[level-membership-for-pediatrics-category]
Chapter 693 Marfan Syndrome
Pathogenesis
Aberrant TGF-β signaling might also play a role in the wider spectrum of manifestations of MFS. Increased TGF-β signaling has been observed in many tissues in MFS mice, including the developing lung, skeletal muscle, mitral valve, and dura. Treatment of these mice with agents that antagonize TGF-β signaling attenuates or prevents pulmonary emphysema, skeletal muscle myopathy, and myxomatous degeneration of the mitral valve. Patients with heterozygous mutations in TGF-β receptors 1 and 2 (TGFβR1 and TGFβR2) have MFS-like manifestations yet normal fibrillin-1. Paradoxically, these mutations also appear to result in increased TGF-β signaling in tissues of these patients. The resulting Loeys-Dietz syndrome (LDS) has much phenotypic overlap with MFS, but it also has many discriminating features (see Differential Diagnosis).
Clinical Manifestations
Skeletal System
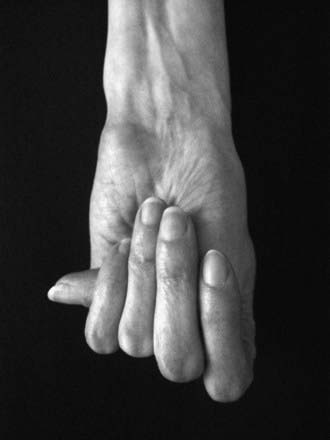
Disproportionate overgrowth of the long bones is often the most striking and immediately evident manifestation of MFS. Anterior chest deformity is caused by overgrowth of the ribs, pushing the sternum anteriorly (pectus carinatum) or posteriorly (pectus excavatum). Overgrowth of arms and legs can lead to an arm span >1.05 times the height or a reduced upper to lower segment ratio (in the absence of severe scoliosis). Arachnodactyly (overgrowth of the fingers) is generally a subjective finding. The combination of long fingers and loose joints leads to the characteristic Walker-Murdoch or wrist sign: full overlap of the distal phalanges of the thumb and fifth finger when wrapped around the contralateral wrist (Fig. 693-1). The Steinberg or thumb sign is present when the distal phalanx of the thumb fully extends beyond the ulnar border of the hand when folded across the palm (Fig. 693-2), with or without active assistance by the patient or examiner.

Thoracolumbar scoliosis is commonly present and can contribute to the systemic score of the diagnosis (Table 693-1). Protrusio acetabuli (inward bulging of the acetabulum into the pelvic cavity), which is generally asymptomatic in young adults, is best identified with radiographic imaging. Pes planus (flat feet) is commonly present and varies from mild and asymptomatic to severe deformity, wherein medial displacement of the medial malleolus results in collapse of the arch and often reactive hip and knee disturbances. Curiously, a subset of patients with the disorder present with an exaggerated arch (pes cavus). Although joint laxity or hypermobility is often identified, joints can be normal or even develop contractures. Reduced extension of the elbows is common and can contribute to the systemic score of the diagnosis (see Table 693-1). Contracture of the fingers (camptodactyly) is commonly observed, especially in children with severe and rapidly progressive MFS. Several craniofacial manifestations are often present and can contribute to the diagnosis (Fig. 693-3). These include a long narrow skull (dolicocephaly), recession of the eyeball within the socket (enophthalmos), recessed lower mandible (retrognathia) or small chin (micrognathia), malar hypoplasia (malar flattening), and downward-slanting palpebral fissures.
Table 693-1 Diagnostic Criteria for Marfan Syndrome (MFS)
In the absence of a family history of MFS, a diagnosis can be reached in 1 of 4 scenarios:
In the absence of a family history of MFS, alternative diagnoses to MFS include:
In the presence of a family history of MFS, a diagnosis can be reached in 1 of 3 scenarios:
SCORING OF SYSTEMIC FEATURES (IN POINTS)†
CRITERIA FOR CAUSAL FBN1 MUTATION
US/LS, upper segment/lower segment ratio.
* Without discriminating features of SGS, LDS, or vEDS (as defined in Table 693-2) and after TGFBR1/2, collagen biochemistry, COL3A1 testing if indicated. Other conditions/genes will emerge with time.
† Maximum total: 20 points; score ≥7 indicates systemic involvement.
From Loeys BL, Dietz HC, Braverman AC, et al: The revised Ghent nosology for the Marfan syndrome, J Med Genet 47:476–485, 2010.
Cardiovascular System
Manifestations of MFS in the cardiovascular system are conveniently divided into those affecting the heart and those affecting the vasculature. Within the heart, the atrioventricular (AV) valves are most often affected. Thickening of the AV valves is common and often associated with prolapse of the mitral and/or tricuspid valves. Variable degrees of regurgitation may be present. In children with early onset and severe MFS, insufficiency of the mitral valve can lead to congestive heart failure, pulmonary hypertension, and death in infancy; this manifestation represents the leading cause of morbidity and mortality in young children with the disorder (Chapter 422.3). Aortic valve dysfunction is generally a late occurrence, attributed to stretching of the aortic annulus by an expanding root aneurysm. Both the aortic and AV valves seem to be more prone to calcification in persons with MFS.
Ventricular dysrhythmia has been described in children with MFS (Chapter 429). In association with mitral valve dysfunction, supraventricular arrhythmia (e.g., atrial fibrillation or supraventricular tachycardia) may be seen. There is also an increased prevalence of prolonged QT interval on electrocardiographic surveys of patients with MFS. Dilated cardiomyopathy, beyond that explained by aortic or mitral valve regurgitation, seems to occur with increased prevalence in patients with MFS, perhaps implicating a role for fibrillin-1 in the cardiac ventricles. However, the incidence seems to be low, and the occurrence of mild to moderate ventricular systolic dysfunction is often attributed to mitral or aortic insufficiency or to the use of β-adrenergic receptor blockade.
Ocular System
Ectopia lentis (dislocation of the ocular lens) of any degree constitutes a major part of the diagnostic criteria for Marfan syndrome (see Table 693-1), although it is not unique to the disorder. Ectopia lentis occurs in around 60-70% of patients with the disorder. When identified, it should prompt further assessment for MFS, although homocystinuria, Weill-Marchesani, and familial ectopia lentis are also associated with this condition. Other manifestations include early and severe myopia, flat cornea, increased axial length of the globe, hypoplastic iris, and ciliary muscle hypoplasia, causing decreased miosis. Patients with MFS can also have retinal detachment and a predisposition for early cataracts or glaucoma.
Family and Genetic History
The current criteria established for the diagnosis of MFS emphasize the need for a first-degree relative to independently meet the criteria before including family history as a major criterion for diagnosis. The criteria that are required to meet a causal mutation in a patient with MFS are described in Table 693-1.
Diagnosis
Given the complexity of the diagnostic evaluation for MFS and the differential diagnosis, the evaluation should be coordinated by an individual or team with extensive experience in the diagnosis of connective tissue disorders. Diagnosis is based on criteria that include both clinical and molecular analyses (see Table 693-1).
Differential Diagnosis
Several disorders are included in the differential diagnosis of MFS on the basis of similar skeletal, cardiac, or ophthalmologic manifestations (Table 693-2). Many patients referred for possible MFS are found to have evidence of a systemic connective tissue disorder, including long limbs, deformity of the thoracic cage, striae atrophicae, mitral valve prolapse, and mild and nonprogressive dilatation of the aortic root, but do not meet diagnostic criteria for MFS. This constellation of features not fulfilling the diagnostic requirements of MFS is referred to by the acronym MASS phenotype, emphasizing the mitral, aortic, skin, and skeletal manifestations. The MASS phenotype can segregate in large pedigrees and remain stable over time. This diagnosis is most challenging in the context of an isolated and young patient. In this setting, careful follow-up is needed to distinguish MASS phenotype from emerging MFS, especially in children.
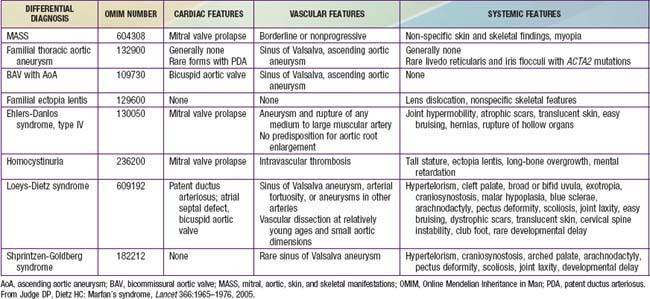
Other fibrillinopathies, such as familial mitral valve prolapse (MVP) syndrome and familial ectopia lentis, also include subdiagnostic manifestations and can be due to mutations in the gene encoding fibrillin-1. Also included in the differential diagnosis of the disorder is homocystinuria, caused by a deficiency of cystathionine β-synthase. Patients with homocystinuria often have tall stature, long-bone overgrowth, and ectopia lentis, but they do not typically have aortic enlargement or dissection. By contrast with MFS, the inheritance of homocystinuria (Chapter 79.3) is autosomal recessive, and affected persons often have mental retardation, a predisposition to thromboembolism, and a high incidence of coronary artery atherosclerosis. Observation of severely elevated concentrations of plasma homocystine is an efficient mechanism to distinguish homocystinuria from MFS.
Current and Future Therapies
Ahimastos AA, Aggarwal A, D’Orsa KM, et al. Effect of perindopril on large artery stiffness and aortic root diameter in patients with Marfan syndrome. JAMA. 2007;298:1539-1546.
Brooke BS, Habashi JP, Judge DP, et al. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787-2795.
Davies RR, Goldstein LJ, Coady MA, et al. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg. 2002;73:17-27.
de Oliveira NC, David TE, Ivanov J, et al. Results of surgery for aortic root aneurysm in patients with Marfan syndrome. JThorac Cardiovasc Surg. 2003;125:789-796.
DePaepe A, Devereux RB, Dietz HC, et al. Revised criteria for Marfan syndrome. Am J Med Genet. 1996;62:417-426.
Faivre L, Masurel-Paulet A, Collard-Béroud G, et al. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. 2009;123:391-398.
Gelb BD. Marfan’s syndrome and related disorders—more tightly connected than we thought. N Engl J Med. 2006;355:841-844.
Glesby MJ, Pyeritz RE. Association of mitral valve prolapse and systemic abnormalities of connective tissue: a phenotypic continuum. JAMA. 1989;262:523-528.
Habashi JP, Judge D, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117-121.
Johnson JN, Hartman TK, Pianosi PT, et al. Cardiorespiratory function after operation for pectus excavatum. J Pediatr. 2008;153:359-364.
Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366:1965-1976.
Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117:2802-2813.
Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275-281.
Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476-485.
Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sport participation for young patients with genetic cardiovascular diseases. Circulation. 2004;109:2807-2816.
Matt P, Schoenhoff F, Habashi J, et al. Circulating transforming growth factor-β in Marfan syndrome. Circulation. 2009;120:526-532.
Meijboom LJ, Vos FE, Timmermans J, et al. Pregnancy and aortic root growth in the Marfan syndrome: a prospective study. Eur Heart J. 2005;26:914-920.
Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407-411.
Rhee D, Solowiejczyk D, Altmann K, et al. Incidence of aortic root dilation in pectus excavatum and its association with Marfan syndrome. Arch Pediatr Adolesc Med. 2008;162:882-885.
Tierney ESS, Feingold B, Printz BF, et al. Beta-blocker therapy does not alter the rate of aortic root dilation in pediatric patients with Marfan syndrome. J Pediatr. 2007;150:77-82.
Yetman AT, Bornemeier RA, McCrindle BW. Usefulness of enalapril versus propranolol or atenolol for prevention of aortic dilation in patients with the Marfan syndrome. Am J Cardiol. 2005;95:1125-1127.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 693 Marfan Syndrome
Pathogenesis
Aberrant TGF-β signaling might also play a role in the wider spectrum of manifestations of MFS. Increased TGF-β signaling has been observed in many tissues in MFS mice, including the developing lung, skeletal muscle, mitral valve, and dura. Treatment of these mice with agents that antagonize TGF-β signaling attenuates or prevents pulmonary emphysema, skeletal muscle myopathy, and myxomatous degeneration of the mitral valve. Patients with heterozygous mutations in TGF-β receptors 1 and 2 (TGFβR1 and TGFβR2) have MFS-like manifestations yet normal fibrillin-1. Paradoxically, these mutations also appear to result in increased TGF-β signaling in tissues of these patients. The resulting Loeys-Dietz syndrome (LDS) has much phenotypic overlap with MFS, but it also has many discriminating features (see Differential Diagnosis).
Clinical Manifestations
Skeletal System
Disproportionate overgrowth of the long bones is often the most striking and immediately evident manifestation of MFS. Anterior chest deformity is caused by overgrowth of the ribs, pushing the sternum anteriorly (pectus carinatum) or posteriorly (pectus excavatum). Overgrowth of arms and legs can lead to an arm span >1.05 times the height or a reduced upper to lower segment ratio (in the absence of severe scoliosis). Arachnodactyly (overgrowth of the fingers) is generally a subjective finding. The combination of long fingers and loose joints leads to the characteristic Walker-Murdoch or wrist sign: full overlap of the distal phalanges of the thumb and fifth finger when wrapped around the contralateral wrist (Fig. 693-1). The Steinberg or thumb sign is present when the distal phalanx of the thumb fully extends beyond the ulnar border of the hand when folded across the palm (Fig. 693-2), with or without active assistance by the patient or examiner.
Thoracolumbar scoliosis is commonly present and can contribute to the systemic score of the diagnosis (Table 693-1). Protrusio acetabuli (inward bulging of the acetabulum into the pelvic cavity), which is generally asymptomatic in young adults, is best identified with radiographic imaging. Pes planus (flat feet) is commonly present and varies from mild and asymptomatic to severe deformity, wherein medial displacement of the medial malleolus results in collapse of the arch and often reactive hip and knee disturbances. Curiously, a subset of patients with the disorder present with an exaggerated arch (pes cavus). Although joint laxity or hypermobility is often identified, joints can be normal or even develop contractures. Reduced extension of the elbows is common and can contribute to the systemic score of the diagnosis (see Table 693-1). Contracture of the fingers (camptodactyly) is commonly observed, especially in children with severe and rapidly progressive MFS. Several craniofacial manifestations are often present and can contribute to the diagnosis (Fig. 693-3). These include a long narrow skull (dolicocephaly), recession of the eyeball within the socket (enophthalmos), recessed lower mandible (retrognathia) or small chin (micrognathia), malar hypoplasia (malar flattening), and downward-slanting palpebral fissures.
Table 693-1 Diagnostic Criteria for Marfan Syndrome (MFS)
In the absence of a family history of MFS, a diagnosis can be reached in 1 of 4 scenarios:
In the absence of a family history of MFS, alternative diagnoses to MFS include:
In the presence of a family history of MFS, a diagnosis can be reached in 1 of 3 scenarios:
SCORING OF SYSTEMIC FEATURES (IN POINTS)†
CRITERIA FOR CAUSAL FBN1 MUTATION
US/LS, upper segment/lower segment ratio.
* Without discriminating features of SGS, LDS, or vEDS (as defined in Table 693-2) and after TGFBR1/2, collagen biochemistry, COL3A1 testing if indicated. Other conditions/genes will emerge with time.
† Maximum total: 20 points; score ≥7 indicates systemic involvement.
From Loeys BL, Dietz HC, Braverman AC, et al: The revised Ghent nosology for the Marfan syndrome, J Med Genet 47:476–485, 2010.
Cardiovascular System
Manifestations of MFS in the cardiovascular system are conveniently divided into those affecting the heart and those affecting the vasculature. Within the heart, the atrioventricular (AV) valves are most often affected. Thickening of the AV valves is common and often associated with prolapse of the mitral and/or tricuspid valves. Variable degrees of regurgitation may be present. In children with early onset and severe MFS, insufficiency of the mitral valve can lead to congestive heart failure, pulmonary hypertension, and death in infancy; this manifestation represents the leading cause of morbidity and mortality in young children with the disorder (Chapter 422.3). Aortic valve dysfunction is generally a late occurrence, attributed to stretching of the aortic annulus by an expanding root aneurysm. Both the aortic and AV valves seem to be more prone to calcification in persons with MFS.
Ventricular dysrhythmia has been described in children with MFS (Chapter 429). In association with mitral valve dysfunction, supraventricular arrhythmia (e.g., atrial fibrillation or supraventricular tachycardia) may be seen. There is also an increased prevalence of prolonged QT interval on electrocardiographic surveys of patients with MFS. Dilated cardiomyopathy, beyond that explained by aortic or mitral valve regurgitation, seems to occur with increased prevalence in patients with MFS, perhaps implicating a role for fibrillin-1 in the cardiac ventricles. However, the incidence seems to be low, and the occurrence of mild to moderate ventricular systolic dysfunction is often attributed to mitral or aortic insufficiency or to the use of β-adrenergic receptor blockade.
[/not-level-membership-for-pediatrics-category]
