory control of complement activation and its proinflammatory effects.132 Additionally, the introduction of H2O2 to RPE cells was shown to reduce the normal interferon-γ-induced stimulation of CFH expression via acetylation of FOXO3, which interrupts STAT1 binding to the CFH promoter.133
The dynamic interplay between oxidative stress and inflammation may contribute to the abnormalities in RPE function evident in AMD. A combination of H2O2 and complement-sufficient serum was shown to disrupt the barrier function of RPE monolayers in culture and evoke polarized secretion of VEGF, although introduction of H2O2 or serum alone did not cause these changes.134 These results suggest that oxidative stress and complement together reduce RPE function. They also point to a common pathway that relates an oxidizing environment and complement activation with VEGF production. Such a pathway could play a role in the neovascularization seen in late AMD.
A close link between oxidative stress and inflammation in AMD is also supported by recent animal work in which mice immunized with mouse serum albumin adducted with the lipid peroxidation product carboxyethylpyrrole developed AMD-like lesions in the retina.135 Specifically, these mice produced antibodies to the hapten, fixed complement component 3 in Bruch’s membrane, and accumulated drusen below the RPE.135 Taken together, these studies demonstrate a molecular link between oxidative stress and complement in RPE cells and suggest that the interaction between oxidative stress and inflammatory mediators plays a key role in the development of AMD.
Oxidative stress-related inflammation may also play a key role in the pathogenesis of diabetic retinopathy. Increased superoxide can promote inflammation by damaging endothelial cells, increasing microvascular permeability, and recruiting neutrophils.38 ROS activate the transcription factor NF-κB, which can increase proinflammatory mediators, such as cytokines, nitric oxide, and prostaglandins. The cytokines interleukin (IL)-1β, IL-6, and IL-8 have been reported to be increased in the vitreous of patients with proliferative diabetic retinopathy.136 Interestingly, it appears that IL-1β increases retinal capillary cell apoptosis.137 Additionally, IL-1 induces the production of cyclooxygenase-2, a catalyst for prostaglandin E2 formation. The inflammatory mediators cyclooxygenase-2 and prostaglandin E2 are reported to contribute to the development of diabetic retinopathy by modulating VEGF-mediated vascular permeability and angiogenesis.138
Retinal therapies targeting oxidative stress
In this section, we will discuss oxidative stress-related therapies for retinal diseases. We will then explore potential avenues for the development of future treatments that protect against oxidative damage in the retina, including antioxidant therapy, AGE inhibitors, and genetic modification. These potential therapies are outlined in Table 22.4.
| Potential avenues for retinal therapy | Specific treatments under investigation |
|---|---|
| Anti-inflammatory agents | |
| Boost in antioxidant defense |
Vitamin A palmitate supplementation to slow rate of retinal degeneration (e.g., RP)143 Supplementation with xanthophylls and omega-3 fatty acids for treatment of AMD in AREDS2 clinical trial27 OT-551 eye drop for geographic atrophy in AMD142 Sulforaphane to protect against retinal oxidative damage via activation of the Nrf2-thioredoxin system152–154 Curcumin to prevent retinal oxidative damage148 via activation of the Nrf2-thioredoxin system148 and increased antioxidant expression (e.g., GST)147 |
| Genetic modification | |
| Inhibition of AGEs | |
| Inhibition of apoptotic pathways |
RPE, retinal pigment epithelium; NF-κB, nuclear factor-κB; RP, retinitis pigmentosa; AMD, age-related macular degeneration; AREDS2, Age-Related Eye Disease Study 2; GST, glutathione-S-transferase; SOD2, superoxide dismutase 2; CAT, catalase; AGEs, advanced glycation end-products; NOS, nitric oxide synthase.
Supplemental antioxidants
Currently, care of patients with dry AMD is centered on antioxidant and zinc supplementation, as there are no direct interventions for drusen or geographic atrophy. AREDS, a multicenter, randomized clinical trial sponsored by the National Eye Institute, demonstrated that daily intake of supplemental antioxidants (β-carotene, vitamin C, vitamin E) and zinc reduced the risk of progression to advanced AMD by 25% over 5 years.22
Supplemental carotenoids have also been associated with decreased AMD risk. Two smaller studies linked xanthophylls (lutein and zeaxanthin) and/or omega-3 fatty acids with improved visual acuity139 and central retinal function (as measured by multifocal ERG).140 In the Rotterdam Study, high dietary intake of zinc, omega-3 fatty acids, β-carotene, and lutein/zeaxanthin corresponded to decreased risk of early AMD onset.141 The AREDS2 study is now investigating the effect of oral supplementation of xanthophylls and omega-3 fatty acids (docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA)) on the risk of AMD progression. In this ongoing clinical trial, scheduled to end in 2013, AMD patients are randomly assigned to four treatment groups: (1) xanthophylls only (10 mg lutein and 2 mg zeaxanthin); (2) omega-3 fatty acids only (350 mg DHA and 650 mg EPA); (3) xanthophylls and omega-3 fatty acids; and (4) placebo.27 The results of this trial may alter the recommended regimen for AMD patients currently treated with antioxidant and zinc supplements.
The topical application of a disubstituted hydroxylamine with antioxidant properties, called OT-551, has been studied for the treatment of geographic atrophy. In a small 10-patient study, patients with bilateral geographic atrophy experienced less visual loss in the eye treated with topical OT-551 compared to the untreated eye, but no significant differences in geographic atrophy area, contrast sensitivity, microperimetry measurements, and total drusen area were found.142 The larger phase II OMEGA study, evaluating the ability of OT-551 to stop geographic atrophy progression and reduce the size of the geographic atrophy lesions, showed no evidence of efficacy in reaching the stated endpoint (P Sternberg, personal communication, 2010). With the lack of promising results in the phase II study, no further clinical trials are planned for this agent to explore its potential efficacy for atrophic AMD.
Studies have suggested the benefit of nutritional and drug supplements in preserving photoreceptor function. The most widely recognized supplement for RP patients is vitamin A palmitate, which has been suggested to slow the rate of retinal degeneration by an unknown mechanism.143 These same studies suggested that vitamin E may be associated with an increase in the deterioration rate of the ERG in RP patients,143 possibly by reducing the availability of other vitamins in the retina. More recently, studies have investigated the role of DHA in the treatment of RP. It was reported that patients initiating vitamin A palmitate supplementation may benefit from the addition of DHA in the first 2 years of treatment.144
Dietary antioxidants
Naturally occurring compounds found in food may protect against retinal degenerations. Curcumin, from the spice turmeric, has been shown to inhibit the NF-κB pathway, which is involved in immune response,145 and to decrease expression of inflammatory mediators.146 Curcumin also increases expression of antioxidants, such as heme oxygenase-1, quinone reductase, and glutathione-S-transferase.147 A 2-week diet supplemented with curcumin protected rats from light-induced retinal damage.148 Similarly, treatment of cultured retinal cells with curcumin protected against H2O2-induced cell death by increasing production of antioxidant enzymes, such as thioredoxin, and activating the Nrf2 antioxidant pathway.148
Another member of the plant-derived polyphenol family, resveratrol, a compound in red wine, has been shown to decrease oxidative damage and RPE proliferation in vitro.149 Evidence suggests that resveratrol may take effect by inhibiting the NF-κB pathway involved in inflammatory response.
The flavonoid quercetin, found in green tea and red onion, is a free radical scavenger and a chelating agent that can reduce iron-driven lipid peroxidation.150 Quercetin was recently reported to protect against H2O2-induced oxidative damage and senescence in cultured RPE cells by inhibiting caspase-3 activity.151
Sulforaphane, an isothiocyanate found in vegetables such as broccoli, appears to exert neuroprotective effects through activation of the Nrf2-thioredoxin system. The antioxidant transcription factor Nrf2 binds to the antioxidant response element (ARE), activating the antioxidant defense system in response to oxidative stress.152 Thioredoxin has been shown to protect against H2O2-induced damage in photoreceptor cells and light damage in mice.153 Sulforaphane induces thioredoxin in light-exposed RPE cells via the Nrf2-ARE pathway,154 thus protecting against photo-oxidative damage to the retina.
Anti-advanced glycation end-product treatment
One possible avenue for treatment of diabetic retinopathy targets the accumulation of AGEs, which can disturb retinal vascular cell function. The AGE inhibitor pyridoxamine protected against diabetes-induced retinal vascular lesions, thickening of the basement membrane, and acellular capillaries in diabetic rats.155 Aminoguanidine, which acts as an inhibitor of inducible nitric oxide synthase and AGE crosslinking, may prevent capillary closure, microaneurysm formation, and nitric oxide synthase depletion.156,157 The efficacy of pyridoxamine and aminoguanidine has yet to be investigated for the treatment of diabetic retinopathy in clinical trials.
Genetic modification
Gene therapy is being explored for the treatment of retinal degeneration. Towards this end, genes involved in the regulation of cellular oxidative stress are being targeted in animal studies to reduce ROS production, bolster antioxidant defenses, and/or increase cellular repair capacity. In mice with ischemia-induced retinal injury, plasmids encoding the human SOD2 (Mn SOD) or CAT (catalase) genes significantly decreased ROS generation and retinal endothelial cell death.158 Another study showed that an adenovirus carrying the CAT gene protected against H2O2-induced damage to RPE cells and light-induced damage to RPE and photoreceptors in mice.159
Conclusions
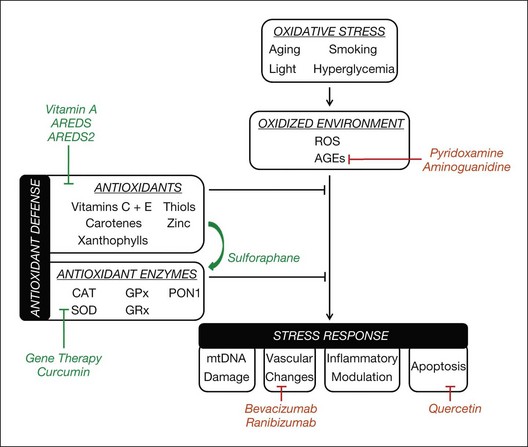
Oxidative stress plays a significant role in the pathogenesis of multiple retinal diseases, including AMD, diabetic retinopathy, and inherited retinal degenerations. ROS, derived primarily from the mitochondria, can adversely affect health and survival of RPE cells, retinal vascular endothelial cells, and photoreceptors. Regardless of a condition’s primary insult, oxidative stress often contributes to retinal cell death via DNA damage, modulation of alternative metabolic pathways, and stimulation of proapoptotic reactions. Because oxidative stress appears to be a common mechanism of retinal cell damage, the development of therapy that protects against retinal oxidative damage has the potential to reduce vision loss from retinal diseases significantly (Fig. 22.1).
1 Beatty S, Koh H, Phil M, et al. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134.
2 Cai J, Nelson KC, Wu M, et al. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221.
3 Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605.
4 Borish ET, Pryor WA, Venugopal S, et al. DNA synthesis is blocked by cigarette tar-induced DNA single-strand breaks. Carcinogenesis. 1987;8:1517–1520.
5 Jones DP, Mody VC, Jr., Carlson JL, et al. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic Biol Med. 2002;33:1290–1300.
6 Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868.
7 Friedman DS, O’Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572.
8 Sarks J, Arnold J, Ho IV, et al. Evolution of reticular pseudodrusen. Br J Ophthalmol. 2011;95:979–985.
9 Zweifel SA, Spaide RF, Curcio CA, et al. Reticular pseudodrusen are subretinal drusenoid deposits. Ophthalmology. 2010;117:303–312. e1
10 Mullins RF, Russell SR, Anderson DH, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–846.
11 Wang L, Clark ME, Crossman DK, et al. Abundant lipid and protein components of drusen. PLoS ONE. 2010;5:e10329.
12 Seddon JM, Cote J, Davis N, et al. Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist–hip ratio. Arch Ophthalmol. 2003;121:785–792.
13 Clemons TE, Milton RC, Klein R, et al. Risk factors for the incidence of advanced age-related macular degeneration in the Age-Related Eye Disease Study (AREDS). AREDS report no. 19. Ophthalmology. 2005;112:533–539.
14 Tan JS, Mitchell P, Kifley A, et al. Smoking and the long-term incidence of age-related macular degeneration: the Blue Mountains Eye Study. Arch Ophthalmol. 2007;125:1089–1095.
15 Cackett P, Wong TY, Aung T, et al. Smoking, cardiovascular risk factors, and age-related macular degeneration in Asians: the Singapore Malay Eye Study. Am J Ophthalmol. 2008;146:960–967. e1
16 Chakravarthy U, Augood C, Bentham GC, et al. Cigarette smoking and age-related macular degeneration in the EUREYE Study. Ophthalmology. 2007;114:1157–1163.
17 Fraser-Bell S, Wu J, Klein R, et al. Smoking, alcohol intake, estrogen use, and age-related macular degeneration in Latinos: the Los Angeles Latino Eye Study. Am J Ophthalmol. 2006;141:79–87.
18 Klein R, Knudtson MD, Cruickshanks KJ, et al. Further observations on the association between smoking and the long-term incidence and progression of age-related macular degeneration: the Beaver Dam Eye Study. Arch Ophthalmol. 2008;126:115–121.
19 Tomany SC, Wang JJ, Van Leeuwen R, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111:1280–1287.
20 Seddon JM, Ajani UA, Sperduto RD, et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA. 1994;272:1413–1420.
21 van Leeuwen R, Boekhoorn S, Vingerling JR, et al. Dietary intake of antioxidants and risk of age-related macular degeneration. JAMA. 2005;294:3101–3107.
22 A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436.
23 Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687.
24 Bone RA, Landrum JT, Mayne ST, et al. Macular pigment in donor eyes with and without AMD: a case-control study. Invest Ophthalmol Vis Sci. 2001;42:235–240.
25 Bernstein PS, Zhao DY, Wintch SW, et al. Resonance Raman measurement of macular carotenoids in normal subjects and in age-related macular degeneration patients. Ophthalmology. 2002;109:1780–1787.
26 Obana A, Hiramitsu T, Gohto Y, et al. Macular carotenoid levels of normal subjects and age-related maculopathy patients in a Japanese population. Ophthalmology. 2008;115:147–157.
27 Krishnadev N, Meleth AD, Chew EY. Nutritional supplements for age-related macular degeneration. Curr Opin Ophthalmol. 2010;21:184–189.
28 Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. 2010;376:124–136.
29 Saaddine JB, Honeycutt AA, Narayan KM, et al. Projection of diabetic retinopathy and other major eye diseases among people with diabetes mellitus: United States, 2005–2050. Arch Ophthalmol. 2008;126:1740–1747.
30 Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890.
31 Kowluru RA, Koppolu P, Chakrabarti S, et al. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res. 2003;37:1169–1180.
32 Kowluru RA, Koppolu P. Diabetes-induced activation of caspase-3 in retina: effect of antioxidant therapy. Free Radic Res. 2002;36:993–999.
33 Kanwar M, Chan PS, Kern TS, et al. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007;48:3805–3811.
34 Kowluru RA, Atasi L, Ho YS. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2006;47:1594–1599.
35 Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011;50:567–575.
36 Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625.
37 Goldin A, Beckman JA, Schmidt AM, et al. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605.
38 Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007:43603.
39 Shiba T, Inoguchi T, Sportsman JR, et al. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Am J Physiol. 1993;265:E783–E793.
40 Williams B, Gallacher B, Patel H, et al. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes. 1997;46:1497–1503.
41 Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820.
42 Bramall AN, Wright AF, Jacobson SG, et al. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu Rev Neurosci. 2010;33:441–472.
43 Ayuso C, Millan JM. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2010;2:34.
44 Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807.
45 Leveillard T, Mohand-Said S, Lorentz O, et al. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36:755–759.
46 Carmody RJ, McGowan AJ, Cotter TG. Reactive oxygen species as mediators of photoreceptor apoptosis in vitro. Exp Cell Res. 1999;248:520–530.
47 Miceli MV, Liles MR, Newsome DA. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp Cell Res. 1994;214:242–249.
48 Yang P, Peairs JJ, Tano R, et al. Oxidant-mediated Akt activation in human RPE cells. Invest Ophthalmol Vis Sci. 2006;47:4598–4606.
49 Yu AL, Fuchshofer R, Kook D, et al. Subtoxic oxidative stress induces senescence in retinal pigment epithelial cells via TGF-beta release. Invest Ophthalmol Vis Sci. 2009;50:926–935.
50 Glotin AL, Debacq-Chainiaux F, Brossas JY, et al. Prematurely senescent ARPE-19 cells display features of age-related macular degeneration. Free Radic Biol Med. 2008;44:1348–1361.
51 Fuchshofer R, Yu AL, Teng HH, et al. Hypoxia/reoxygenation induces CTGF and PAI-1 in cultured human retinal pigment epithelium cells. Exp Eye Res. 2009;88:889–899.
52 Winkler BS, Boulton ME, Gottsch JD, et al. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32.
53 Roehlecke C, Schaller A, Knels L, et al. The influence of sublethal blue light exposure on human RPE cells. Mol Vis. 2009;15:1929–1938.
54 Howes KA, Liu Y, Dunaief JL, et al. Receptor for advanced glycation end products and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:3713–3720.
55 Ma W, Lee SE, Guo J, et al. RAGE ligand upregulation of VEGF secretion in ARPE-19 cells. Invest Ophthalmol Vis Sci. 2007;48:1355–1361.
56 Sreekumar PG, Kannan R, de Silva AT, et al. Thiol regulation of vascular endothelial growth factor-A and its receptors in human retinal pigment epithelial cells. Biochem Biophys Res Commun. 2006;346:1200–1206.
57 Kannan R, Zhang N, Sreekumar PG, et al. Stimulation of apical and basolateral VEGF-A and VEGF-C secretion by oxidative stress in polarized retinal pigment epithelial cells. Mol Vis. 2006;12:1649–1659.
58 Klettner A, Roider J. Constitutive and oxidative-stress-induced expression of VEGF in the RPE are differently regulated by different Mitogen-activated protein kinases. Graefes Arch Clin Exp Ophthalmol. 2009;247:1487–1492.
59 Byeon SH, Lee SC, Choi SH, et al. Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol Vis Sci. 2010;51:1190–1197.
60 Kuroki M, Voest EE, Amano S, et al. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J Clin Invest. 1996;98:1667–1675.
61 Platt DH, Bartoli M, El-Remessy AB, et al. Peroxynitrite increases VEGF expression in vascular endothelial cells via STAT3. Free Radic Biol Med. 2005;39:1353–1361.
62 Abou-Mohamed G, Johnson JA, Jin L, et al. Roles of superoxide, peroxynitrite, and protein kinase C in the development of tolerance to nitroglycerin. J Pharmacol Exp Ther. 2004;308:289–299.
63 Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343.
64 Duh E, Aiello LP. Vascular endothelial growth factor and diabetes: the agonist versus antagonist paradox. Diabetes. 1999;48:1899–1906.
65 Fernandes R, Hosoya K, Pereira P. Reactive oxygen species downregulate glucose transport system in retinal endothelial cells. Am J Physiol Cell Physiol. 2011;300:C927–C936.
66 Eichler W, Reiche A, Yafai Y, et al. Growth-related effects of oxidant-induced stress on cultured RPE and choroidal endothelial cells. Exp Eye Res. 2008;87:342–348.
67 Hoffmann S, Friedrichs U, Eichler W, et al. Advanced glycation end products induce choroidal endothelial cell proliferation, matrix metalloproteinase-2 and VEGF upregulation in vitro. Graefes Arch Clin Exp Ophthalmol. 2002;240:996–1002.
68 Yu DY, Cringle SJ, Su EN, et al. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Invest Ophthalmol Vis Sci. 2000;41:3999–4006.
69 Yu DY, Cringle S, Valter K, et al. Photoreceptor death, trophic factor expression, retinal oxygen status, and photoreceptor function in the P23H rat. Invest Ophthalmol Vis Sci. 2004;45:2013–2019.
70 Padnick-Silver L, Kang Derwent JJ, Giuliano E, et al. Retinal oxygenation and oxygen metabolism in Abyssinian cats with a hereditary retinal degeneration. Invest Ophthalmol Vis Sci. 2006;47:3683–3689.
71 Komeima K, Rogers BS, Lu L, et al. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2006;103:11300–11305.
72 Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605.
73 Tso MO, Zhang C, Abler AS, et al. Apoptosis leads to photoreceptor degeneration in inherited retinal dystrophy of RCS rats. Invest Ophthalmol Vis Sci. 1994;35:2693–2699.
74 Carmody RJ, Cotter TG. Oxidative stress induces caspase-independent retinal apoptosis in vitro. Cell Death Differ. 2000;7:282–291.
75 Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in vivo and differential expression of apoptotic protease activating factor-1 and caspase-3 during retinal development. Cell Death Differ. 2002;9:1220–1231.
76 Doonan F, Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in mouse models of retinal degeneration. J Neurosci. 2003;23:5723–5731.
77 Kunchithapautham K, Rohrer B. Autophagy is one of the multiple mechanisms active in photoreceptor degeneration. Autophagy. 2007;3:65–66.
78 Sanvicens N, Gomez-Vicente V, Masip I, et al. Oxidative stress-induced apoptosis in retinal photoreceptor cells is mediated by calpains and caspases and blocked by the oxygen radical scavenger CR-6. J Biol Chem. 2004;279:39268–39278.
79 Doonan F, Donovan M, Cotter TG. Activation of multiple pathways during photoreceptor apoptosis in the rd mouse. Invest Ophthalmol Vis Sci. 2005;46:3530–3538.
80 Ahuja P, Caffe AR, Ahuja S, et al. Decreased glutathione transferase levels in rd1/rd1 mouse retina: replenishment protects photoreceptors in retinal explants. Neuroscience. 2005;131:935–943.
81 Ahuja-Jensen P, Johnsen-Soriano S, Ahuja S, et al. Low glutathione peroxidase in rd1 mouse retina increases oxidative stress and proteases. Neuroreport. 2007;18:797–801.
82 Hill JW, Hu JJ, Evans MK. OGG1 is degraded by calpain following oxidative stress and cisplatin exposure. DNA Repair (Amst). 2008;7:648–654.
83 Shen J, Yang X, Dong A, et al. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol. 2005;203:457–464.
84 Komeima K, Rogers BS, Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007;213:809–815.
85 Usui S, Oveson BC, Lee SY, et al. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J Neurochem. 2009;110:1028–1037.
86 Leveillard T, Sahel JA. Rod-derived cone viability factor for treating blinding diseases: from clinic to redox signaling. Sci Transl Med. 2010;2:26ps16.
87 Cronin T, Raffelsberger W, Lee-Rivera I, et al. The disruption of the rod-derived cone viability gene leads to photoreceptor dysfunction and susceptibility to oxidative stress. Cell Death Differ. 2010;17:1199–1210.
88 Jarrett SG, Lewin AS, Boulton ME. The importance of mitochondria in age-related and inherited eye disorders. Ophthalmic Res. 2010;44:179–190.
89 Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488.
90 Jin GF, Hurst JS, Godley BF. Rod outer segments mediate mitochondrial DNA damage and apoptosis in human retinal pigment epithelium. Curr Eye Res. 2001;23:11–19.
91 Feher J, Kovacs I, Artico M, et al. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006;27:983–993.
92 Nordgaard CL, Berg KM, Kapphahn RJ, et al. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:815–822.
93 Nordgaard CL, Karunadharma PP, Feng X, et al. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2848–2855.
94 Karunadharma PP, Nordgaard CL, Olsen TW, et al. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51:5470–5479.
95 Jiang S, Moriarty-Craige SE, Orr M, et al. Oxidant-induced apoptosis in human retinal pigment epithelial cells: dependence on extracellular redox state. Invest Ophthalmol Vis Sci. 2005;46:1054–1061.
96 Justilien V, Pang JJ, Renganathan K, et al. SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci. 2007;48:4407–4420.
97 Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003;44:5327–5334.
98 Kowluru RA, Kowluru V, Xiong Y, et al. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006;41:1191–1196.
99 Jones MM, Manwaring N, Wang JJ, et al. Mitochondrial DNA haplogroups and age-related maculopathy. Arch Ophthalmol. 2007;125:1235–1240.
100 SanGiovanni JP, Arking DE, Iyengar SK, et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE. 2009;4:e5508.
101 Canter JA, Olson LM, Spencer K, et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE. 2008;3:e2091.
102 Udar N, Atilano SR, Memarzadeh M, et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2966–2974.
103 Kofler B, Mueller EE, Eder W, et al. Mitochondrial DNA haplogroup T is associated with coronary artery disease and diabetic retinopathy: a case control study. BMC Med Genet. 2009;10:35.
104 Rivera A, Fisher SA, Fritsche LG, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14:3227–3236.
105 Jakobsdottir J, Conley YP, Weeks DE, et al. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77:389–407.
106 Kanda A, Chen W, Othman M, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2007;104:16227–16232.
107 Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40:892–896.
108 Schmidt S, Hauser MA, Scott WK, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006;78:852–864.
109 Schaumberg DA, Hankinson SE, Guo Q, et al. A prospective study of 2 major age-related macular degeneration susceptibility alleles and interactions with modifiable risk factors. Arch Ophthalmol. 2007;125:55–62.
110 Wang G, Spencer KL, Court BL, et al. Localization of age-related macular degeneration-associated ARMS2 in cytosol, not mitochondria. Invest Ophthalmol Vis Sci. 2009;50:3084–3090.
111 Yang D, Elner SG, Lin LR, et al. Association of superoxide anions with retinal pigment epithelial cell apoptosis induced by mononuclear phagocytes. Invest Ophthalmol Vis Sci. 2009;50:4998–5005.
112 Zhang B, Hu Y, Ma JX. Anti-inflammatory and antioxidant effects of SERPINA3K in the retina. Invest Ophthalmol Vis Sci. 2009;50:3943–3952.
113 Patel M, Chan CC. Immunopathological aspects of age-related macular degeneration. Semin Immunopathol. 2008;30:97–110.
114 Edwards AO, Ritter R, 3rd., Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424.
115 Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421.
116 Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389.
117 Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232.
118 Zareparsi S, Branham KE, Li M, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet. 2005;77:149–153.
119 Souied EH, Leveziel N, Richard F, et al. Y402H complement factor H polymorphism associated with exudative age-related macular degeneration in the French population. Mol Vis. 2005;11:1135–1140.
120 Simonelli F, Frisso G, Testa F, et al. Polymorphism p.402Y>H in the complement factor H protein is a risk factor for age related macular degeneration in an Italian population. Br J Ophthalmol. 2006;90:1142–1145.
121 Despriet DD, Klaver CC, Witteman JC, et al. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA. 2006;296:301–309.
122 Seitsonen S, Lemmela S, Holopainen J, et al. Analysis of variants in the complement factor H, the elongation of very long chain fatty acids-like 4 and the hemicentin 1 genes of age-related macular degeneration in the Finnish population. Mol Vis. 2006;12:796–801.
123 Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462.
124 Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561.
125 Fagerness JA, Maller JB, Neale BM, et al. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100–104.
126 Ennis S, Gibson J, Cree AJ, et al. Support for the involvement of complement factor I in age-related macular degeneration. Eur J Hum Genet. 2010;18:15–16.
127 Hageman GS, Hancox LS, Taiber AJ, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med. 2006;38:592–604.
128 Hughes AE, Orr N, Esfandiary H, et al. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–1177.
129 Scholl HP, Charbel Issa P, Walier M, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. 2008;3:e2593.
130 Reynolds R, Hartnett ME, Atkinson JP, et al. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci. 2009;50:5818–5827.
131 Huang SJ, Costa DL, Gross NE, et al. Peripheral drusen in membranoproliferative glomerulonephritis type II. Retina. 2003;23:429–431.
132 Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res. 2007;84:635–645.
133 Wu Z, Lauer TW, Sick A, et al. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J Biol Chem. 2007;282:22414–22425.
134 Thurman JM, Renner B, Kunchithapautham K, et al. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009;284:16939–16947.
135 Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–198.
136 Yuuki T, Kanda T, Kimura Y, et al. Inflammatory cytokines in vitreous fluid and serum of patients with diabetic vitreoretinopathy. J Diabetes Complications. 2001;15:257–259.
137 Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–1347.
138 Wilkinson-Berka JL. Vasoactive factors and diabetic retinopathy: vascular endothelial growth factor, cycoloxygenase-2 and nitric oxide. Curr Pharm Des. 2004;10:3331–3348.
139 Cangemi FE. TOZAL Study: an open case control study of an oral antioxidant and omega-3 supplement for dry AMD. BMC Ophthalmol. 2007;7:3.
140 Parisi V, Tedeschi M, Gallinaro G, et al. Carotenoids and antioxidants in age-related maculopathy Italian study: multifocal electroretinogram modifications after 1 year. Ophthalmology. 2008;115:324–333. e2
141 Ho L, van Leeuwen R, Witteman JC, et al. Reducing the genetic risk of age-related macular degeneration with dietary antioxidants, zinc, and ω-3 fatty acids: the Rotterdam study. Arch Ophthalmol. 2011;129:758–766.
142 Wong WT, Kam W, Cunningham D, et al. Treatment of geographic atrophy by the topical administration of OT-551: results of a phase II clinical trial. Invest Ophthalmol Vis Sci. 2010;51:6131–6139.
143 Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111:761–772.
144 Berson EL, Rosner B, Sandberg MA, et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Arch Ophthalmol. 2004;122:1306–1314.
145 Singh S, Aggarwal BB. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected]. J Biol Chem. 1995;270:24995–25000.
146 Abe Y, Hashimoto S, Horie T. Curcumin inhibition of inflammatory cytokine production by human peripheral blood monocytes and alveolar macrophages. Pharmacol Res. 1999;39:41–47.
147 Scapagnini G, Colombrita C, Amadio M, et al. Curcumin activates defensive genes and protects neurons against oxidative stress. Antioxid Redox Signal. 2006;8:395–403.
148 Mandal MN, Patlolla JM, Zheng L, et al. Curcumin protects retinal cells from light-and oxidant stress-induced cell death. Free Radic Biol Med. 2009;46:672–679.
149 King RE, Kent KD, Bomser JA. Resveratrol reduces oxidation and proliferation of human retinal pigment epithelial cells via extracellular signal-regulated kinase inhibition. Chem Biol Interact. 2005;151:143–149.
150 Murota K, Mitsukuni Y, Ichikawa M, et al. Quercetin-4’-glucoside is more potent than quercetin-3-glucoside in protection of rat intestinal mucosa homogenates against iron ion-induced lipid peroxidation. J Agric Food Chem. 2004;52:1907–1912.
151 Kook D, Wolf AH, Yu AL, et al. The protective effect of quercetin against oxidative stress in the human RPE in vitro. Invest Ophthalmol Vis Sci. 2008;49:1712–1720.
152 Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260.
153 Tanito M, Agbaga MP, Anderson RE. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic Biol Med. 2007;42:1838–1850.
154 Tanito M, Masutani H, Kim YC, et al. Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Invest Ophthalmol Vis Sci. 2005;46:979–987.
155 Stitt A, Gardiner TA, Alderson NL, et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes. 2002;51:2826–2832.
156 Edelstein D, Brownlee M. Mechanistic studies of advanced glycosylation end product inhibition by aminoguanidine. Diabetes. 1992;41:26–29.
157 Friedman EA. Advanced glycosylated end products and hyperglycemia in the pathogenesis of diabetic complications. Diabetes Care. 1999;22(Suppl 2):B65–B71.
158 Chen B, Caballero S, Seo S, et al. Delivery of antioxidant enzyme genes to protect against ischemia/reperfusion-induced injury to retinal microvasculature. Invest Ophthalmol Vis Sci. 2009;50:5587–5595.
159 Rex TS, Tsui I, Hahn P, et al. Adenovirus-mediated delivery of catalase to retinal pigment epithelial cells protects neighboring photoreceptors from photo-oxidative stress. Hum Gene Ther. 2004;15:960–967.