Chapter 31 Management of Tumors of the Fourth Ventricle
Anatomy
The fourth ventricle is a broad tent-shaped cerebrospinal fluid (CSF) cavity located behind the brain stem and in front of the cerebellum in the center of the posterior fossa (Fig. 31-1). CSF enters through the cerebral aqueduct, which opens into the fourth ventricle at its rostral end. The ventricle widens caudally until its maximum width at the level of the lateral recesses, from which CSF exits through the two foramina of Luschka into the cerebellopontine cisterns on either side. The ventricle narrows again to its caudal terminus at the obliterated central canal of the spinal cord, called the obex from the Latin for “barrier.” The foramen of Magendie is just posterior to the obex and allows CSF to exit into the cerebellomedullary cistern, which is continuous with the cisterna magna. There are no arteries or veins within the cavity of the fourth ventricle. All of the vessels associated with this region are in the fissures located just outside the fourth ventricular roof.
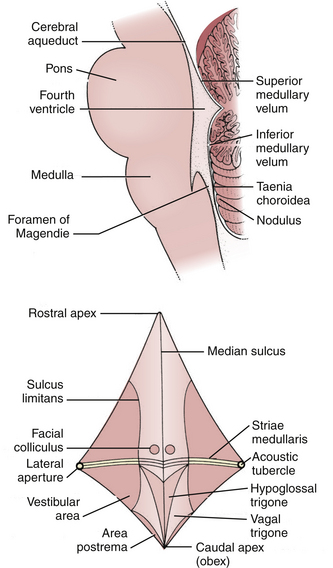
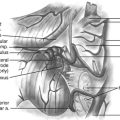
The glistening white floor of the fourth ventricle is the posterior surface of the brain stem (Fig. 31-2). The border between the pons and medulla occurs approximately at the level of the foramina of Luschka. The superior (pontine) part of the floor begins at the aqueduct and expands to the lower margin of the cerebellar peduncles. The inferior (medullary) part of the floor begins just below the lateral recesses at the attachment of the tela choroidea to the taenia choroidea and extends to the obex, limited laterally be the taeniae, which mark the inferolateral margins of the floor. Between these is the intermediate part, which extends into the lateral recesses on either side. There is a longitudinal midline sulcus in the fourth ventricular floor called the median sulcus. On either side of the median sulcus is the sulcus limitans, which also runs longitudinally parallel to the median sulcus. The sulcus limitans is an important landmark for functional anatomy of nuclei beneath the ventricular floor, as motor nuclei are medial and sensory nuclei lateral to the sulcus limitans. Medial to the sulcus limitans on either side of the median sulcus is the median eminence, a collection of four paired elevations in the fourth ventricular floor that are collectively referred to as the calamus scriptorius since they resemble the head of a fountain pen (Fig. 31-1). Rostral to caudal, the median eminence consists of the facial colliculus, which overlies the facial nucleus; the hypoglossal triangle, which overlies the hypoglossal nucleus; the vagal triangle, which overlies the dorsal nucleus of the vagus; and the area postrema, a tongue-shaped structure that is part of the brain-stem emetic center. Lateral to the sulcus limitans is the vestibular area, so named because is overlies the vestibular nuclei. This area is widest in the neighborhood of the lateral recess, where the striae medullaris cross transversely across the inferior cerebellar peduncles to disappear into the median sulcus. The auditory tubercle in the lateral part of the vestibular area overlies the dorsal cochlear nucleus and cochlear nerve.
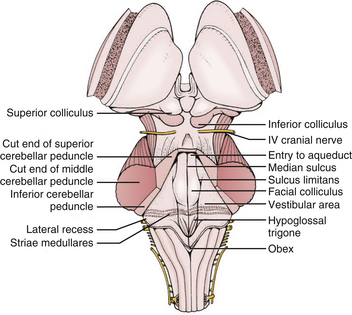
FIGURE 31-2 Fourth ventricle after removal of the cerebellum.
(Modified from Cohen AR. Surgical Disorders of the Fourth Ventricle. Cambridge, MA: Blackwell Science; 1996.)
The roof of the fourth ventricle is tent-shaped, rising to an apex called the fastigium that divides the superior roof from the inferior roof. The median part of the superior roof, called the superior medullary velum, consists of a thin lamina of white matter between the cerebellar peduncles. Just behind its outer surface is the lingula, the uppermost division of the vermis. The lateral walls of the superior roof are formed by the superior and inferior cerebellar peduncles, which lie between the fourth ventricle and the middle cerebellar peduncle. The rostral midline of the inferior roof is formed by the nodule, which lies directly in front of the uvula, the lower part of the vermis that hangs down between the tonsils (mimicking the appearance of the pharynx). Lateral to the nodule is the inferior medullary velum, a thin sheet of neural tissue that stretches over the fourth ventricle to connect the nodule to the flocculi on either side just superior to the outer extremity of the lateral recess. The inferior medullary velum is thus part of the primitive flocculonodular lobe of the cerebellum. The caudal inferior roof consists of the tela choroidea, two thin arachnoid-like membranes sandwiching a vascular layer of choroidal vessels to which the choroid plexus is attached. The junction between the tela choroidea and the nodule/inferior medullary velum (telovelar junction) is at the level of the lateral recess. The tela choroidea is attached to the ventricular floor at narrow white ridges called taeniae choroidea, which meet at the obex and extend upward to turn laterally over the inferior cerebellar peduncles into each lateral recess, forming its lower border. As a result, the choroid plexus (extending from the ventricular surface of the tela) forms an upside-down L shape on either side of midline. There is a medial segment of choroid plexus that extends longitudinally from the foramen of Magendie up to the nodule and a lateral segment that extends transversely from the rostral ends of the medial segments out to the foramen of Luschka. The three fourth ventricular outlet foramina (Magendie and Luschka) are located in the tela choroidea itself, and frequently choroid plexus protrudes from these foramina.
The cerebellopontine fissures are intimately related to the lateral recesses of the fourth ventricle. They are produced by the folding of the cerebellum laterally around the sides of the pons and middle cerebellar peduncles. Each cerebellopontine fissure is shaped like a V in the coronal plain with the point facing laterally. The outer surface of the V is made up of the petrosal surfaces of the cerebellum, and the inner surface is made up of the middle cerebellar peduncles. The lateral recess and foramen of Luschka open into the medial part of the inferior limb of the V near the flocculus. Several cranial nerves run through the cerebellopontine fissure, including the trigeminal (through the superior limb) and the facial, glossopharyngeal, and vagus (through the inferior limb). The anterior inferior cerebellar arteries (AICA) also run through these fissures. Each AICA courses posteriorly around the pons then sends branches to nerves of the acoustic meatus and choroid plexus protruding from the foramen of Luschka before passing around the flocculus on the middle cerebellar peduncle to supply the petrosal surface of the cerebellum. Venous blood from the cerebellopontine fissure and lateral recess primarily drains into the superior petrosal sinus. The vein of the cerebellopontine fissure is formed by the convergence of several veins on the apex of the fissure, including the vein of the middle cerebellar peduncle into which the vein of the inferior cerebellar peduncle drains. This vein courses near the superior limb of the fissure to drain into the superior petrosal sinus rostral to the facial and glossopharyngeal nerves.
Surgical Technique
Surgical Approach
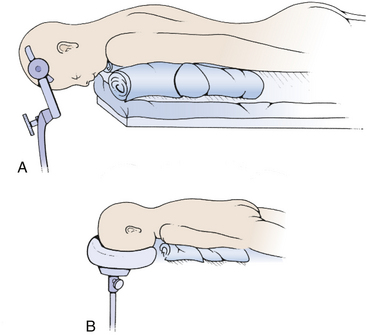
The most commonly used position for the midline suboccipital approach (especially in very young patients) is the prone position, in which the patient is rolled after induction of anesthesia so that the face is toward the floor (Fig. 31-3). There are many advantages to this position: the anatomy is clearly visualized, it is easy for two to work together since one operator can stand on either side, and the multiple complications of the sitting position do not occur. The most significant disadvantage of the prone position is venous congestion that can lead to more significant blood loss, pooling of blood in the operative field, and soft tissue swelling of the face. This congestion is much worse if the head is rotated and flexed, and is improved somewhat by elevating the head above the level of the heart. Also, nasotracheal rather than orotracheal intubation can minimize compression of the base of tongue and impairment of venous drainage of the tongue and pharynx. The weight is distributed to minimize pressure points that can lead to skin breakdown and neuropathy, especially at the ulnar nerve at the elbow, common peroneal nerve across fibular head, and lateral femoral cutaneous nerve at the iliac crest. Two longitudinal padded roles are placed under the patient, and the knees and ankles are padded. The neck is placed in the “military tuck position” with moderate flexion of the upper cervical spine (to open up the space between the foramen magnum and the arch of C1) and less flexion of the lower cervical spine (to bring the occiput parallel with the patient’s back). The chin and chest at least two fingers apart. Finally, the table is positioned so that the neck is parallel to floor and the head is above the heart. The shoulders can be gently retracted toward the feet with some tape, and a strap under the buttocks is helpful to prevent sliding. The surgeon and assistant then operate from either side using the microscope, and the scrub nurse’s Mayfield table can be placed over the patient’s back.
The third option for positioning is the sitting position, in which the patient is positioned sitting upright so that the operative corridor is parallel to the floor (Fig. 31-4). The sitting position offers a very clear operative field since blood and cerebrospinal fluid drain out of the operative site. However, there are many risks to the sitting position.1 The most significant dangers are cardiovascular instability and hypotension, air embolism, and subdural hematoma. All patients should have an agitated saline echocardiogram to exclude right to left shunt through a patent foramen ovale that could complicate air embolism and presence of such a shunt is an absolute contraindication for the sitting position. Precordial Doppler ultrasonic flow and end-tidal CO2 should be monitored throughout the case. The risk of subdural hematoma is greatly increased by presence of a shunt, and if possible the shunt should be occluded prior to attempting an operation in the sitting position. Other risks of the sitting position include tension pneumocephalus, cervical myelopathy, thermal loss (especially in children), surgeon fatigue, and sudden loss of CSF from enlarged lateral and third ventricles after removal of a fourth ventricle mass lesion. When applying the head holder, the pin sites must be covered with Vaseline gauze to minimize entry of air2 and the head taped to the head-holder for extra support in case the pins become dislodged. The patient is elevated slowly into the sitting position so that the foramen magnum is at the surgeon’s eye level with both of the patient’s legs flexed at the knees to prevent postoperative sciatica. The instrument table is placed over the patient’s head. Infants too young for pins may be taped to a padded headrest to support the forehead and chin, but it is probably safer to use the prone position. Throughout the case the patient should be carefully monitored for signs of hypotension or air embolism. If air embolism occurs, the wound should be packed with a saline-soaked sponge, and anesthesia should aspirate the atrial catheter to attempt to remove the embolus from the left atrium. If the embolus is severe, the patient should be placed in left decubitus position; otherwise, as soon as the patient is stable, the wound may be slowly exposed while covering the potential source of air with Gelfoam and Surgicel. If careful preparation is undertaken and complications dealt with promptly, the sitting position can be relatively safe.3,4
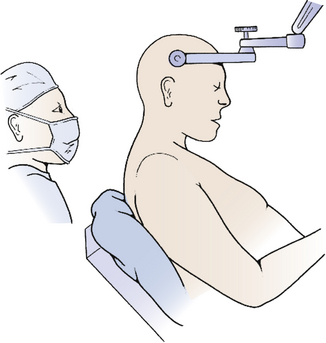
FIGURE 31-4 Sitting position. Skeletal fixation is maintained using a pin head holder with the neck moderately flexed.
(Modified from Cohen AR. Surgical Disorders of the Fourth Ventricle. Cambridge, MA: Blackwell Science; 1996.)
The incision is made with a number 10 blade applying firm digital compression, and bleeding points are coagulated (Fig. 31-5). The incision should be midline, but if the tumor is lateral, a hockey-stick incision can be used to allow for a wider craniectomy. The skin is undermined superficial to the fascia on both sides of the superior half of incision in preparation to create a fascial flap for closure (Fig. 31-6). The skin is then elevated with toothed forceps or a skin hook and a plane of dissection developed with knife or monopolar coagulation, sparing the occipital artery and nerve whenever possible. Even a slight deviation off midline will produce brisk bleeding from the muscles once deeper tissues are exposed. When anatomical landmarks are identified to confirm that the operative course is truly midline, cerebellar or Weitlaner retractors are placed to maintain exposure. As deeper layers are exposed, curved retractors may be used.
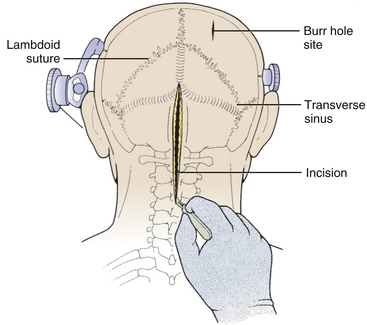
FIGURE 31-5 Skin Incision. The midline linear incision extends from just above the inion to the midcervical region.
(Modified from Cohen AR. Surgical Disorders of the Fourth Ventricle. Cambridge, MA: Blackwell Science; 1996.)
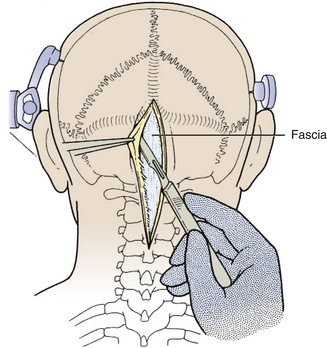
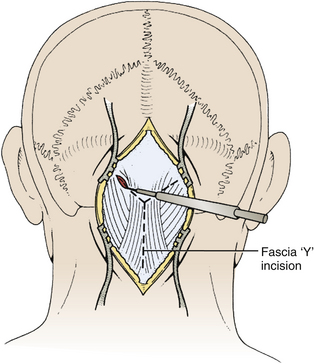
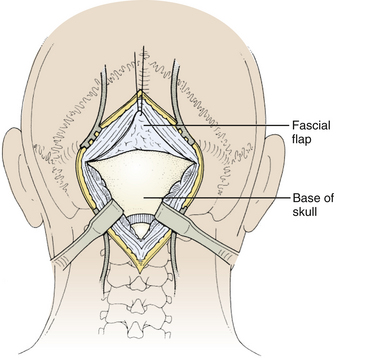
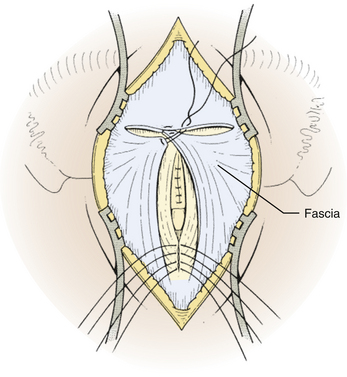
Next, the fascia is incised using a Y-shaped incision, keeping the lateral ends of the Y below the ligamentous insertion (Fig. 31-7). While a linear midline fascial incision without the upper limbs of the Y allows use of the avascular plane between the splenius capitus and semispinalis capitis muscles, it is often difficult to reapproximate such an incision tightly at the superior nuchal line. Muscle flaps are then developed with monopolar cautery and periosteal elevators, stripping the muscle from the bone as far as the mastoid emissary vein. This exposure is maintained with two curved cerebellar retractors and the rostral flap is placed under tension using a 3-0 silk suture to reflect it rostrally (Fig. 31-8). The muscle insertions are stripped off the spinous process and laminae of C2. Finally, the junction between the pericranium and dura at the foramen magnum is sharply dissected, and then the posterior fossa dura separated from the inner table of the occipital bone using a curette.
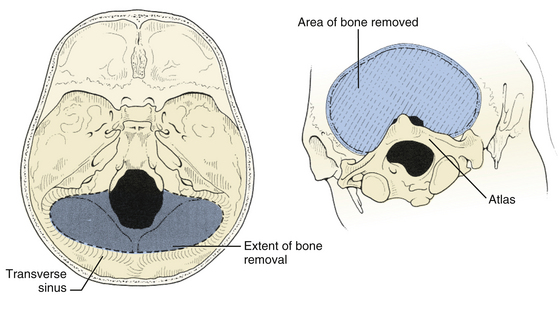
The suboccipital craniotomy is begun with burr holes on either side of midline just below the transverse sinuses, about three centimeters from midline (Fig. 31-9). A third burr hole can be placed below the torcular in older patients. In children, the dura is not firmly adherent to the skull so it is safe to drill close to or even on top of the sinuses, but more caution must be used with adults. The dura near the burr hole is then stripped using a Penfield and the bone removed using a high speed. The superior and lateral limits of the craniotomy are the transverse and sigmoid sinuses (Fig. 31-10). Inferiorly, the craniotomy should always include the posterior edge of the foramen magnum to prevent laceration of the brain against the closed bony rim when cerebellar elements are retracted downward and minimize damage from herniation if hematoma or swelling should occur postoperatively. The midline bone is removed last since it is often very vascular and contains a keel that can be quite deep. This keel must be stripped of dura with a Penfield, using extreme caution near the occipital sinus in the midline and the annular sinus near the foramen magnum. All exposed bone edges should be waxed, especially in the sitting position. Because of the irregular contour of the inner bone surface in adult patients, it is sometimes necessary perform a craniectomy rather than a craniotomy, removing the bone in a piecemeal fashion.
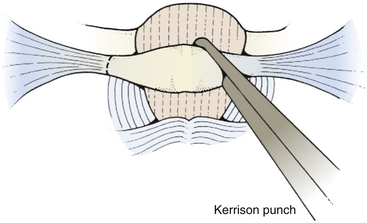
To expose the posterior arch of C1, the soft tissues overlying it are reflected laterally using a small periosteal elevator, stripping the inferior arch first since the vertebral artery is on its superior aspect. It is sometimes easier to do this after C2 has been exposed. The periosteum can sometimes be swept off the arch of C1 using an index finger covered with gauze. Monopolar cautery should be used with caution when dissecting the soft tissue over C1 (especially at the superolateral surface) to prevent injury to vertebral artery. It is important to remember that C1 can be bifid and is often cartilaginous in infants and young children. C1 laminectomy is helpful for lesions that herniate beneath the foramen magnum. To remove the lamina, small angled curettes can be used to strip the deep surface of the bone, and then the bone itself removed with an angled Kerrison punch or Leksell rongeur (Fig. 31-11). Because extending a laminectomy below C2 in young children increases the risk of swan neck deformity,5 it is prudent to remove the smallest amount of bone possible. For most tumors, it is usually only necessary to remove as far as one level above the most caudal aspect of the tumor.
Prior to the dural incision, the wound should be irrigated and retractor systems and microscope prepared (Fig. 31-12). If the dura is tense, the intracranial pressure can be reduced with external ventricular drainage (if available), hyperventilation, or mannitol, although mannitol should be used with caution in the sitting position as it has been implicated in the development of subdural hematomas. All techniques for dural incision require crossing the occipital and annular sinuses, which may be very large in infants under age 2 years and can persist until 25 years of age. A Y-shaped incision allows wide visualization and can be extended if necessary (Fig. 31-13). One superior limb should be incised first with a number-15 blade. The incision should start just inferior to the transverse sinus and travel obliquely to the midline, stopping short of the occipital sinus. The other superior limb is incised next, and then they are connected over the midline. If there is significant bleeding from the midline occipital sinus, it should be controlled with obliquely placed hemostatic clips or suture ligatures (Fig. 31-14). Either way, both the superficial and deep layer of the dura must be incised or the sinus will be tented open. The vertical limb of the Y is opened last using scissors so that the dura can be tented if bleeding is seen. The vertical incision extends to the foramen magnum so that it will extend below the falx cerebelli, which is occasionally present in childhood. If bleeding is very troublesome, the dura can be opened paramidline. The dura is then covered with a moist collagen sponge or wet Gelfoam sandwich to prevent desiccation and anchored to the fascia with 4-0 neurolon suture. This allows wide exposure of the cerebellar vermis and hemispheres (Figs. 31-15 and 31-16). The arachnoid is opened next over the cisterna magna to allow drainage of CSF (Fig. 31-17). If the tumor is in the cerebellar hemisphere, another dural incision can be extended laterally to more fully expose the involved cerebellum.
FIGURE 31-12 Dural exposure after removal of bone flap. The arch of C1 has been preserved in this patient.
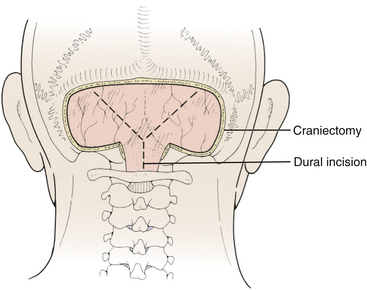
FIGURE 31-13 Dural incision. The vertical limb of the Y-shaped incision, which overlies the occipital sinus, is opened last.
(Modified from Cohen AR. Surgical Disorders of the Fourth Ventricle. Cambridge, MA: Blackwell Science; 1996.)
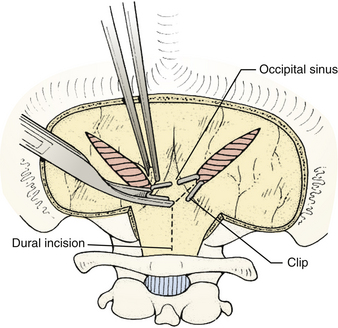
FIGURE 31-14 Control of bleeding from occipital sinus with clips.
(Modified from Cohen AR. Surgical Disorders of the Fourth Ventricle. Cambridge, MA: Blackwell Science; 1996.)
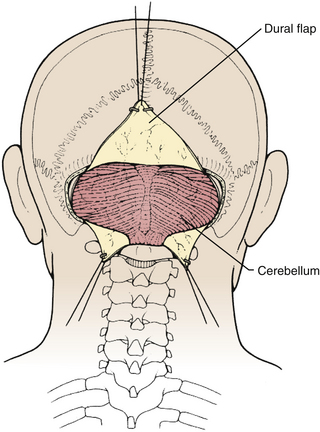
FIGURE 31-15 Exposure of cerebellar hemispheres and vermis.
(Modified from Cohen AR. Surgical Disorders of the Fourth Ventricle. Cambridge, MA: Blackwell Science; 1996.)
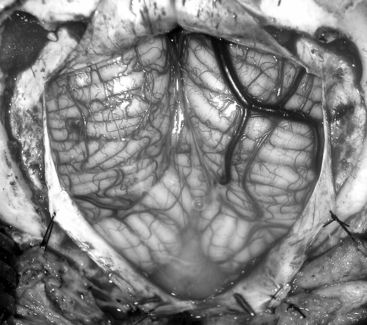
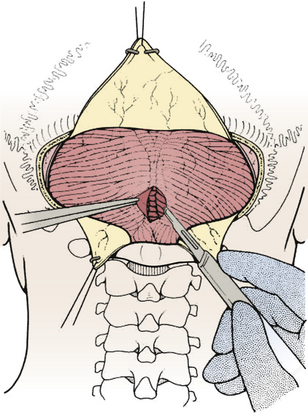
FIGURE 31-17 Opening the cisterna magna. This allows drainage of CSF and relaxes the posterior fossa.
(Modified from Cohen AR. Surgical Disorders of the Fourth Ventricle. Cambridge, MA: Blackwell Science; 1996.)
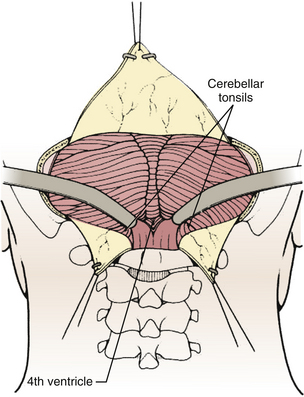
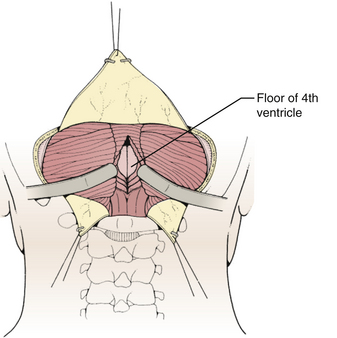
Techniques for intradural exposure and resection of the tumor will vary depending upon the location and size of the tumor, and will be discussed in more detail for each individual tumor. Gentle separation of the cerebellar tonsils will expose the cerebellomedullary fissure through the opened vallecula giving an unimpeded view of the inferior roof of the fourth ventricle (Fig. 31-18). Narrow malleable automatic retractors can be used to maintain separation of the tonsils; the retractor system should be kept close to the patient so as not to interfere with the subsequent operation. The operating microscope is brought into the field and the anatomy is identified. In particular, the location of the caudal loops of PICA should be carefully noted since they are often tethered to the tonsils and the walls of the cerebellomedullary fissure by small perforating branches. The foramen of Magendie and the small tuft of choroid plexus protruding from it will be clearly seen, as well as any tumor that protrudes from the foramen. The thin layers forming the lower part of the roof can be opened to expose the cavity of the fourth ventricle. Often this will provide sufficient exposure, but if not, it is sometimes helpful to retract the inferior vermis rostrally or incise the caudal vermis, avoiding the gutter between the vermis and the hemisphere to prevent injury to the inferior vermian veins there (Fig. 31-19). Lateral lesions may require removal of one tonsil by dividing the pedicle attaching the superolateral margin of the tonsil to the biventral lobule. To reach the lateral roof or lateral recess, part of the cerebellar hemisphere can be resected without significant morbidity as long as the dentate nuclei are not violated. If the tumor is not adherent to the floor of the fourth ventricle, cottonoid patties should be placed beneath the tumor to protect the delicate brain-stem structures just beneath the floor. These cottonoids should be placed under direct vision and never used as a tool to dissect the tumor from the floor of the fourth ventricle. After the tumor has been removed, the glistening white floor of the fourth ventricle should be clearly visible. The retractors are then removed and the cerebellar hemispheres allowed to fall back into place. If there is extension of the tumor through one of the foramina of Luschka into the cerebellopontine cistern, the ipsilateral tonsil and cerebellar hemisphere can be retracted medially to expose it. Sometimes it is necessary to do a secondary retromastoid approach to completely resect the tumor.
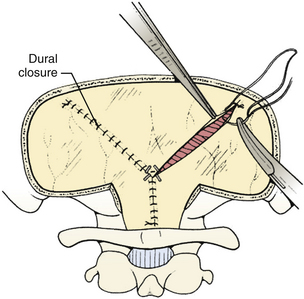
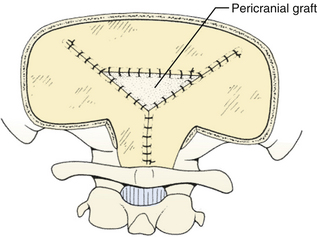
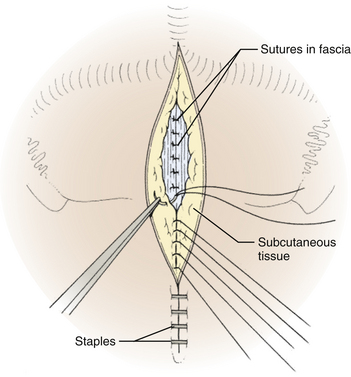
The dura is closed using a running 4-0 neurolon or polypropylene after approximating the dural edges with interrupted sutures (Fig. 31-20). A Valsalva maneuver will identify potentially dangerous venous bleeding. The dural closure should be watertight if possible, starting peripherally then working centrally to gradually overcome the tension. If the dura is not watertight, there is increased risk of pseudomeningocele due to a ball-valve effect or hydrocephalus from arachnoid adhesions produced by blood from the muscles. Sometimes the dura will be dried and shrunken by the end of the case, especially if measures have been taken to obliterate the occipital sinus. In this case, the remaining defect can be covered with a pericranial or fascial graft (Fig. 31-21). Freeze-dried bovine pericardium or human allograft dura can also be used, but use of autogenous material is less likely to produce postoperative aseptic meningitis.6 If clips were used on the midline occipital sinus, they can be removed as the dura is sutured. The suture line may be covered with thrombin-soaked Gelfoam. If a craniotomy was performed, the bone flap can be secured with wires, plates and screws, or sutures. Alternatively, the defect can be covered with a titanium screen held in place by gently compressing the screen and allowing it to insert itself between the dura and inner margins of the bony defect. The fascia is closed with interrupted absorbable sutures to approximate the muscle and fascia (Fig. 31-22). If the fascia is dried and difficult to approximate, the skeletal fixation apparatus can be loosened and the neck extended to facilitate closure. An adequate amount of tissue must be left at the superior fascial flap to prevent buttonholes at superior nuchal line. The scalp is then closed in layers, ending with a subcutaneous reapproximation using interrupted absorbable sutures with inverted knots. If in the sitting position, all layers should start from the caudal end of the wound so that the tails do not hang in the way. The wound is then closed with sutures or staples (Fig. 31-23). The wound is covered with a sterile dressing and the patient extubated in a supine position.
Complications
Hydrocephalus is common with fourth ventricular tumors, and is one of the most significant causes of morbidity and mortality associated with these tumors.7,8 In the past, many patients with tumors and hydrocephalus underwent temporizing preoperative shunting to treat hydrocephalus and prevent pseudomeningocele, CSF leak, and meningitis from fistula. However, more recently it has been observed that shunting is associated with many complications, and the increased incidence of subdural hematoma, infection, and brain-stem compression from upward herniation may outweigh its benefits.9–12 Also, the advent of advanced radiographic imaging has allowed diagnosis of fourth ventricular tumors much earlier than before, when patients were frequently moribund with dehydration and malnutrition from vomiting and hydrocephalus needed to be urgently treated. Today, only about 10% to 20% of patients with cerebellar and posterior fossa tumors require permanent shunting,7,8,13 and most of these have slow-growing tumors such as astrocytoma since more acute tumors distend the ventricles for a short period of time and do not allow outlet adhesions to form. Risk factors for shunt dependence include younger age, larger preoperative ventricle size, and more extensive tumors. In many cases, preoperative high dose steroids will produce satisfactory improvement in hydrocephalus. Otherwise, an appropriate alternative to shunting is perioperative external ventricular drainage,14 especially if a patient presents lethargic or obtunded. This allows for precise pressure monitoring and control of drainage rate to prevent upward herniation and, if continued postoperatively, clearance of debris, proteinaceous blood, and air from the operation. Although external ventricular drainage does reduce the necessity to use permanent shunts, the infection rate may be as high as 10%, so it should be used judiciously. If a shunt is required for a malignant tumor, there may be an increased risk of extraneural metastasis through the shunt tubing (especially to the peritoneum),15 although some studies have suggested that such metastases may occur as often in patients without shunts.16
Pneumocephalus in the ventricles and subdural space is common after fourth ventricular surgery, especially when patients are operated in the sitting position,17 although it also occurs after prone operations. It is much more common when patients have preoperative hydrocephalus, and frequently results from overzealous drainage of CSF through an external ventricular drain intraoperatively. Since nitrous oxide can diffuse into air filled spaces, it is possible that nitrous oxide contributes to tension pneumocephalus, although this is controversial. If tension pneumocephalus is recognized intraoperatively, the patient should be placed in Trendelenburg position and the operative bed irrigated to replace air with the irrigating fluid. Symptomatic postoperative tension pneumocephalus can be treated with a small frontal burr hole to relieve the pressure caused by the trapped air. Intraventricular air may cause ventriculoperitoneal shunt malfunction due to airlock.
Aseptic meningitis, also called posterior fossa fever, is a rare occurrence after posterior fossa surgery, especially for epidermoids or dermoids that rupture intraoperatively leaking cholesterol cyst fluid, although it also occurs after resection of astrocytoma or medulloblastoma. It may be a presenting symptom preoperatively but much more common as a postoperative complication.18,19 Patients usually present about 1 week after surgery with fever, headache, irritability, and CSF pleocytosis. It can be difficult in some cases to differentiate aseptic meningitis from true bacterial meningitis, which should always be carefully excluded before treating for aseptic meningitis. The condition resolves with steroid or anti-inflammatory treatment and serial lumbar punctures to remove CSF.
The “posterior fossa syndrome,” also called posterior fossa mutism or pseudobulbar palsy, is characterized by the delayed onset of mutism, emotional lability, and supranuclear lesions that occurs within a few days after midline posterior fossa operations.20 The syndrome has been seen in as many as 15% of intraventricular approaches to lesions near the brain stem, but has also been described with supracerebellar infratentorial approach to the pineal region and retromastoid lateral cerebellar approach to the side or front of the brain stem. Patients present with global confusion, disorientation, combativeness, paranoia, or visual hallucinations. They are generally alert and will follow simple commands, but will sometimes refuse to speak or present scanning speech. Orofacial apraxia, drooling, dysphagia, pharyngeal dysfunction, and flat affect are common, but there is no actual weakness, hence the term pseudobulbar palsy. Because of the delay in onset, it has been suggested that edema from operative manipulation may play a role, for example through transmission of retractor pressure from the medial cerebellum through fiber pathways along the middle and superior cerebellar peduncles into the upper pons and midbrain. There are no consistent neuropathologic findings, and most patients have some improvement over several weeks to months.20–22
Ipsilateral limb ataxia, dysmetria, dysdiadokinesis, and hypotonia usually results from damage to the cerebellar hemisphere, especially the dentate nucleus, which is located along the superolateral margin of the roof of the fourth ventricle adjacent to the upper pole of the tonsil. Most injuries to the dentate nucleus occur during dissection of a hemispheric tumor. Retraction during dissection of the superior vermis can injure the superior cerebellar peduncle (which) producing similar symptoms. Unless the dentate is completely ablated, most patients recover well within a few months with only minor residual intention tremor that does not interfere with motor development.
Acute urinary retention is an uncommon complication of dissection of the fourth ventricular floor near the striae medullaris, presumably due to injury to the pontine micturition center in the pontine tegmentum, the structure that integrates the cortex with sacral and pelvic sensory pathways that apprise bladder filling status.23 Patients with this condition demonstrate inability to initiate voiding in spite of a full bladder with high intravesicular pressure. Since the pontine micturition center is deep in the pons near the reticular activating system, this symptom is usually associated with a disturbance in sensorium, but can occur in conscious patients. It is usually reversible but does not respond to detrusor augmenting agents or alpha-adrenergic blockers. Patients are best managed by intermittent catheterization.
Patients treated with radiation sometimes have significant learning disabilities,24 and should undergo follow-up neuropsychiatric evaluation. Radiation treatment has also been associated with endocrine dysfunction, growth dysfunction, hypothyroidism, delayed or precocious puberty, and secondary malignancy.25 Patients that have extensive laminectomies are predisposed to development of swan-neck deformity, and should be kept in a soft cervical collar for 6 to 8 weeks until the paraspinal muscles reattach and monitored with cervical spine x-rays every few months for a few years to check for spinal deformities.
Specific Tumors
Medulloblastoma
The term “medulloblastoma” was initially introduced by Bailey and Cushing who noted the highly cellular architecture of small round basophilic cells that showed various degrees of differentiation along neuronal and glial lines. They assumed that the cell of origin (the “medulloblast”) was a primitive cell capable of both neural and glial differentiation.26 More recently, it has been theorized that these are related to tumors with similar histology in other locations, such as the pineal gland (pineoblastoma), ependyma (ependymoblastoma), retina (retinoblastoma), and elsewhere (neuroblastoma). The term “primitive neuroectodermal tumor” (PNET) was used to describe these tumors. A medulloblastoma would therefore be described as a PNET of the fourth ventricle.
Medulloblastoma is the most common malignant primary brain tumor in children, accounting for 20% to 25% of all childhood primary brain tumors and 40% of all childhood posterior fossa tumors.27,28 Peak incidence is 3 to 5 years of age; half of all medulloblastoma patients are under 10 years of age at diagnosis, and three quarters are under age 15. Medulloblastoma is uncommon in infancy, and less than 5% of patients present under 1 year of age. There is a second peak between 20 and 40 years so that medulloblastoma accounts for 5% of all adult posterior fossa tumors and 1% of all adult brain tumors. Adult medulloblastomas are more likely to be hemispheric than midline, likely due to the lateral migration of cells of the granular layer of cerebellum from the inferior medullary velum. Adult medulloblastomas are also more likely to be cystic or necrotic, have poorly defined margins and less contrast enhancement.29 They may even involve the cerebellar surface and resemble meningiomas. There is a slight male preponderance in most clinical series, and for reasons that are unclear, medulloblastomas have a significantly higher incidence in North America than elsewhere in the world.30 Medulloblastomas frequently metastasize in the subarachnoid space, and some dissemination is evident in 20% to 30% of all patients and 50% of young patients at diagnosis.31,32 Familial medulloblastoma has been reported.33
Medulloblastomas grow quickly, so onset of symptoms is usually fairly acute; most patients are symptomatic less than 2 months before the tumor is diagnosed, and very few report symptoms for longer than 6 months. Most patients initially experience symptoms of increased intracranial pressure from CSF obstruction. Symptoms typically begin with intermittent headache (often worst in the morning) followed by vomiting and eventually gait problems.34 Gait difficulties include wide-based gait and inability to tandem walk; these findings are often subtle and not appreciated by the child or the parents but frequently alert the physician to the presence of a neurologic lesion. Clinical signs include ataxia for midline tumors or dysmetria/dysdiadokinesia for lateral tumors. Most patients have papilledema by the time they present for evaluation. Less common signs include diplopia from abducens palsy, facial paresis or lower cranial nerve palsy from tumor invasion, or head tilt from tumor extension into the upper spinal canal or impaction of the cerebellar tonsils at the foramen magnum against the first two cervical nerve roots. Since medulloblastomas can metastasize along the subarachnoid space, some patients present with cranial or spinal nerve root symptoms from distant metastases or even seizures from cortical metastases. Patients who present with signs of metastatic disease have a limited life expectancy.
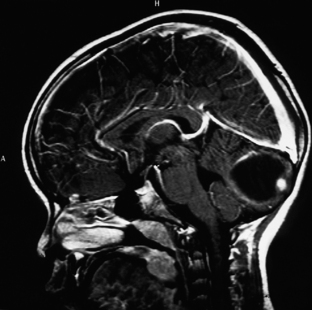
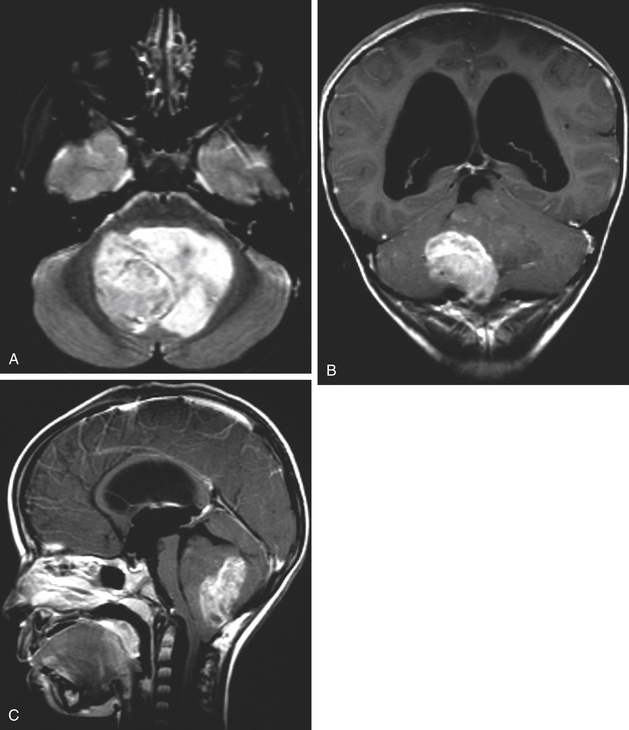
Medulloblastomas usually appear as midline solid tumors on neuroimaging, although cystic change is sometimes observed. Eighty-five percent are midline vermian lesions, usually arising from the vermis or inferior medullary velum and growing into the fourth ventricle, sometimes appearing to be entirely intraventricular.32 Adult lesions are much more likely to be in a lateral location. CT demonstrates a homogeneous hyperdense lesion that enhances intensely and diffusely after contrast administration with occasional heterogeneously enhancing regions due to necrosis, although rarely medulloblastomas do not enhance at all on CT.35 Calcification will be apparent in 10% of medulloblastomas,36 but presence of calcium or cystic change is more typical of ependymomas. On MRI, the lesion is hypointense to isointense to the brain on T1 and hyperintense or hypointense on T2 with heterogeneous signal due to microcysts, necrotic cavities, tumor vessels, or calcification (Fig. 31-24). The tumor displays irregular enhancement with MRI contrast material,36,37 and enhanced MRI will disclose small cortical or basal metastases in 5% to 10% of cases. The sagittal MRI can help delineate the relationship of the tumor to the vermis, midbrain tectum, vein of Galen, and cervicomedullary junction. Additionally, sagittal MRI can differentiate true intraventricular tumors from extraventricular vermian tumors: intraventricular tumors will widen the aqueduct and displace the quadrigeminal plate posterosuperiorly, while dorsal lesions will kink the quadrigeminal plate giving it a C-shaped appearance.38 Infrequently, the tumor will be seen to extend out of foramen of Luschka, but this is far more typical of ependymomas. In young children, medulloblastomas are often radiographically indistinguishable from ependymomas by radiographic appearance alone. Because of the frequency of craniospinal metastases, all patients should get a contrasted MRI of the entire craniospinal axis.
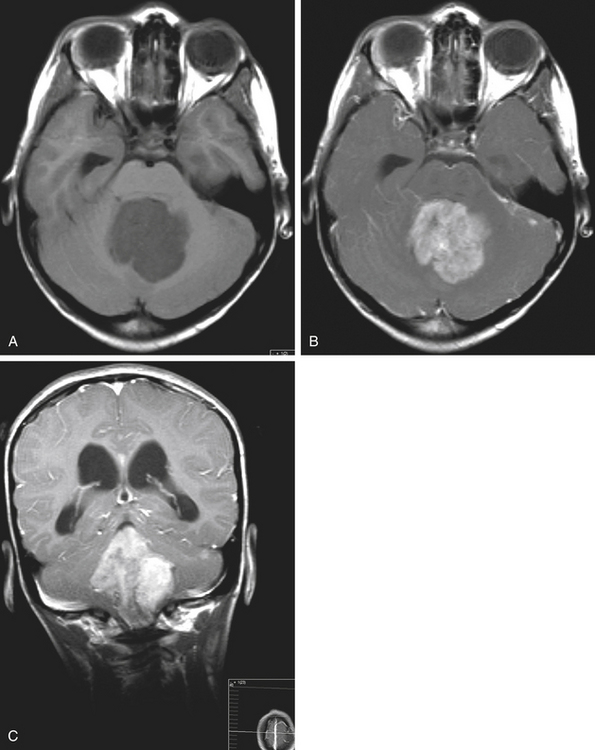
Grossly, medulloblastomas are discrete, soft tumors, although adult lesions tend to be firmer and more adherent to leptomeninges. Medulloblastomas can be divided into two broad histologic patterns: classical and desmoplastic. Classical medulloblastomas, which make up three quarters of the total, are seen to have dense, diffusely monotonous sheets of cells with intensely basophilic nuclei and scant cytoplasm (“small round blue cells”). There is regional variability in the size and shape of cells, number of mitoses, and appearance of nuclei, and necrosis is common. Desmoplastic medulloblastomas have a higher proportion of fibrous stroma associated with the perivascular collagen skeleton of tumor. Sometimes there are uniform compact lines of cells around islands of relative hypocellularity; when this is seen, the compact rims stain heavily with reticulin and the hypocellular areas stain for GFAP. Occasionally individual cells resembling oligodendrocytes are seen, having perinuclear halos and staining with tubulin and synaptophysin. The desmoplastic variant is more common in older patients. In young patients location is not associated with histology, but older patients are more likely to have desmoplastic histology in more lateral tumors.39 In both histologic patterns there are occasionally neuroblastic areas with histology similar to neuroblastomas with Homer-Wright rosettes (rings of nuclei surrounding a central zone of fibrillary processes) and perivascular pseudorosettes that resemble those in ependymomas except that they do not stain for GFAP. Mature ganglion cells are sometimes seen, although it is controversial whether these represent further neuronal differentiation or engulfing of deep cerebellar nuclei by the tumor. Also seen are islands of glial development characterized by clusters of GFAP-staining cells with pink cytoplasm commonly seen as circular whirls of cells with bipolar processes. These may represent entrapped or reactive astrocytes.
Even with a vermian incision, it is seldom possible to expose the entire tumor. Therefore, the next step is to debulk the central portion of the tumor using blunt or sharp dissectors with microscissors and microsuction aspiration. Desmoplastic tumors cannot be aspirated with microsuction, so ultrasonic aspiration can be used for these tumors, but this must be used with caution around the brain stem because of increased destruction of tissue. Bleeding is controlled with bipolar cautery. To expedite removal of the tumor and minimize blood loss, the tumor can be divided into four quadrants; as soon as bleeding becomes troublesome from one quadrant, a micropatty is placed there and attention turned to a different quadrant, and so on. By the time the dissection returns to the first quadrant the bleeding will have slowed enough to allow continued resection. After the tumor has been debulked, the shell of tumor is carefully stripped off the brain stem from inferior to superior using a small brain retractor and separated from the brain stem using cottonoid patties. Cottonoid patties should be placed along the fourth ventricular floor as early as possible to protect the brain stem, and it is important to constantly ensure that the trajectory is correct to prevent diving into the brain stem at an angle. When the brain stem is protected with a cottonoid patty, the inferior half of the tumor can then be safely removed. The tumor is rarely adherent or invasive into the brain stem, but when it is, gross total resection should not be attempted due to the risk of permanent cranial nerve defects. Rather, any focal areas of adhesion are separated using bipolar cautery and covered with a cottonoid. Later, when the tumor is entirely removed, the residual tumor tissue can be carefully aspirated parallel to the plane of the fourth ventricular floor until only a thin lining remains, and this lining can be coagulated with bipolar cautery to reduce the viability of remaining cells.
The next step is to resect the lateral and anterior portions of the tumor. The lateral and superior attachments of the tumor usually blend with the paravermian brain so there is seldom a plane between tumor and normal brain. As a result, most residual tumor fragments are left in this area. Since there are minimal postoperative neurologic deficits that result from removing a thin rim of cerebellum, the dissection should be carried out on the brain side of the brain-tumor interface so that all tumor is resected and hemostasis is easier to obtain. At the end of the dissection there will be a bed of clean white brain. The lateral dissection is extended onto the ependymal surface, where the tumor attaches to the cerebellum, and this margin is defined upward and forward until the dilated caudal aqueduct is reached, producing a conical dissection field. Finally, the anterosuperior tumor pole is removed, leaving the tip of the tumor covering the opening of the caudal aqueduct until the end of the operation so that no blood from the dissection will enter the lateral or third ventricles.
Since medulloblastoma often spreads through the subarachnoid space and the tumors are highly radiosensitive, radiotherapy to the entire neuraxis is considered standard even when there are no obvious lesions on postoperative imaging.31 The best survival rates are obtained with 3600 to 4000 cGy to whole craniospinal axis supplemented to 5400 to 5600 cGy to the primary site and 1800 to 3000 cGy extra to any area of lump disease. Young children have a high incidence of postradiation neurocognitive deficits, so radiation should be delayed or eliminated in the very young; various studies have suggested lower dosage40 or chemotherapy until the brain is mature enough to handle radiation.28 Trials using various chemotherapy regimens have demonstrated improved survival with chemotherapy, especially for locally extensive or widely disseminated tumors.28,41,42
Prognosis for patients with medulloblastoma is related to size, invasiveness, and dissemination of tumor at diagnosis; age of the patient; and postoperative residual tumor. Large tumors have been shown to have a lower 5-year disease-free survival.42 Brain-stem invasion also carries a poorer prognosis, and is problematic for preoperative staging because it cannot be predicted based on MRI.42,43 Presence of dissemination through the neuraxis is the single most significant predictor of outcome for all histologic types of medulloblastoma. Even microscopic dissemination (determined by lumbar puncture performed at least 10 days postoperatively) carries a significantly lower 5-year survival.28 Extraneural metastases to bone and even lymph nodes have been reported, but the incidence is too low to warrant screening all medulloblastoma patients.44 Young patients are much more likely to have dissemination, but patients younger than 4 years of age have a worse prognosis out of proportion to the increased dissemination rates.41 Finally, completeness of resection is thought to impact subsequent behavior of the tumor,43,45 and presence of more than a cubic millimeter of residual tumor has been associated with worse survival. Histology has not been shown to impact prognosis except for rare tumor subtypes at the extreme ends of the histologic spectrum; specifically, medulloblastomas with extensive nodularity and large cell/anaplastic medulloblastomas are associated with better and worse clinical outcomes, respectively. However, histologic features have not been shown to correlate with clinical outcome for the vast majority of medulloblastomas that lie between these extremes.46
Overall, with surgery and radiation, 5-year disease-free survival rates approach 80%.28 Medulloblastomas have classically been said to follow Collin’s law, which says that a cure has been obtained if there is no tumor recurrence over period equal to the age at diagnosis plus 9 months. However, one third of survivors at 5 years have recurrence, and one third of the recurrences are outside of the period predicted by Collin’s law. Most recurrence occurs in the cerebellum or along CSF pathways. Surveillance imaging has been shown to improve survival.32,47
Atypical Teratoid/Rhabdoid Tumor
Atypical teratoid/rhabdoid tumors share many clinical and pathologic features with medulloblastomas but appear to be unique entity with a more aggressive course and worse prognosis.48 They may occur anywhere in the neuraxis but are most commonly found in the cerebellum, and clinical presentation is similar to that of medulloblastomas. Most patients are under 2 years of age at diagnosis. One third of patients have subarachnoid dissemination upon presentation. Histologically they are composed at least partly of rhabdoid cells but also have areas typical for medulloblastoma and malignant mesenchymal or epithelial tissue. They stain for epithelial membrane antigen, vimentin, and smooth muscle actin. They are associated with abnormalities of chromosome 22 whereas medulloblastomas typically have an i(17q) abnormality.48 Prognosis is dismal, and most patients die within 1 year of diagnosis regardless of treatment.49
Astrocytoma
In the pediatric population, astrocytomas account for about one fourth of all brain tumors, and one third of posterior fossa tumors. These tumors occur most often during the first 20 years of life, with peak incidence from 5 to 8 years of age, and very few patients are younger than 1 year or older than 40. Males and females are affected equally.50,51 Unlike astrocytomas of the cerebrum, they are chronic and slowly progressive tumors that usually present subacutely and can be resected with minimal morbidity with a very high rate of cure.
Because they are slow-growing tumors, onset of symptoms is usually far more insidious than medulloblastomas and ependymomas. In 1931, Cushing remarked with amazement that “tumors of such high magnitude in such a critical situation and so certain to produce early hydrocephalus can be tolerated for so many years with no comparatively insignificant symptoms.”52 Most patients have had symptoms for many months by the time the tumor is diagnosed, and some undergo extensive gastrointestinal or neuropsychological investigation before the tumor is diagnosed. Although outside the lumen of the fourth ventricle, astrocytomas frequently disrupt the flow of CSF, so the most common presenting symptoms are those of increased intracranial pressure such as headache, vomiting, abducens palsy, and papilledema. Other common symptoms include altered gait, clumsiness, and head tilt. Common signs include papilledema, ataxia that is often unilateral, appendicular dysmetria, nystagmus, and macrocephaly that occasionally dates back several years even to infancy. Severe neck pain, opisthotonus, bradycardia, hypertension, and altered neurologic function are less common but require immediate attention, and rarely patients will present with signs of acute hydrocephalus such as stupor or coma, projectile vomiting, and oculomotor and facial palsies. Blindness was once a very common presenting symptom (present on presentation in 23 of the 76 patients reviewed by Cushing) but is rarely seen today.50
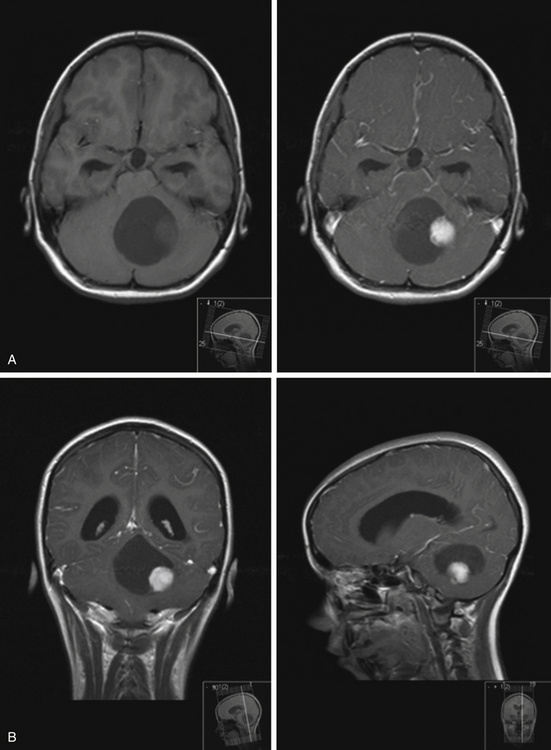
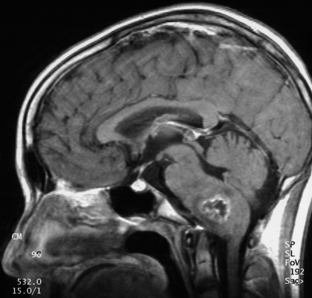
On imaging, astrocytomas are solid, cystic, or mixed lesions that arise from the medial hemisphere or vermis;53,54 one third are entirely in one cerebellar hemisphere (Fig. 31-25). When in the midline, they can be difficult to differentiate from medulloblastomas and ependymomas. MRI will usually disclose a round to oval mass with well-defined margins and of mixed intensity, hypointense to surrounding brain on T1 and hyperintense on T2. On CT the tumor is hypodense or isodense to surrounding brain. Unlike low-grade supratentorial astrocytomas, the tumor enhances brightly with CT and MRI contrast material. If the tumor is cystic, there may be an enhancing mural nodule and the cyst fluid will be slightly denser than CSF on T2-weighted images. The cyst wall is sometimes denser than surrounding brain but does not contain tumor unless it enhances (Figs. 31-26, 31-27, and 31-28). Calcification and peritumoral edema are infrequently observed.54 Skull x-rays are generally not helpful, although they may show signs of chronically increased intracranial pressure such as thinning or asymmetric bulging of occipital squama, chronic splitting of cranial sutures, or demineralization of sella turcica.
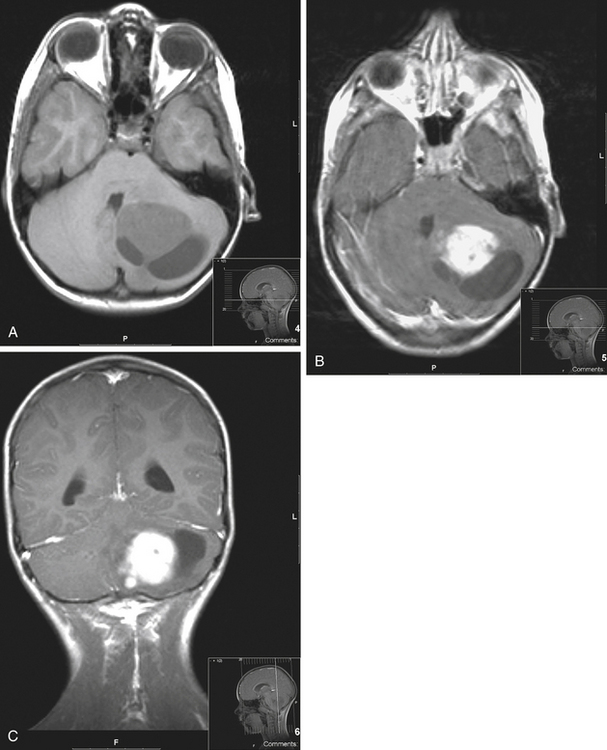
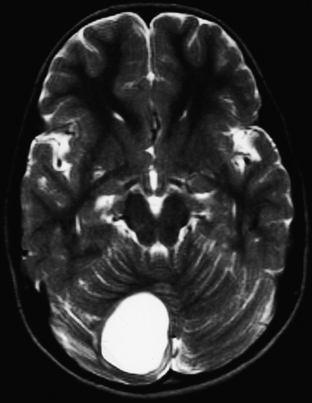
FIGURE 31-26 Astrocytoma, T2. The cyst associated with this tumor is often much larger than the mural nodule.
Some posterior fossa astrocytomas have histology and clinical behavior that deviates from the typical pilocytic astrocytoma described above. These tumors resemble low-grade astrocytomas of the cerebral hemispheres.55–57 They have diffusely homogeneous histology, lack microcysts and Rosenthal fibers, and are characterized by pseudorosettes, high cell density, necrosis, mitoses, and calcification. Incidence of these tumors is much lower than pilocytic astrocytomas and they are more common in older patients, especially those who have previously received radiation.55,58 They have a much higher rate of malignant degeneration,51 and high-grade anaplastic astrocytomas and even glioblastomas have been reported in the posterior fossa.59–61 Even when histologically low grade, they may infiltrate into cerebellar nuclei or peduncles and often lack clear tumor margins making gross tumor resection difficult. Survival rates with these tumors is lower than the typical astrocytomas,59 and there are reports of diffuse leptomeningeal spread55 and even spread to the muscles of the neck requiring several local surgical procedures to control.62
After exposing the posterior fossa, the tumor is usually immediately apparent as enlargement of the vermis or unilateral hemisphere with distended folia. The cerebellar tonsils may be pushed down on the side of the tumor. Intraoperative ultrasonography can be used transdurally to be certain bony exposure is adequate or after dura has been opened to demonstrate the closest location of tumor to cerebellar surface to minimize the amount of cerebellar tissue that must be removed to provide adequate exposure of the tumor. After incising the vermis or hemisphere, the tumor is removed with the ultrasonic aspirator until normal cerebellar tissue is seen. Very large tumors will cause the compressed cerebellum to relax inward after debulking so self-retaining retractors are helpful to keep the tumor bed open. If there is a cystic component to the tumor, the cyst is entered and cyst fluid quickly suctioned, supporting the hemisphere with self-retaining retractors to prevent collapse away from the tentorium with rupture of bridging veins. The mural nodule is then identified and removed using the ultrasonic aspirator. If the tumor is mixed or enhancement of the cyst wall is seen, the entire cyst wall should be removed and normal cerebellar tissue exposed; otherwise, simple removal of the mural nodule is curative. To prevent contamination of the CSF with blood and cyst fluid (which can produce chemical meningitis that increases the likelihood of postoperative communicating hydrocephalus), the cisterna magna and fourth ventricle should not be opened unless it is necessary to do so to achieve gross total resection. All walls of the tumor bed are then carefully inspected to ensure there is no residual tumor. Finally, before closing, it is important to ensure that bridging tentorial veins are not stretched. These veins can be safely sacrificed but tearing one later can lead to a life-threatening hematoma. All patients should get a postoperative contrast-enhanced scan within the first twenty-four hours to evaluate the extent of resection, provide a baseline if hydrocephalus should develop, and evaluate for cerebellar/brain-stem retraction edema or clinically silent hematomas. If more than one cubic centimeter has been left behind, early re-exploration to remove the residual tumor is indicated.
True pilocytic astrocytomas are almost always resectable by modern techniques and have excellent prognosis with no adjuvant therapy if gross total resection is possible.53,56 Postoperative MRI is particularly poor at differentiating residual astrocytoma from postsurgical changes so early postoperative imaging to obtain a baseline is probably not helpful.63 If there is a small amount of residual tumor, serial imaging can be used to follow its growth. Although there are several well-documented cases of low-grade astrocytoma undergoing malignant degeneration several years after surgical resection, this is probably quite rare and each of the documented cases had received radiation therapy. Radiation therapy has not been shown to be successful in the management of these tumors,64 and is associated with significant side effects, although focused radiation may be helpful for surgically inaccessible or rapidly growing lesions.65 Combined chemotherapy and radiotherapy for high-grade cortical astrocytomas has been shown to decrease rates of metastasis for children with high-grade astrocytomas. As with pilocytic astrocytomas, patients with more complete resections of infiltrative tumors fare better.
Recurrence of posterior fossa astrocytomas is related to the extent of resection, location, invasiveness, and histology of the original tumor.51,67 Age of the patient does not appear to have any effect on outcome. Recurrence is most common at the primary site, and if any tumor is left behind, symptoms almost invariably recur. Cystic tumors can usually be totally extirpated, but solid tumors tend to regrow after prolonged remission even if grossly resected. Solid midline tumors are most likely to recur since they more often extend toward the cerebellar peduncles, aqueduct, or brain stem, which may prevent total excision. Invasion of the subarachnoid space often occurs but this does not indicate a poor prognosis as with most other tumors. Astrocytomas do not follow Collin’s law for tumor recurrence and can have recurrences long after complete resection.68 Overall 10-year survival for astrocytomas is close to 100% with complete resection, and 20-year survival is above 70%.56,67
Ependymoma
Ependymoma is the third most common infratentorial tumor of childhood, representing 10% to 20% of all posterior fossa brain tumors, although in children under 3 years of age they represent 30% of all posterior fossa tumors. They occur very rarely in adults.69 Since ependymomas arise from the cells lining the ventricles, they can appear in any of the ventricles or ependymal streaks (obliterated portions of ventricles). Sixty-five percent of these tumors are infratentorial, and the most common location is in the middle to lower fourth ventricle or at the junction of the lateral inferior medullary velum and foramen of Luschka. Males are affected about twice as often as females, and 60% of all patients are under 5 years old at diagnosis.70 Recently, it has been suggested that ependymomas may have a viral etiology, since DNA sequences identical to segments of the SV40 virus have been identified in childhood ependymomas,71,72 and ependymomas can be induced in rodents by intracerebral inoculation of SV40.73 For unclear reasons, they are much more common in India than elsewhere in the world.30
Ependymomas are relatively fast growing tumors and present more acutely than astrocytomas but not as acutely as medulloblastomas. Median duration of symptoms is 2 to 3 months. The most common presenting symptoms are nausea and vomiting from increased intracranial pressure, ataxia, and nystagmus. Some patients have repeated vomiting in the absence of hydrocephalus due to direct stimulation of the brain-stem emetic center. At presentation, one third to one half of ependymomas will have grown through the foramen of Luschka or central canal with direct extension into the upper cervical spine, and this can produce nuchal rigidity, torticollis, posterior neck pain, or head tilt. Infiltration of the brain stem or direct tumor involvement within lateral brain-stem recesses can produce cranial nerve deficits, and this is associated with a particularly poor outcome.70,74 Finally, ependymomas do metastasize (although not as often as medulloblastomas), and patients rarely present with signs of metastatic spinal cord or nerve root compression.74,75
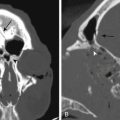
Ependymomas usually appear isodense to brain on both MRI and CT, although they may be hypodense on CT, hypointense on T1-weighted MRI, and hyperintense on T2.76 They are almost always seen on the floor of the fourth ventricle with the body of the ventricle expanded around the tumor, and are often quite large by the time they are discovered (Fig. 31-29). They frequently extend through the foramen of Luschka into the cerebropontine angle or through the foramen of Magendie occasionally compressing the upper cervical cord. This extraforaminal extension is an important diagnostic feature since other tumors in the differential (medulloblastoma, astrocytoma, and choroid plexus papilloma) almost never do this (Fig. 31-29B). Calcifications are seen in one quarter of ependymomas on CT, although a greater percentage of tumors have microcalcification;77 these calcifications are usually soft and do not pose a problem during tumor removal, so preoperative CT is not necessary. MRI appearance is usually heterogeneous from calcification, cysts, blood products, necrotic foci, and tumor vascularity. Enhancement on both CT and MRI is more heterogeneous than most other tumors in this area, and peritumoral edema is often seen on T2-weighted images.76
Grossly, the tumor is gray, well-circumscribed, homogeneous or less often lobulated, and is seen to arise directly from the floor of the fourth ventricle. Microscopically, ependymomas are quite heterogeneous, and can have highly variable histology within a single tumor. The two most common ependymoma histologic patterns are cellular and epithelial. Cellular (or “glial”) ependymomas consist of sheets of glial-appearing cells interrupted by perivascular pseudorosettes, in which fusiform cells with tapered fibrillary processes that stain for GFAP surround blood vessels. The pseudorosettes produce perivascular eosinophilic zones free of nuclei, giving the tumor a “leopard skin” appearance under low magnification. Epithelial ependymomas have cells with discernible boundaries that are arranged in canals and form true rosettes, reflecting the cell’s origin as ventricular lining. As with normal ependyma, these cells have basal bodies that anchor cilia and intracytoplasmic spherical or rod-shaped structures called blepharoplasts. Other rare histologic variants include papillary in which tumor cells are seen to cover glial tissue, tanocytic with elongated cells that resemble astrocytes, and clear cell in which cell have perinuclear halos like oligodendrocytes. Regardless of histologic pattern, ependymomas frequently sometimes have small cystic areas, regions of necrosis, evidence of acute or chronic hemorrhage, and calcification. More than half of infratentorial ependymomas have evidence of microcalcification, which is important to differentiate the tumor from medulloblastoma,77 and the chromatin is much denser than is seen with astrocytomas. Occasionally focal areas of anaplastic degeneration are seen with increased mitoses, cellular anaplasia, vascular proliferation, necrosis, and fewer perivascular pseudorosettes, although this happens much more often with supratentorial ependymomas. Unlike astrocytomas and medulloblastomas, in which histologic grade and proliferative potential correlate with prognosis, it is controversial whether histologic grade affects prognosis; this is likely to be because of the diversity of pathology seen. High mitotic index and presence of focal areas of dense cellularity are associated with poor prognosis, but calcium and ependymal rosettes have no impact.
The so-called ependymoblastoma, which has been reclassified as an entirely separate embryonal PNET, usually occurs supratentorially in young children and consists of sheets of poorly differentiated cells with frequent mitoses and distinctive rosettes of pseudostratified cells surrounding a lumen and surrounded by terminal bars. Median survival for ependymoblastoma is a few months, and patients frequently have diffuse spread through the subarachnoid space by presentation.79
Since ependymomas usually arise from the floor of the fourth ventricle and are frequently adherent to the lower cranial nerves, it is often quite difficult to achieve gross total resection, and in the past surgery was limited to removing enough of the tumor to allow free flow of CSF. More recently, the benefits of total resection have been shown to outweigh the risks, as there is a significant difference in survival depending on extent of resection.70,74 The posterior fossa is exposed by standard techniques. If there is extension of the tumor through the foramen of Magendie, it will be apparent in the cisterna magna immediately after the dura is opened. Any tumor tissue extending into the cervical canal is gently aspirated from the surface of the cord with suction or ultrasonic aspiration. This is usually easy to do since the tumor does not invade the pia. The cerebellar hemispheres are then retracted laterally and resection continued along the floor of the fourth ventricle until the point of origin is determined (usually caudal to the stria medullaris). Ependymomas rarely submerge deep into the brainstem, so a 1-mm thick carpet of tumor should be left at its point of attachment, and resection continued parallel to the floor of the fourth ventricle using the ultrasonic aspirator. It is vitally important to identify the glistening white fourth ventricular floor before attempting to remove tumor from its point of attachment. If the tumor extends through the foramen of Luschka, the area of origin will usually be found at the junction of the lateral medullary velum and the medial foramen of Luschka. In this case, the tumor should be resected from within the fourth ventricle as far laterally as possible and then the retractors removed from the fourth ventricle and placed underneath the cerebellar tonsil to expose the cerebellomedullary cistern where the rest of the tumor may be visualized and resected. Ependymomas are quite soft and can usually be safely aspirated off the lower cranial nerves, but it is important not to violate PICA and its lateral medullary branches, although the lateral medullary veins can be safely sacrificed if inadvertently entered. Any changes in cardiac rate or rhythm (more common when dissecting on the left side) can be abolished by soaking a micropatty in 1% lidocaine and placing it on the cranial nerve near the root entry zone for a few minutes. Removing tumors from deep within the cerebellopontine angle may require partial resection of the lateral cerebellar hemisphere to facilitate exposure. Occasionally the tumor arises from the roof rather than the floor of the fourth ventricle and will invade the roof and inferior vermis without attachment to the floor of the fourth ventricle. This often allows for complete tumor removal, but excessive resection of the inferior vermis or nodulus increases the risk of damage to the superior cerebellar peduncle producing postoperative ataxia or the posterior fossa syndrome. Recurrent ependymomas are usually invasive or adherent, so it is challenging to distinguish invasive disease from normal anatomy. As a result, resection of recurrent disease is almost always incomplete.74
Because recurrence is most common at the primary site, radiotherapy with 4500 to 5600 cGy to the primary site has shown to significantly improve overall rate and duration of disease-free survival in patients with incompletely resected ependymoma in several retrospective studies.80,81 Most authors therefore recommend local radiotherapy even if gross total resection is confirmed by postoperative MRI and staging is negative for leptomeningeal spread,80 although one recent study recommends withholding further treatment unless there is evidence for tumor recurrence.74 If there are no radiographically evident metastases, craniospinal irradiation probably does not improve outcome, since only 3% of patients with low-grade tumors later develop have clinical evidence of metastatic disease (although one third have dissemination at autopsy). Also, most spinal seeding occurs only after recurrence at the primary site, and craniospinal irradiation has not been shown to prevent spinal metastases.82 However, anaplastic ependymomas are much more likely to be disseminated and should be treated with craniospinal irradiation,74 although it may not help.83 If disseminated disease is confirmed, most authors recommend 3600 cGy to entire axis with an additional boost of 1980 cGy to the local site and areas of macroscopic dissemination.80
There is not much data concerning chemotherapy for ependymomas. Although chemotherapy does transiently reduce tumor bulk and stabilize tumor growth in patients with recurrent disease, no chemotherapeutic regimen has been shown to be effective after radiation at the time of initial diagnosis.81,84
Prognosis for patients with ependymoma is poor compared with medulloblastomas and astrocytomas, although survival studies are difficult to interpret because most include anaplastic histology and supratentorial tumors. Most studies show that long-term survival is strongly correlated to amount of residual tumor as judged by postoperative MRI. For example, 5-year disease-free survival approaches 70% to 90% if there is no evidence of residual tumor compared with 0% to 30% if residual tumor is untreated.70,80,85 Other studies show no effect.81 Because ependymomas tend to insinuate into crevices within and about the fourth ventricle, the surgeon’s assessment overestimates completeness of resection as much as one third of the time, and so MRI should be used to determine completeness of resection. Therefore, any presence of residual enhancing tissue on imaging studies following completion of radiation should be explored and resected if possible. Overall 5-year survival is variously quoted at 15% to 50% for children and 50% to 75% for adults with fourth ventricle ependymomas.70,85 Histology and age of the patient do not significantly impact survival,70,74 although anaplastic ependymomas have a worse outcome,68 and female patients do better.80 Midline tumors have a better 5-year survival than lateral ependymomas out of proportion to their ease of resection.86 Disseminated ependymomas, about 5% of low-grade and 15% of anaplastic tumors,82 have a particularly poor prognosis, with 5-year survival rates near 12%.
Brain-Stem Glioma
Brain-stem gliomas are much more common in children than adults, and make up about 10% to 20% of intracranial pediatric tumors, and 15% to 30% of all posterior fossa tumors.87,88 They are a heterogeneous group of tumors with many distinct clinical and pathologic varieties. Duration of symptoms prior to presentation varies widely but is generally 3 to 5 months with insidious onset. Unlike the tumors described above, increased intracranial pressure is infrequent except as very late manifestation. Most patients present with a triad of a cerebellar deficit, pyramidal tract deficit, and involvement of cranial nerve nuclei; more than half have clinically detectable facial weakness, pharyngeal weakness, trigeminal deficit, or paresis of conjugate gaze by admission. The vast majority of brain-stem gliomas are astrocytomas with gangliogliomas and oligodendrogliomas making up the remainder. Histologically, they resemble fibrillary or pilocytic astrocytomas of cerebellar hemispheres, although they can show anaplastic components and cytoarchitecture of glioblastoma multiforme. Brain-stem tumors can be classified into three general categories based on location: tumors of the midbrain (including tectal gliomas and focal midbrain tumors, not discussed here); dorsally exophytic brain-stem tumors; and cervicomedullary tumors.
Pontine tumors have the highest prevalence in the first decade but can present in adults as old as forty. They tend to be infiltrative with indistinct borders and are always malignant regardless of histology at diagnosis89 and spread early through the leptomeningeal space90 and even extraneurally.91 Pontine tumors usually present with cranial nerve palsies followed by pyramidal tract signs, ataxia, and hydrocephalus in the most advanced stages. Imaging demonstrates diffuse pontine enlargement with hypodense signal to brain on CT, hypointense on T1-weighted MRI and hyperintense on T2-weighted MRI. Calcification and hemorrhagic foci are unusual. They are often seen to extend into the midbrain, medulla, or cerebellum, and can encircle the basilar artery.92 They rarely enhance on CT (occasionally there are areas of focal enhancement) but do enhance with MRI (Fig. 31-30). These tumors are surgically inaccessible, and surgical intervention does not alter survival.93,94 Radiation therapy is standard treatment, and many patients initially exhibit a striking response in terms of alleviation of symptoms and even radiographic resolution of tumor. However, within 6 to 12 months of starting radiation, there is nearly always recurrence with diffuse leptomeningeal spread. Chemotherapy has not been shown to improve survival time. Most patients are dead within 2 years of diagnosis, and overall 5-year survival rates are less than 10%.94
A subset (about 20%) of patients with pontine gliomas will have dorsal exophytic growth in which the tumor has eroded the ependyma to produce a wide-based, lobulated intraventricular mass that does not involve the cerebellum.95,96 These patients have more chronic and insidious symptoms such as failure to thrive, headaches, and loss of balance, but hydrocephalus is often an earlier finding. They have a lower rate of malignancy than diffuse pontine tumors and a much better prognosis. These tumors may be amenable to surgical resection using microsurgical techniques to achieve maximal subtotal removal.
Focal pontine astrocytomas have been described that are confined to half or part of half of the pons, sometimes extending into the fourth ventricle. These are more common in type one neurofibromatosis and have a variable natural history.97–99 They are amenable to surgical debulking and may not require immediate intervention after surgery; some patients do well with an aggressive surgical approach.
Cervicomedullary tumors generally present in young children, with average age of onset at 6 to 7 years. They almost never extend above the pontomedullary junction because their superior growth is limited by crossing fibers in the lower medulla and pontomedullary junction.100 They generally grow outward and extend into the fourth ventricle as a cystic or solid exophytic growth, and can eventually produce hydrocephalus from compression of the fourth ventricle or its outlet foramina. The symptoms are insidious and are often present for months or years. Most patients initially experience dysfunction of the lower cranial nerves manifested by difficulty swallowing, nasal speech, or recurrent aspiration. Sometimes they have torticollis or neck pain, and half will have some spinal cord dysfunction, usually insidious motor findings or paresthesias. MRI demonstrates the lesions quite well, but CT is of little utility because of the artifact created by the bones of the posterior fossa. These tumors are surgically accessible.101
Since most surgically accessible brain-stem gliomas extend to posterior aspect of brain stem or bulge into the fourth ventricle or cervicomedullary cistern, the standard fourth ventricle approach is useful to address them. For cervicomedullary tumors, intraoperative ultrasound should be used to identify the rostral and caudal poles of neoplasm prior to opening dura, and the initial myelotomy should initially be made in the middle of the tumor.102 In all cases, the tumor should be carefully removed from the inside out using ultrasonic aspiration, laser, microsuction, or irrigating bipolar forceps. It is possible to work to the brain interface when resecting pontine or cervical lesions, but when resecting medullary tumors, it is prudent to stop after only half to three quarters of the tumor is removed so that there is no chance that normal medullary structures will be violated. In particular, the cranial nerve nuclei should be carefully avoided. Monitoring (especially SSEP) is helpful to ensure important structures are not inadvertently damaged. It is important to avoid excessive manipulation and to consider every deviation in pulse or blood pressure to be a warning sign. Postoperatively, the most common threat to good recovery is respiratory difficulties, especially for medullary lesions. Therefore, patients should be monitored carefully and extubated only after fully awake. Postoperative radiation therapy may be helpful. If the tumor is focal and benign and gross total resection is obtained, many patients can have extended disease-free survival.
Choroid Plexus Papilloma
Choroid plexus papillomas are rare tumors, accounting for 2% to 3% of pediatric intracranial tumors and 0.5% of adult tumors.103,104 Unlike most fourth ventricular brain tumors that are more common in the pediatric population, fourth ventricular papillomas are more common in adults and most pediatric choroid plexus papillomas are in the lateral ventricles. The third ventricle is an uncommon location at any age. Males slightly outnumber females in most clinical series. When in the fourth ventricle, they usually occur in the cavity, although rarely they are found in the lateral recess105 or in the cerebellopontine angle arising from the small tuft of choroid plexus that extends through the foramen of Luschka.106,107 Carcinomas of the choroid plexus account for about 20% of choroid plexus tumors and are usually found in the lateral ventricles,103,108,109 but they occasionally involve the fourth ventricle. These malignant tumors almost invariably occur in young children, often during the first few days of life, and carry a grave prognosis with wide dissemination at or shortly after presentation.
Most choroid plexus papillomas present with symptoms of increased intracranial pressure such as headache, gait disturbances, and abducens palsy. Excessive formation of CSF is responsible for much of the hydrocephalus, although obstruction to CSF flow by the tumor or at the level of the arachnoid granulations due to hemorrhage is probably a more common cause. Because of CSF overproduction, affected infants often have papilledema in spite of an open fontanelle and macrocephaly disproportional to the size of the tumor. When in the cerebellopontine angle, dysfunction of lower cranial nerves and cerebellar ataxia are the main clinical findings.106,107 In adults, these tumors usually do not produce excessive CSF and are sometimes discovered incidentally at autopsy.
On imaging, choroid plexus papillomas are usually seen as intraventricular masses. On CT they are hyperdense and enhance brightly and homogeneously with contrast.110 Calcifications are common in childhood papillomas but are rare during infancy.111 MRI demonstrates hypointensity on T1 and heterogeneous hyperintensity on T2. The tumor can be highly vascular, and angiography is useful for preoperative planning when faced with an extremely brightly enhancing tumor or one that demonstrates high vascularity on MRI to identify feeding vessels and develop a plan for early devascularization of the tumor. There is often hypertrophy of the feeding artery, which is usually PICA, although AICA supplies some tumors arising from choroid plexus far in the lateral recess. An important diagnostic clue is engulfing of the choroid glomus by tumor, which usually suggests choroid plexus papilloma.112 Choroid plexus carcinomas are less homogeneous on neuroimaging due to necrosis, intratumoral hemorrhage, and cysts.113
Grossly, the papilloma is well-defined, lobulated, mulberry-like, and reddish purple, with a firm and vascular basal portion. Microscopically, these tumors strongly resemble normal choroid with a single layer of cuboidal epithelium that occasionally appears somewhat crowded and taller seated on a simple fibrovascular stroma with little or no abnormal mitotic activity. Calcification is common in these tumors and is extensive in 10%. Some atypical histologic features such as enlargement, irregularity, hyperchromasia, mitoses, and loss of papillary growth pattern is present in half of these tumors but have no prognostic significance. In infants there is occasionally evidence of ependymal differentiation with piling of ependymal cells or presence of cilia and blepharoplast. In these patients, it can be difficult to differentiate the tumor from papillary ependymomas. Rarely the tumors have a cystic portion within the tumor or in the brain parenchyma filled with CSF presumably secreted by the papilloma. The so-called “oncocytic variant of papilloma” resembles oncocytomas in other organs, with cytoplasm packed with mitochondria. Carcinoma of the choroid plexus is recognizable by heterogeneity, necrosis, invasiveness, cellular pleomorphism, and mitotic figures. The tumor has a tendency to form multilayered epithelium and invades the parenchyma.111 This malignant tumor may be confused with metastatic adenocarcinoma, especially when it occurs in adults, and sometimes the only indication that it is a chroroid tumor is the presence of cilia, microvilli, and zonula adherens on electron microscopy.
Complete surgical resection of fourth ventricular choroid plexus papillomas is frequently possible, since the tumor typically does not invade the parenchyma or floor of the fourth ventricle. Because they are very vascular tumors, it is important to prepare for significant blood loss. The fourth ventricle is exposed by standard techniques. Because of their extreme vascularity, it is important to identify feeding vessels during the resection. If the tumor is small, it may be possible to identify its point of attachment to normal choroid plexus and devascularize the tumor by coagulating and sectioning the tonsillar and vermian choroidal branches of PICA. Larger tumors may have feeding arteries embedded in the core of the tumor and occasionally envelope PICA, so the intraventricular papillary portion (which is less vascular than the core) is first shrunk with bipolar cautery or ultrasonic aspirator, and then the feeding branches identified and coagulated. Forceful retraction of the tumor should be avoided since there is sometimes a major draining vein on the dome of the papilloma and inferior vermian veins may be arterialized due to intratumoral shunting; these can produce significant hemorrhage if violated. It may be necessary to section the inferior vermis to reach the rostral end of the tumor. If there is extension through the foramen of Luschka into the cerebellopontine angle, the intraventricular part is resected first then the rest of the tumor is exposed by elevating the ipsilateral tonsil and hemisphere. There may be displacement of cranial nerves and brain stem but the papilloma can usually be easily be separated from these structures.106
At one time radiation therapy was commonly used for unresectable or recurrent papillomas, but it is controversial whether it is effective.114–116 Since there are risks associated with radiation (especially in children), adjuvant radiotherapy is not warranted for these tumors, although some authors assert that radiotherapy may be effective in treating invasive benign tumors that could not be resected.104 Choroid plexus carcinomas, on the other hand, should receive radiation to the entire craniospinal axis since these tumors often disseminate along CSF pathways; most long-term survivors of chroroid plexus carcinoma were treated with radiation.109,115 Chemotherapy may be appropriate for young patients in which radiation is too risky.
Benign papillomas have a good prognosis. Total resection is usually curative without recurrence, and symptoms generally do not recur even following subtotal resection. Overall, 5-year survival rates for papillomas are nearly 100%.103,104,114–116 Choroid plexus carcinomas have a much poorer prognosis because gross total resection is usually impossible due to frequent infiltration of brain stem and cerebellar peduncles and extreme vascularity, the tumor often disseminates through CSF pathways, and it usually occurs in very young children which limits use of adjuvant radiotherapy. While there are reports of gross total resection of choroid plexus carcinomas without recurrence,103,109 they tend to recur even after total resection. Most authors agree that the degree of resection is the most important variable, followed by histology (cellular atypia, microscopic invasion, or mitosis).116 Five-year survival rates vary significantly among reports, but are generally less than 50%, with carcinomas of the fourth ventricular choroid plexus having a worse prognosis than those in the lateral ventricles.109,114
Hemangioblastoma
Hemangioblastomas are benign, slow-growing vascular tumors that are found exclusively in the neuraxis.117 The most common location is the cerebellum, followed by the brain stem (usually on the floor of the fourth ventricle near the cervicomedullary junction) and spinal cord. They rarely occur supratentorially, but when they do and are dural based, it is difficult to distinguish them from angioblastic meningiomas. They account for 1% to 2% of all intracranial neoplasms, and 7% to 12% of adult postfossa tumors. Isolated sporadic hemangioblastomas are more common in middle-aged adults 30 to 40 years of age. Males slightly outnumber females.
Ten percent to 20% of hemangioblastomas occur as part of the von Hippel-Lindau syndrome. This syndrome was initially described in 1904 when von Hippel identified two patients with vascular retinal tumors. In 1926, Lindau noted an association with retinal tumors, cerebellar tumors, and visceral cysts.118 It is now known that von Hippel-Lindau syndrome is an autosomal dominant condition with varying degrees of penetrance, similar to neurofibromatosis and other neurocutaneous syndromes.119,120 In addition to multiple hemangioblastomas (which occur in 40% of patients with von Hippel-Lindau), there are multiple angiomatoses of the retina, visceral cysts and tumors especially of kidney and pancreas, pheochromocytoma, and papillary cystadenoma of epididymis or mesosalpinx (called “tubular adenomata” by Lindau). Recently the gene for von Hippel-Lindau has been mapped to a small region of chromosome 3p25-p26.121 and a protein called pVHL identified that is a moderator of mRNA elongation and may be responsible for the syndrome. Whether associated with von Hippel-Lindau or not, hemangioblastomas are always benign tumors, and although there is occasional local subarachnoid seeding after surgery, distant metastases have never been reported.
As with most other tumors of the posterior fossa, the most common presenting symptoms are increased intracranial pressure and ataxia. Since the most common location is in the fourth ventricle at the level of the obex, some patients present with intractable nausea and vomiting from direct irritation of the area postrema in the fourth ventricular floor. The mass effect is usually due to cystic enlargement rather than the solid tumor itself.122 In 10% of patients with hemangioblastoma, there will be a secondary polycythemia due to erythropoietin secreted by stromal tumor cells.123,124 This is more common with solid tumors, but cyst fluid from hemangioblastomas often contains erythropoietin. Resection of the tumor usually improves this polycythemia. Any patient known to have retinal angioma and polycythemia should be scanned to rule out hemangioblastoma. If there is evidence of von Hippel-Lindau, it is necessary to image the entire neuraxis to search for multiple lesions. First degree relatives should also be screened since up to 20% will have the disease.
On CT and MRI, most fourth ventricular hemangioblastomas appear cystic with a mural nodule that is located next to the pial surface of the brain (usually inferior or lateral), but they can be solid or mixed (supratentorial and spinal lesions tend to be solid). The nodule (which contains the tumor) invariably enhances but the cyst wall does not. CT demonstrates an isodense tumor with hypodense cystic fluid. On MRI the solid portion of the tumor is hypointense to brain tissue on T1 and slightly hyperintense on T2, and the cyst fluid is isointense or hyperintense to CSF on T1 and hyperintense on T2 to CSF (Fig. 31-31). Most hemangioblastomas produce peritumoral edema visible on T2 and there are often flow voids that indicate enlarged draining veins (Fig. 31-32). There may be hemorrhage with a hemorrhagic fluid level within the tumor cyst or blood products such as hemosiderin within the solid tumor.126 Tumors can be multiple with additional lesions in the brain stem and spinal cord; when in the spinal cord, they strongly resemble a syrinx except that they enhance dramatically with contrast. Leptomeningeal hemangioblastosis has been reported.127 Because the tumors are extremely vascular, conventional or magnetic resonance angiography is essential in preoperative planning for successful removal of hemangioblastomas to identify the location of feeding vessels and develop a plan for early devascularization to avoid serious hemorrhage. For very large or vascular tumors, preoperative embolization of major feeding vessels may reduce intraoperative hemorrhage, although revascularization of the tumor occurs rapidly so the embolization should be carried out no more than 1 to 2 days prior to the operation. Patients with multiple small hemangioblastomas may be treated with embolization alone.
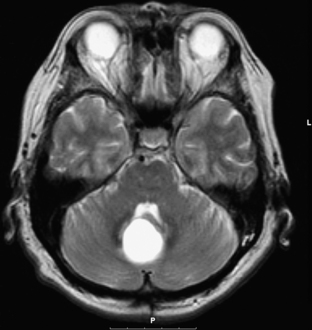
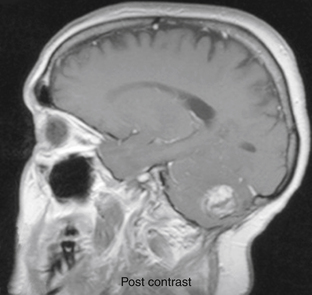
Because hemangioblastomas are always benign and total removal is curative, surgical removal is the treatment of choice for fourth ventricle hemangioblastomas.117 Perioperative steroids are helpful because of the extensive swelling, and sitting position may make hemorrhage easier to deal with intraoperatively. The fourth ventricle is exposed by standard techniques, and then different techniques are used to remove the tumor itself depending on whether it is cystic or solid. If the tumor is cystic, the cyst should be entered and the fluid drained. The mural nodule is then identified and dissected away from the cyst cavity using bipolar coagulating forceps. Occasionally the preoperative angiogram will identify a large feeding artery on the external surface of the cyst that must be divided prior to resecting the tumor; otherwise, the cyst wall should be left intact since it is not made up of tumor tissue but compressed gliotic cerebellum. If the tumor is solid, it is essential to avoid violating the tumor itself since this will lead to brisk hemorrhage. The dissection should be carried out between the external surface of the tumor and adjacent compressed gliotic cerebellum. Even after the major arterial supply has been controlled, there will frequently be many small perforating arteries or arterioles feeding the tumor that require coagulation. As soon as the dissection planes meet behind the tumor, it can be rolled out of the cerebellar bed and bleeding controlled. If the tumor tissue is violated prior to separation from the adjacent cerebellum, the tumor should be rapidly dissected from the cerebellar bed and hemorrhage controlled afterwards; attempting hemostasis within the center of tumor is futile and only leads to continued hemorrhage and delayed swelling.
All patients should get a postoperative enhanced MRI to determine if complete resection has been achieved. If no residual tumor remains, most patients have no recurrence, except in the case of von Hippel-Lindau. Partial resection usually leads to recurrence. Adjuvant radiotherapy is generally ineffective; although gamma knife can sometimes cause the solid portion of the tumor to stop growing or even to shrink, the cystic component does not respond and usually requires surgical treatment.128
Epithelial Cysts: Epidermoids and Dermoids
Epidermoid and dermoid cysts occur intracranially as result of nests of epithelial cells remaining intracranially during embryogenesis probably due to failure of separation between neural and cutaneous ectoderm at the time of closure of the neural groove.129 Epidermoids, also called pearly tumors or cholesteatomas, include only ectodermal elements. They are usually lateral in location; 50% are parapontine, and they frequently affect the cerebellopontine angle, suprasellar cistern, and cranial base.130,131 By contrast, dermoid cysts include elements of all three germ layers and include skin appendages. They tend to be located along the central neuraxis anywhere from the pituitary to distal spinal cord, although the most common location is in the posterior fossa, especially the fourth ventricle. Dermoids are sometimes associated with a dermal sinus tract that extends from the skin toward the tumor with an associated suboccipital skull defect,132 and there will usually be a cutaneous marker such as hair, telangiectasia, pigmentation, or increased subcutaneous tissue. Together, epidermoids and dermoids account for 1% of intracranial masses, and epidermoids are somewhat more common in most clinical series. Males and females are affected equally.
Although congenital in origin, epidermoids and dermoids can become symptomatic at any age, although dermoids tend to manifest earlier, usually in childhood. Both tumors grow very slowly and by linear rather than exponential growth since they enlarge by deposition of stratified squamous epithelium and its products (such as keratin and cholesterol) into the center rather than mitotic proliferation as with true neoplasms. Dermoids grow somewhat faster since, in addition to growth by desquamation, they also fill with secretions of sebaceous glands, sweat glands, and hair follicles. Symptoms are related to site of the cyst and are not specific. Most become symptomatic due to mass effect, although epidermoids occasionally burrow into the brain stem. Epidermoids occasionally rupture causing sterile meningitis. Dermoids associated with a dermal sinus most often present with localized swelling, redness, tenderness, and purulent drainage from inflammation or infection of the dermal sinus. This often occurs several times before neurosurgical consult is obtained. Some patients present with meningitis or cerebellar abscess since the sinus tract can act as a portal for bacterial entry into the subarachnoid space,133,134 and subsequent scarring can lead to hydrocephalus.
On CT, an epidermoid cyst appears as a nonenhancing hypodense mass with frond-like margins that interdigitate into normal brain structures. Dermoids have a similar appearance but are somewhat higher density and may also contain calcification or fat, and sometimes the cyst wall enhances with contrast. Both epidermoids and dermoids have well-defined margins and contain a central low-density area similar to CSF but with frequently nonhomogeneous contents (“dirty CSF”). On MRI, epidermoids appear homogeneous, hypointense to brain on T1-weighted images and hyperintense to CSF on T2. Dermoids appear more heterogeneous because of a greater variety of constituents (keratin debris has high attenuation and dermal elements have low attenuation), so their appearance varies considerably on different T-weighted images, but they are sometimes hyperintense to brain on T1 due to the presence of fatty tissue. Sometimes it is possible to see a dermal sinus tract. Both cyst types conform to adjacent anatomy but may produce some mass effect; they almost never produce edema.135 Hydrocephalus can occur but is rarer than with other fourth ventricular tumors. Because the imaging appearance is so variable (especially for dermoids), the differential diagnosis can be extensive and includes abscess, cystic astrocytoma, hemangioblastoma, and even medulloblastoma or cystic ependymoma.
Morbidity and degree of invasiveness of the tumor dictate decisions regarding surgical treatment. If surgically excised, these cysts must be totally removed since subtotal resection usually leads to recurrence. The capsule may separate easily or be densely adherent (especially if infected due to dermal sinus tract); either way, every effort should be made to keep the capsule intact to prevent spillage of cyst contents or purulent material into subarachnoid space, which can lead to severe chemical meningitis. If the cyst does rupture intraoperatively, the area should be copiously irrigated with corticosteroid irrigant and the patient should receive intravenous steroids postoperatively. Cysts that are densely adherent to the floor of the fourth ventricle or cervicomedullary junction can be gently coagulated with the bipolar forceps to minimize the risk of recurrence. If a dermal sinus tract is present, it must be explored even if imaging does not indicate any abnormality, although absence of bony defect on exploration excludes the possibility of intracranial extension. An elliptical incision is made around the opening of the dermal sinus tract and the bone is removed inferiorly since the tract always extends inferiorly below the torcular. The tract is then removed en toto. The sinus tract is often intimately related to torcula so it is important to be prepared for major venous bleeding. Postoperatively, all patients should receive an MRI scan with contrast to obtain a baseline; if residual cyst is seen immediate re-exploration is warranted. Overall, the long-term prognosis for patients with fourth ventricular dermoid or epidermoid cysts is quite good.136
In addition to epidermoids and dermoids, neuroepithelial and endodermal cysts can occur in the region of the fourth ventricle. Neuroepithelial cysts result from embryologic folding of primitive ventricular lining into or out of ventricles. These include ependymal cysts and colloid cysts that usually involve the anterior third ventricle but can rarely occur in the fourth ventricle.137–139 They secrete solid or viscous exudate that results in gradual enlargement. Endodermal cysts are slow growing, endothelial-lined cysts that are most often located in the spinal canal but can occur in the fourth ventricle. They are likely due to an error in embryogenesis, probably early in gastrulation since their location tends to follow the location of the primitive notochord.140,141 Each of these are very slow growing but can become symptomatic from mass effect due to local compression or obstruction of the ventricular system; many remain asymptomatic and are discovered only incidentally or at autopsy. Treatment options for symptomatic cysts include excision, fenestration, or shunting.
Meningioma
Meningiomas can rarely produce a fourth ventricular mass. Overall, meningiomas are quite common and account for more than 15% of all primary intracranial tumors, but intraventricular meningiomas account for only 0.5% to 2% of all meningiomas, and most intraventricular meningiomas are in the trigone of the lateral ventricle. Fourth ventricular meningiomas without dural attachment are quite rare.142–145 The first reported case of a fourth ventricular meningioma that was removed by Ernest Sachs in 1936 in a patient with diminished hearing, leading Cushing to suggest coexistent neurofibromatosis.146 Since then, there have been only a few dozen additional cases reported. Meningiomas are believed to arise from arachnoid cap cells, which are specialized cells found on outer aspect of the arachnoid layer particularly at arachnoid granulations, because the distribution of location of meningiomas parallels the frequency of arachnoid cap cells in normal meninges.147 It is possible that these cells are dragged into ventricles by vessels piercing the ependymal layer during choroid plexus development. Most fourth ventricular meningiomas arise from the choroid plexus or inferior tela choroidea, although they can extend into the posterior fossa from the posterior petrous ridge, clivus, tentorium, foramen magnum, or cerebellar hemisphere convexity. The blood supply is usually from PICA.
Meningiomas primary affect adults, and peak occurrence is during the fifth decade. Females are affected more often than males. Typically fourth ventricular meningiomas are asymptomatic until large enough to obstruct flow of CSF. The vast majority are larger than three centimeters by the time of presentation. The most common symptoms are headache, vomiting, nystagmus, cerebellar dysfunction such as ataxia and dysmetria, cranial nerve palsies, and behavioral change.144
Grossly, meningiomas are firm, well-circumscribed, and globular or lobulated yellow to pink-gray tumors. They are usually homogeneous in consistency and can be cystic or gritty from calcification. Microscopically, meningiomas are divided into four categories: meningothelial, fibroblastic, transitional (between meningothelial and fibroblastic), and angioblastic. Meningothelial meningiomas have uniform sheets of cells with indistinct borders, occasional orientation into whorls, foamy yellow areas (call xanthomatous), and basophilic calcifications called psammoma (“sand”) bodies. Fibroblastic meningiomas have spindle cells with elongated interwoven bundles of cell bodies with collagen and reticulin fibers. Angioblastic meningiomas have pathology very similar to hemangiopericytoma and hemangioblastoma, and some believe that they are not meningiomas at all. Among fourth ventricular meningiomas, pathologic analysis has been roughly equally divided between meningothelial, fibroblastic, and transitional.143 A few have been largely psamommatous, one osteoblastic,148 one associated with Sturge-Weber syndrome,149 and one with an associated inflammatory reaction.150
Subependymoma
Fourth ventricular subependymomas are rare neoplasms that account for less than 1% of all tumors in adults.151,152 Peak incidence is 40 to 60 years of age, and males are affected more often than females. They are usually found on the caudal fourth ventricular floor or roof of the fourth ventricle, but have also been described in the lateral recess of the fourth ventricle, the lateral ventricles, and rarely the third ventricle. They are frequently asymptomatic and usually found incidentally at autopsy, but can grow large enough to produce hydrocephalus or mass effect causing visual abnormalities, ataxia, or cranial nerve palsies.153 They have been associated with the Chiari II malformation.154
On CT, subependymomas are assymmetric, lobulated, often calcified intraventricular masses that enhance moderately with contrast. MRI reveals a mass that is hypointense on T1 and hyperintense on T2 with little or no surrounding edema. Compared with lateral and third ventricular subependymomas, fourth ventricular subependymomas are more likely to have calcification and enhance (heterogeneously) with contrast155 (Fig. 31-33). Grossly they can be firm or friable but are usually gray and soft with occasional cystic change and hemorrhage.151 Small subependymomas often have a small area of attachment to the ventricular wall with sharp demarcation from the underlying brain, but larger tumors will often have several secondary sites of attachment.151 Microscopically, subependymomas have a lobulated appearance with clusters of astrocytic nuclei in a dense fibrillary background of neuroglial fibers.156 There are no cytologic features of malignancy such as mitoses but the tumor can infiltrate the brain.
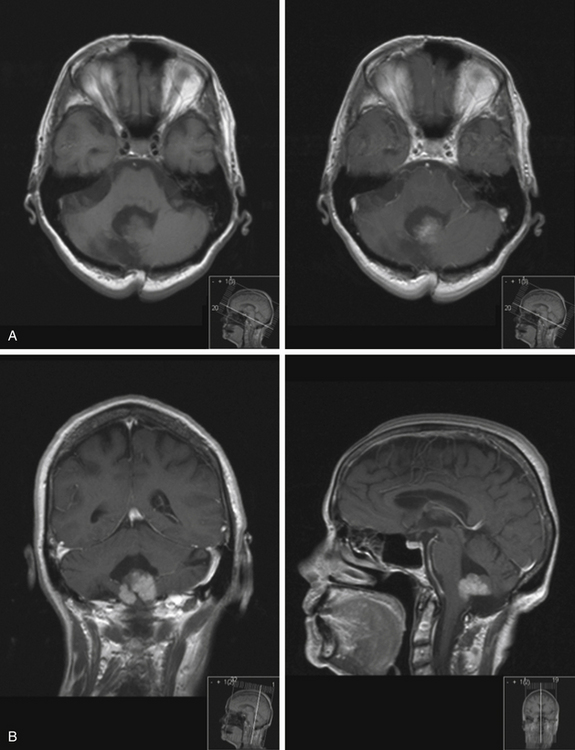
It is controversial whether these tumors arise from subependymal glia, astrocytes of the subependymal plate, ependyma, a mixture of astrocytes and ependymal cells, or simply a non-neoplastic reaction to another process such as meningitis. Initially it was believed that the subependymomas originated from subependymal glia because microscopically the cells resemble fibrillary subependymal astrocytes. However, mixed ependymoma/subependymoma tumors have been described in which areas typical for subependymoma are adjacent to areas typical for ependymoma.152 Also, electron microscopic analysis of subpendymoma reveals blepharoplasts, intermediate junctions, and microvilli that suggest ependyma, along with intermediate filaments that suggest astrocytoma.157
If the patient is asymptomatic and there is no sign of brain-stem compression, it is appropriate to follow with serial MRI scans to monitor growth of the tumor. Subependymomas occasionally become symptomatic and warrant surgical treatment. After internal debulking with ultrasonic aspiration or laser, the tumor is gradually folded in on itself and dissected from the surrounding brain. There is usually a well-defined plane between the tumor and normal brain, but occasionally the tumor will be infiltrative and adherent, presumably due to an ependymal component of the tumor. In these cases, it is extremely important not to pull on the capsule attached to the fourth ventricle, since respiratory failure from brain-stem manipulation is the most common perioperative cause of death in patients with subependymoma.152 If the patient is elderly and presents with obstructive hydrocephalus without brain-stem compression from a mass with radiologic appearance consistent with subependymoma, insertion of a ventriculoperitoneal shunt and observation with MRI may be a good alternative to removing the tumor surgically.
Most patients who survive surgery to completely resect a pure subependymoma do well with no recurrence,151 although the natural history of incomplete resection is not known.153 Presence of an ependymal component carries a worse prognosis and should be treated as a true ependymoma.
Lhermitte-Duclos Disease
Lhermitte-Duclos disease is characterized by a cerebellar mass of abnormal ganglion cells producing circumscribed regions of enlarged cerebellar folia.36,158 It is unclear whether it represents a congenital cerebellar malformation, hamartoma, phakomatosis, arrest of cell migration, graded hypertrophy of granular cell neurons, or true neoplasm. Immunohistochemical analysis confirms that the cells are derived from granule cells with a minor population of Purkinje cells, and they have no significant proliferative activity, which suggests that the disease is a non-neoplastic malformation predominantly derived from granule cells.159 Because little is known about its pathogenesis, many names have been used to describe this lesion,158 including dysplastic gangliocytoma, hamartoma, hamartoblastoma, neurocytic blastoma, diffuse ganglioneuroma, myelinated neurocytoma, granulomolecular cerebellar hypertrophy, gangliocytoma myelinicum diffusum, and purkinjeoma.
Most patients with Lhermitte-Duclos also have megencephaly. Other associated malformations include hydromyelia, cortical heterotopia, leontiasis ossea, neurofibromatosis, microgyria, hemihypertrophy, numerous hemangiomata, and polydactyly.160 It is known to be familial in some cases,161 and there is an association between Lhermitte-Duclos and Cowden disease,162–165 an autosomal dominant condition characterized by multiple cutaneous trichilemmomas, oral papillomatosis, and increased incidence of breast, colon, adnexa malignancies. The association suggests that both conditions may represent a single disorder of cellular development, perhaps a phakomatosis involving all three germ layers with a tendency for malignant degeneration of lesions.163
Peak incidence is between 20 and 40 years of age,158 but patients can present at any age, even at birth.166 There is no sex predominance. In most cases, the lesion grows slowly and produces few symptoms until the abnormal bulk of the cerebellum becomes large enough to obstruct flow of CSF and produce brain-stem compression. Most patients have a history of several months of progressive headache, vomiting, gait disturbance, and cranial nerve dysfunction. The lesion almost always becomes progressively larger and is rarely an incidental finding at autopsy.
Imaging demonstrates unilateral cerebellar enlargement with a gyriform pattern within the lesion corresponding to the enlarged folia (Fig. 31-34). Most patients have hydrocephalus from fourth ventricular compression at the time of presentation. On CT, the mass is hypodense with ill-defined borders and no enhancement. There is sometimes thinning of occipital squama and focal areas of calcification.167–170 On MRI, the lesion is well defined, hypointense on T1 and hyperintense on T2. The lesion usually does not enhance on CT or MRI,168,171 but there is occasionally peripheral enhancement due to proliferation of veins near the edge of the tumor in the molecular layer and leptomeninges.172,173 Except for the lack of enhancement, the mass can closely resemble a low-grade astrocytoma.170,174 The preserved folial pattern and lack of enhancement produces a striated “tiger-striped cerebellum” pattern that is characteristic of this tumor.175,176
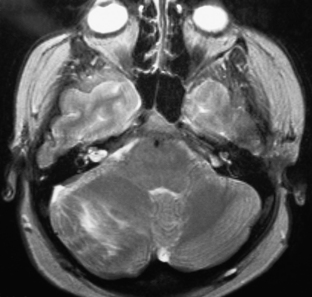
Grossly, Lhermitte-Duclos appears as broad, pale cerebellar folia confined to one hemisphere or a portion of one hemisphere.36,177 Microscopically, there is a thick outer layer of well-developed myelinated nerve fibers, encompassing an inner layer of abnormal densely packed neurons that superficially resemble Purkinje cells. There is no layering of Purkinje cells and granular neurons, the folia central white matter is often absent, and that transition between normal and abnormal areas is gradual.
As the lesion almost always continues to enlarge progressively, patients who become symptomatic from Lhermitte-Duclos invariably require surgery. Upon exposure of the cerebellum, the large pale broadened gyri should be immediately apparent. The affected tissue is usually hypovascular and easily removed with ultrasonic aspiration.177 Since there is no clear plane between involved and normal cerebellum, the preoperative MRI should be used to determine the extent of resection. Recurrence years after initial resection have been reported,159,178–180 so complete resection should be attempted and patients should get periodic follow-up MRI scans. Otherwise no adjuvant treatment is necessary.
Metastasis
Brain metastases occur in about one quarter of all patients with metastatic cancer, so metastases outnumber all other brain tumors in adults. This is true in the posterior fossa as well, where 15% of metastases are located (equal to the proportional weight of brain tissue in the posterior fossa).181,182 In adults, the most common metastases to brain tissue are lung (30%), breast (20%), melanoma (10%), kidney (10%), and gastrointestinal tract (5%). Some cancers, like melanoma, metastasize to the brain very commonly, so melanoma brain metastases are often found even though it is a relatively rare tumor. Others, like gastrointestinal tumors, rarely metastasize to the brain but are so common that they are frequently found in the brain. Any tumor that metastasizes to the brain can be found in the posterior fossa, but gastrointestinal, bladder, and uterine tumors do so slightly more often than the others do, and lymphoma sometimes presents as a subependymal mass that extends into the ventricle. When they occur in the posterior fossa, metastatic tumors often become symptomatic from fourth ventricular compression, and most patients present with signs of increased intracranial pressure and hydrocephalus. In children, the most common metastatic tumors are neuroblastoma, rhabdomyosarcoma, Wilms’ tumor, and ALL (chloromas).
The radiographic appearance of metastatic tumors is highly variable. CT may show hyperdensity (from dense cellularity of most metastatic tumors) or hypodensity, and MRI may be hyperintense, hypointense, or isointense on both T1 and T2. Metastases usually occur at the gray-white junction and are often multiple, although renal and gastrointestinal metastases tend to be solitary. Because they have no blood-brain barrier, virtually all metastatic tumors enhance on CT and MRI, either diffusely or at the tumor margins (“ring-enhancing”); there are sometimes nonenhancing central areas consistent with necrosis.183,184 Hemorrhage is more characteristic of certain tumors, especially melanoma, choriocarcinoma, renal cell cancer, and lung cancer. Adjacent tissue usually has significant vasogenic edema that follows white matter tracts that is often so extensive that the tumor will be obscured on CT or T2-weighted MRI. The histology of metastases is similar to that of the tumor of origin. They differ from primary brain tumors in that they contain more collagen and have more distinct borders with adjacent normal brain.
Brain metastases are usually tertiary metastases from previously metastatic locations in the lung, liver, or lymph nodes; therefore, presence of metastases in the brain suggests advanced cancer, and the prognosis is very poor. Survival rates vary with different tumors, but few patients survive more than 1 year. Surgery for metastatic disease can prolong patients’ survival and quality of life, especially if the metastasis is solitary, which is true in about 50%.181 Radiation is often used as primary or adjunctive therapy.
Other Tumors of the Fourth Ventricle
Teratomas rarely occur in the fourth ventricle or cerebellar vermis, either along or in combination with the more common pineal location.185–189 Clival chordomas, arising from the remnants of the primitive notochord, can spread along the floor of the posterior fossa to compress the brain stem and cranial nerves, and require adjuvant radiotherapy since complete surgical excision is rarely possible. Nerve root fibromas of the lower cranial nerves often affect the cerebellopontine angle, especially in neurofibromatosis type II, which includes bilateral vestibular schwannomas and silent fibromas of the vagus and trigeminal nerves. Gangliogliomas have been described in the cerebellum and brain stem.190,191 Finally, single cases of fourth ventricular myxofibroxanthoma192 and hemangioma calcificans193 have been reported.
Molecular Biology and Cytogenetics
Medulloblastoma
Studies of the molecular biology and genetics of medulloblastomas have led to a better understanding of the disease, and to novel treatments. Medulloblastomas are difficult to cure, with some subtypes carrying a poorer prognosis. Various genetic alterations and molecular aberrations have been identified in these tumors, including mutations in the hedgehog (Hh), Wnt, and Notch pathways, which are active during embryogenesis as well as cellular proliferation and/or differentiation.193–195 This understanding has led to important clinical translations. Other pathways are also implicated. More recently, several groups have tried to stratify medulloblastomas into molecular subgroups, currently four to five based on genetic profiles, each subgroup with predominant aberrations in the Wnt, Hh, and other pathways.193,196–198 Though we are beginning to understand that some subgroups have a better prognosis and some a poorer one, more studies are needed to understand the clinical behavior of tumors with various molecular profiles active.193
In addition, understanding the molecular pathways active in medulloblastomas has led to novel treatments. Cyclophosphamide has been recognized as an Hh pathway inhibitor, with promising results in vitro and in vivo.193–195 Small-molecule inhibitors have also been used to block the Hh pathway. ErbB pathway inhibition may also be a promising target, as well as HDAC inhibitors that act on histone deacetylases, which are involved in gene expression control.193–195 New targets and pharmacologic agents are under investigation.
Ependymoma
Ependymomas are common tumors encountered in the pediatric and adult population, and can occur in the posterior fossa, the spine, and the supratentorial regions. Though histologically they are often indistinguishable, current studies suggest that ependymomas from these different locations may have very distinct molecular and genetic profiles.193 Frequent chromosomal abnormalities include losses of 6q, 17p, and 22q, and gains of 1q and 9q.193 More recently amplifications of MYCN, EGFR, NOTCH1, NOTCH4, VAV1, YAP1, and deletion of CDKN2A and SULT4A1 have also been observed.193 The search for driver genes involved in tumorigenesis is still ongoing. Despite our growing understanding of the molecular factors involved in the formation of these tumors, prognosis remains poor, and translation of genetic profiles into therapeutic targets is still needed.
Pilocytic Astrocytoma
Though pilocytic astrocytomas in the posterior fossa can have a good outcome with complete surgical resection, those in locations near the brain stem can have a less favorable prognosis and outcome. Some chromosomal changes noted in pilocytic astrocytomas have been trisomy of 5 and 7, and gain of 7q.193 More recently, smaller chromosomal aberrations have been identified, such as duplication of chromosome 7q34, containing the locus for the BRAF gene, which is beginning to be identified as an important oncogene in the development of these tumors.193 Other gene alterations also have been noted. For instance, BRAF-KIAA1549 gene fusion but no mutation of IDH1 or IDH2 has been noted in up to 70% of pilocytic astrocytomas as compared to diffuse astrocytomas, further helping to stratify glial tumor subtypes occurring in this location as separate clinical entities for prognosis and treatment.193
Albrecht S., Haber R.M., Goodman J.C., et al. Cowden syndrome and Lhermitte-Duclos disease. Cancer. 1991;70:869-876.
Bambakidis N.C., Robinson S., Cohen M., et al. Atypical teratoid/rhabdoid tumors of the central nervous system: clinical, radiographic and pathologic features. Pediatr Neurosurg. 2002;37(2):64-70.
Berger M.S., Wilson C.B. Epidermoid cyst of the posterior fossa. J Neurosurg. 1985;62:214-219.
Bertolone S.J., Yates A.J., Boyett J.M. Combined modality therapy for poorly differentiated gliomas of the posterior fossa in children: a Children’s Cancer Group report. J Neurooncol. 2003;63(1):49-54.
Breslow N., Langholz B. Childhood cancer incidence: geographical and temporal variations. Int J Cancer. 1983;32:702-716.
Burkhard C., Di Patre P.L., Schuler D., et al. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg. 2003;98(6):1170-1174.
Campbell J.W., Pollack I.F. Cerebellar astrocytomas in children. J Neurooncol. 1996;28(2):223-231. (3)
Carlotti C.G., Smith C., Rutka J.T., et al. The molecular genetics of medulloblastoma: an assessment of new therapeutic targets. Neurosurg Rev. 2008;31:359-369.
Desai K.I., Nadkarni T.D., Muzumdar D.P., et al. Prognostic factors for cerebellar astrocytomas in children: a study of 102 cases. Pediatr Neurosurg. 2001;35(6):311-317.
Dubuc A.M., Northcott P.A., Mack S., et al. The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep. 2010;10:215-223.
Duffner P.K., Cohen M.E., Thomas P.R.M., et al. The long-term effects of cranial irradiation in the central nervous system. Cancer. 1985;56:1841-1847.
Ellenbogen R.G., Winston K.R., Kupsky W.J. Tumors of the choroid plexus in children. Neurosurgery. 1989;25:327-335.
Epstein F., McCleary E.L. Intrinsic brain-stem tumors of childhood: surgical indications. J Neurosurg. 1986;64:11-15.
Farmer J.P., Montes J.L., Freeman C.R., et al. Brainstem gliomas. A 10-year institutional review. Pediatr Neurosurg. 2001;34(4):206-214.
Fernandez C., Figarella-Branger D., Girard N., et al. Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery. 2003;53(3):544-553.
Guyotat J., Signorelli F., Desme S., et al. Intracranial ependymomas in adult patients: analyses of prognostic factors. J Neurooncol. 2002;60(3):255-268.
Northcott P.A., Rutka J.T., Taylor M.D., et al. Genomics of medulloblastoma: from Giemsa-banding to next-generation sequencing in 20 years. Neurosurg Focus. 2010;28(1):E6.
Onvani S., Etame A., Smith C.A., et al. Genetics of medulloblastoma: clues for novel therapies. Exp Rev. 2010;10(5):811-823.
Packer R.J., Sutton L.N., D’Angio G., et al. Management of children with primitive neuroectodermal tumors of the posterior fossa/medulloblastoma. Pediatr Neurosci. 1985-86;12:272-282.
Smyth M.D., Horn B.N., Russo C., et al. Intracranial ependymomas of childhood: current management strategies. Pediatr Neurosurg. 2000;33(3):138-150.
Tarbell N.J., Loeffler J.S. Recent trends in the radiotherapy of pediatric gliomas. J Neurooncol. 1996;28(2):233-244. (3)
Thompson M.C., Fuller C., Hogg T.L., et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924-1931.
van Veelen-Vincent M.L., Pierre-Kahn A., Kalifa C., et al. Ependymoma in childhood: prognostic factors, extent of surgery, and adjuvant therapy. J Neurosurg. 2002;97(4):827-835.
Wanebo J.E., Lonser R.R., Glenn G.M., et al. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):82-94.
Weil R.J., Lonser R.R., DeVroom H.L., et al. Surgical management of brainstem hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):95-105.
1. Porter J.M., Pidgeon C., Cunningham A.J. The sitting position in neurosurgery: a critical appraisal. Br J Anaesth. 1999;82(1):117-128.
2. Pang D. Air embolism associated with wounds from a pin-type head-holder. Case report. J Neurosurg. 1982;57:710-713.
3. Harrison E.A., Mackersie A., McEwan A., et al. The sitting position for neurosurgery in children: A review of 16 years’ experience. Br J Anaesth. 2002;88(1):12-17.
4. Orliaguet G.A., Hanafi M., Meyer P.G., et al. Is the sitting or the prone position best for surgery for posterior fossa tumours in children? Paediatr Anaesth. 2001;11(5):541-547.
5. Steinbok P., Boyd M., Cochrane D. Cervical spine deformity following craniotomy and upper cervical laminectomy for posterior fossa tumors in children. Childs Nerv Syst. 1989;5:25-28.
6. Culley D.J., Berger M.S., Shaw D., et al. An analysis of factors determining the need for ventriculoperitoneal shunts following posterior fossa tumor surgery in children. Neurosurg. 1994;34(3):402-448.
7. Gnanalingham K.K., Lafuente J., Thompson D., et al. The natural history of ventriculomegaly and tonsillar herniation in children with posterior fossa tumours—an MRI study. Pediatr Neurosurg. 2003;39(5):246-253.
8. Raimondi A.J., Tomita T. Hydrocephalus and infratentorial tumors. Incidence, clinical picture, and treatment. J Neurosurg. 1981;55:174-182.
9. Epstein F., Murali R. Pediatric posterior fossa tumors: hazards of the “preoperative” shunt. Neurosurgery. 1978;3(3):348-350.
10. Waga S., Shimizu T., Shimosaka S., et al. Intratumoral hemorrhage after a ventriculoperitoneal shunting procedure. Neurosurgery. 1981;9(3):249-252.
11. Vaquero J., Cabezudo J.M., De Sola R.G., et al. Intratumoral hemorrhage in posterior fossa tumors after ventricular drainage. Report of two cases. J Neurosurg. 1981;54:406-408.
12. McLaurin R.L. Disadvantages of the preoperative shunt in posterior fossa tumors. Clin Neurosurg. 1983;30:286-294.
13. Dias M.S., Albright A.L. Management of hydrocephalus complicating childhood posterior fossa tumors. Pediatr Neurosci. 1989;15:283-290.
14. Rappaport Z.H., Shalit M.N. Perioperative external ventricular drainage in obstructive hydrocephalus secondary to infratentorial brain tumours. Acta Neurochir. 1989;85:118-121.
15. Magtibay P.M., Friedman J.A., Rao R.D., et al. Unusual presentation of adult metastatic peritoneal medulloblastoma associated with a ventriculoperitoneal shunt: a case study and review of the literature. Neuro-oncol. 2003;5(3):217-220.
16. Berger M.S., Baumeister B., Geyer J.R., et al. The risks of metastases from shunting in children with primary central nervous system tumors. J Neurosurg. 1991;74:872-877.
17. Suri A., Mahapatra A.K., Singh V.P. Posterior fossa tension pneumocephalus. Childs Nerv Syst. 2000;16(4):196-199.
18. Carmel P.W., Frazer R., Stein B. Aseptic meningitis following posterior fossa surgery in children. J Neurosurg. 1974;41:44-48.
19. De Chadarevian J.P., Becker W.J. Mollaret’s recurrent aseptic meningitis: relationship to epidermoid cysts. J Neuropathol Exp Neurol. 1980;39:661-669.
20. Wisoff J.H., Epstein F.J. Pseudobulbar palsey after posterior fossa operation in children. Neurosurgery. 1984;15:707-709.
21. Pollack I.F., Polinko P., Albright A.L., et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery. 1995;37(5):885-893.
22. Pollack I.F. Posterior fossa syndrome. Int Rev Neurobiol. 1997;41:411-432.
23. McGuire E.J. The innervation and function of the lower urinary tract. J Neurosurg. 1986;65:278-285.
24. Packer R.J., Sutton L.N., Atkins T.E., et al. A prospective study of cognitive function in children receiving whole-brain radiotherapy and chemotherapy: 2-year results. J Neurosurg. 1989;70:707-713.
25. Duffner P.K., Cohen M.E., Thomas P.R.M., et al. The long-term effects of cranial irradiation in the central nervous system. Cancer. 1985;56:1841-1847.
26. Bailey P., Cushing H. Medulloblastoma cerebelli. A common type of midcerebellar glioma ot childhood. Arch Neurol Psychiatry. 1931;14:192-224.
27. Farwell J.R., Dohrmann G.J., Flannery J.T. Medulloblastoma in childhood: an epidemiological study. J Neurosurg. 1984;61:657-664.
28. Packer R.J., Cogen P., Vezina G., Rorke L.B. Medulloblastoma: clinical and biologic aspects. Neuro-oncol. 1999;1(3):232-250.
29. Malheiros S.M., Carrete H.Jr., Stavale J.N. MRI of medulloblastoma in adults. Neuroradiology. 2003;45(7):463-467.
30. Breslow N., Langholz B. Childhood cancer incidence: geographical and temporal variations. Int J Cancer. 1983;32:702-716.
31. Scheurlen W., Kuhl J. Current diagnostic and therapeutic management of CNS metastasis in childhood primitive neuroectodermal tumors and ependymomas. J Neurooncol. 1998;38(2-3):181-185.
32. Koeller K.K., Rushing E.J. From the archives of the AFIP: medulloblastoma. A comprehensive review with radiologic-pathologic correlation. Radiographics. 2003;23(6):1613-1637.
33. von Koch C.S., Gulati M., Aldape K. Familial medulloblastoma: case report of one family and review of the literature. Neurosurgery. 2002;51(1):227-233.
34. Park T.S., Hoffman H.J., Hendrick E.B., et al. Medulloblastoma: clinical presentation and management—experience at the Hospital for Sick Children, Toronto 1950–1980. J Neurosurg. 1983;58:543-552.
35. Segall H.D., Zee C.S., Naidich T.P., et al. Computed tomography in neoplasms of the posterior fossa in children. Radiol Clin North Am. 1982;20:237-253.
36. Koeller K.K., Henry J.M. From the archives of the AFIP: superficial gliomas: radiologic-pathologic correlation. Armed Forces Institute of Pathology. Radiographics. 2001;21(6):1533-1556.
37. Meyers S.P., Kemp S.S., Tarr R.W. MR imaging features of medulloblastoma. Am J Roentgenol. 1992;158:859-865.
38. Nemoto Y., Inoue Y., Fukuda T., et al. Displacement of the quadrigeminal plate in tumors of the fourth ventricle. J Comput Assist Tomogr. 1989;13:769-772.
39. Arseni C., Ciurea A.V. Statistical survey of 276 cases of medulloblastoma (1935-1978). Acta Neurochir. 1981;57:159-162.
40. Packer R.J., Sutton L.N., D’Angio G., et al. Management of children with primitive neuroectodermal tumors of the posterior fossa/medulloblastoma. Pediatr Neurosci. 1985-86;12:272-282.
41. Evans A., Jenkin D., Sposto R., et al. The treatment of medulloblastoma. J Neurosurg. 1990;72:572-582.
42. Packer R.J., Sutton L.N., Goldwein J.W., et al. Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg. 1991;74:433-440.
43. Chan A.W., Tarbell N.J., Black P.M., et al. Adult medulloblastoma: prognostic factors and patterns of relapse. Neurosurgery. 2000;47(3):623-631.
44. Lefkowitz I., Packer R., Ryan S., et al. Late recurrence of primitive neuroectodermal tumor/medulloblastoma. Cancer. 1988;62:826-830.
45. Packer R.J., Rood B.R., MacDonald T.J., et al. Medulloblastoma: present concepts of stratification into risk groups. Pediatr Neurosurg. 2003;39(2):60-67.
46. Perry A. Medulloblastomas with favorable versus unfavorable histology: how many small blue cell tumor types are there in the brain? Adv Anat Pathol. 2002;9(6):345-350.
47. Saunders D.E., Hayward R.D., Phipps K.P., et al. Surveillance neuroimaging of intracranial medulloblastoma in children: how effective, how often, and for how long? J Neurosurg. 2003;99(2):280-286.
48. Rorke L.B., Packer R.J., Biegel J.A., et al. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85(1):56-65.
49. Bambakidis N.C., Robinson S., Cohen M., et al. Atypical teratoid/rhabdoid tumors of the central nervous system: clinical, radiographic and pathologic features. Pediatr Neurosurg. 2002;37(2):64-70.
50. Campbell J.W., Pollack I.F., et al. Cerebellar astrocytomas in children. J Neurooncol. 1996;28(2-3):223-231.
51. Schneider J.H.Jr., Raffel C., McComb J.G., et al. Benign cerebellar astrocytomas of childhood. Neurosurgery. 1992;30:58-63.
52. Cushing H. Experiences with the cerebellar astrocytomas. A critical review of 76 cases. Surg Gynecol Obstet. 1931;52:129-204.
53. Morreale V.M., Ebersold M.J., Quast L.M., et al. Cerebellar astrocytoma: experience with 54 cases surgically treated at the Mayo Clinic, Rochester, Minnesota, from 1978 to 1990. J Neurosurg. 1997;87(2):257-261.
54. Lee Y.Y., Van Tassel P., Bruner J.M., et al. Juvenile pilocytic astrocytomas: CT and MR characteristics. Am J Neuroradiol. 1989;10:363-370.
55. Endo H., Kumabe T., Jokura H., et al. Leptomeningeal dissemination of cerebellar malignant astrocytomas. J Neurooncol. 2003;63(2):191-199.
56. Fernandez C., Figarella-Branger D., Girard N., et al. Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery. 2003;53(3):544-553.
57. Gilles F.H., Winston K., Fulchiero A., et al. Histological features and observational variation in cerebellar gliomas in children. J Natl Cancer Inst. 1977;58:175-181.
58. Griffin T.W., Beaufait D., Blasko J.C. Cystic cerebellar astrocytomas in childhood. Cancer. 1979;44:276-280.
59. Gupta V., Goyal A., Sinha S., et al. Glioblastoma of the cerebellum. A report of 3 cases. J Neurosurg Sci. 2003;47(3):157-165.
60. Kuroiwa T., Numaguchi Y., Rothman M.I., et al. Posterior fossa glioblastoma multiforme: MR findings. AJNR Am J Neuroradiol. 1995;16(3):583-589.
61. Obana W., Cogen P., Davis R., et al. Metastatic juvenile pilocytic astrocytoma. J Neurosurg. 1991;75:972-975.
62. Kepes J.J., Lewis R.C., Vergara G.G. Cerebellar astrocytoma invading the musculature and soft tissues of the neck. J Neurosurg. 1980;52:414-418.
63. Rollins N.K., Nisen P., Shapiro K.N. The use of early postoperative MR in detecting residual juvenile cerebellar pilocytic astrocytoma. AJNR Am J Neuroradiol. 1998;19(1):151-156.
64. Burkhard C., Di Patre P.L., Schuler D., et al. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg. 2003;98(6):1170-1174.
65. Tarbell N.J., Loeffler J.S. Recent trends in the radiotherapy of pediatric gliomas. J Neurooncol. 1996;28(2-3):233-244.
66. Bertolone S.J., Yates A.J., Boyett J.M. Children’s Cancer Group. Combined modality therapy for poorly differentiated gliomas of the posterior fossa in children: a Children’s Cancer Group report. J Neurooncol. 2003;63(1):49-54.
67. Desai K.I., Nadkarni T.D., Muzumdar D.P., et al. Prognostic factors for cerebellar astrocytomas in children: a study of 102 cases. Pediatr Neurosurg. 2001;35(6):311-317.
68. Austin E., Alvord E. Recurrences of cerebellar astrocytomas: a violation of Collin’s law. J Neurosurg. 1988;68:41-47.
69. Guyotat J., Signorelli F., Desme S., et al. Intracranial ependymomas in adult patients: analyses of prognostic factors. J Neurooncol. 2002;60(3):255-268.
70. van Veelen-Vincent M.L., Pierre-Kahn A., Kalifa C., et al. Ependymoma in childhood: prognostic factors, extent of surgery, and adjuvant therapy. J Neurosurg. 2002;97(4):827-835.
71. Croul S., Otte J., Khalili K. Brain tumors and polyomaviruses. J Neurovirol. 2003;9(2):173-182.
72. Bergsagel D., Finegold M., Batel J., et al. DNA sequences similar to those of simian virus 40 in ependymoma and choroid plexus tumors of childhood. N Engl J Med. 1992;326(15):989.
73. Kirschstein R., Gerger P. Ependymomas produced after intracerebral innoculation of SV40 into newborn hamsters. Nature. 1962;195:299.
74. Nazar G.B., Hoffman H.J., Becker J.E., et al. Infratentorial ependymomas in childhood: prognostic factors and treatment. J Neurosurg. 1990;72:408-417.
75. Nakasu S., Ohashi M., Suzuki F., et al. Late dissemination of fourth ventricle ependymoma: a case report. J Neurooncol. 2001;55(2):117-120.
76. Lefton D.R., Pinto R.S., Martin S.W. MRI features of intracranial and spinal ependymomas. Pediatr Neurosurg. 1998;28(2):97-105.
77. Swartz J.D., Zimmerman R.A., Bilaniuk L.T. Computed tomography of intracranial ependymomas. Radiology. 1982;143:97-101.
78. Chiu J., Woo S., Ater J., et al. Intracranial ependymoma in children: analysis of prognostic factors. J Neurooncol. 1992;13:283.
79. Ross G., Rubinstein L. Lack of histopathological correlation of malignant ependymomas with postoperative survival. J Neurosurg. 1989;70:31-36.
80. Oya N., Shibamoto Y., Nagata Y., et al. Postoperative radiotherapy for intracranial ependymoma: analysis of prognostic factors and patterns of failure. J Neurooncol. 2002;56(1):87-94.
81. Goldwein J.W., Glauser T.A., Packer R.J., et al. Recurrent intracranial ependymomas in children—survival, patterns of failure, and prognostic factors. Cancer. 1990;66(3):557.
82. Vanuytsel L., Brada M. The role of prophylactic spinal irradiation in localized intracranial ependymoma. Int J Radiat Oncol Biol Phys. 1991;21:825-830.
83. Goldwein J.W., Corn B.W., Rinlay J.L., et al. Is craniospinal irradiation required to cure children with malignant (anaplastic) intracranial ependymomas? Cancer. 1991;67:2766-2771.
84. Paulino A.C. Radiotherapeutic management of intracranial ependymoma. Pediatr Hematol Oncol. 2002;19(5):295-308.
85. Smyth M.D., Horn B.N., Russo C., et al. Intracranial ependymomas of childhood: current management strategies. Pediatr Neurosurg. 2000;33(3):138-150.
86. Kiyonobu I., Matsushima T., Inoue T.Y.N., et al. Correlation of microanatomical localization with postoperative survival in posterior fossa ependymomas. Neurosurgery. 1993;32:38-44.
87. Berger M., Edwards M., Lamasters D., et al. Pediatric brain stem tumors: radiographic, pathological, and clinical correlations. Neurosurgery. 1983;12:298-302.
88. Littman P., Jarret P., Bilanuk L., et al. Pediatric brainstem gliomas. Cancer. 1980;45:2787-2792.
89. Mantravadi R., Phatak R., Bellur S., et al. Brainstem gliomas: an autopsy study of 25 cases. Cancer. 1982;49:1294-1296.
90. Packer R.J., Alien J.C., Deck M., et al. Brainstem glioma: clinical manifestation of meningeal gliomatosis. Ann Neurol. 1983;14:177-182.
91. Yanagawa Y., Miyazawa T., Ishihara S., et al. Pontine glioma with osteoblastic skeletal metastases in a child. Surg Neurol. 1996;46(5):481-484.
92. Packer R.J., Zimmerman R.A., Luerssen T.G., et al. Brainstem gliomas of childhood: magnetic resonance imaging. Neurology. 1985;35:397-401.
93. Epstein F., McCleary E.L. Intrinsic brain-stem tumors of childhood: surgical indications. J Neurosurg. 1986;64:11-15.
94. Farmer J.P., Montes J.L., Freeman C.R., et al. Brainstem gliomas: a 10-year institutional review. Pediatr Neurosurg. 2001;34(4):206-214.
95. Pollack I.F., Hoffman H.J., Humphreys R.P., et al. The long-term outcome after surgical treatment of dorsally exophytic brain-stem gliomas. J Neurosurg. 1993;78(6):859-863.
96. Stroink A.R., Hoffman H.J., Hendrick E.B., et al. Transependymal benign dorsally exophytic brain stem gliomas in childhood: diagnosis and treatment recommendations. Neurosurgery. 1987;20:439-444.
97. Bilaniuk L.T., Molloy P.T., Zimmerman R.A., et al. Neurofibromatosis type 1: brain stem tumours. Neuroradiology. 1997;39(9):642-653.
98. Milstein J.M., Geyer J.R., Berger M.S., et al. Favorable prognosis for brainstem gliomas in neurofibromatosis. J Neurooncol. 1989;7:367-371.
99. Vandertop W.P., Hoffman H.J., Drake J.M., et al. Focal midbrain tumors in children. Neurosurg. 1992;31:186-194.
100. Epstein F.J., Farmer J.P. Brain stem glioma growth patterns. J Neurosurg. 1993;78:408-412.
101. Young Poussaint T., Yousuf N., Barnes P.D., et al. Cervicomedullary astrocytomas of childhood: clinical and imaging follow-up. Pediatr Radiol. 1999;29(9):662-668.
102. Epstein F., Wisoff J. Intra-axial tumors of the cervicomedullary junction. J Neurosurg. 1987;67:483-487.
103. Ellenbogen R.G., Winston K.R., Kupsky W.J. Tumors of the choroid plexus in children. Neurosurgery. 1989;25:327-335.
104. Tomita T., McLone D., Flannery A. Choroid plexus papillomas of neonates, infants, and children. Pediatr Neurosci. 1988;14:23-30.
105. Ken J.G., Sobel D.F., Copeland B., et al. Choroid plexus papillomas of the foramen of Luschka: MR appearance. AJNR Am J Neuroradiol. 1991;12:1201-1203.
106. Picard C., Copty M., Lavoie G., et al. A primary choroid plexus papilloma of the cerebellopontine angle. Surg Neurol. 1979;12:123-127.
107. Talacchi A., De Micheli E., Lombardo C., et al. Choroid plexus papilloma of the cerebellopontine angle: a twelve patient series. Surg Neurol. 1999;51(6):621-629.
108. Shinoda J., Kawaguchi M., Matsuhisa T., et al. Choroid plexus carcinoma in infants: report of two cases and review of the literature. Acta Neurochir (Wien). 1998;140(6):557-563.
109. Packer R., Perilongo G., Johnson D., et al. Choroid plexus carcinoma in childhood. Cancer. 1992;69:580-585.
110. Shin J.H., Lee H.K., Jeong A.K., et al. Choroid plexus papilloma in the posterior cranial fossa: MR, CT, and angiographic findings. Clin Imaging. 2001;25(3):154-162.
111. Ho D.M., Wong T.T., Liu H.C., et al. Choroid plexus tumors in childhood. Histopathologic study and clinicopathological correlation. Childs Nerv Syst. 1991;7:437-441.
112. Coates T.L., Hinshaw D.B., Peckman N., et al. Pediatric choroid plexus neoplasms: MR, CT, and pathologic correlation. Radiology. 1989;173:81-88.
113. Vasquez E., Ball W.S.Jr., Prenger E.C., et al. Magnetic resonance imaging of fourth ventricular choroid plexus neoplasms in childhood. Pediatr Neurosurg. 1991-92;17:48-52.
114. McEvoy A.W., Harding B.N., Phipps K.P., et al. Management of choroid plexus tumours in children: 20 years experience at a single neurosurgical centre. Pediatr Neurosurg. 2000;32(4):192-199.
115. Wolff J.E., Sajedi M., Brant R., et al. Choroid plexus tumours. Br J Cancer. 2002;87(10):1086-1091.
116. McGirr S.J., Ebersold M.J., Scheithauer B.W., et al. Choroid plexus papillomas: long-term follow-up results in a surgically treated series. J Neurosurg. 1988;69:843-849.
117. Weil R.J., Lonser R.R., DeVroom H.L., et al. Surgical management of brainstem hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):95-105.
118. Lindau A. Studien uber Kleinhirncysten. Bau, Pathologenese und Beziehungen zur Angiomatosis Retinae. Acta Pathol Microbiol Scand Suppl. 1926;1:1-128.
119. Friedrich C.A. Von Hippel-Lindau syndrome. A pleomorphic condition. Cancer. 1999;86(suppl 11):2478-2482.
120. Neumann H.P.H., Eggert H.R., Weigel K., et al. Hemangioblastomas of the central nervous system. A 10-year study with special reference to von Hippel-Lindau syndrome. J Neurosurg. 1989;70:24-30.
121. Seizinger B.R., Smith D.I., Filling-Katz M.R., et al. Genetic flanking markers refine diagnostic criteria and provide insights into the genetics of Von Hippel Lindau disease. Proc Natl Acad Sci U S A. 1991;88(7):2864-2868.
122. Wanebo J.E., Lonser R.R., Glenn G.M., et al. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):82-94.
123. Waldmann T.A., Levin E.H., Baldwin M., et al. The association of polycythemia with a cerebellar hemangioblastoma: the production of an erythropoiesis stimulating factor by the tumor. Am J Med. 1961;31:318-324.
124. Tachibana O., Yamashima T., Yamashita J., et al. Immunohistochemical study of erythropoietin in cerebellar hemangioblastomas associated with secondary polycythemia. Neurosurgery. 1991;28:24-26.
125. Elster A.D., Arthur D.W., et al. Intracranial hemangioblastomas: CT and MR findings. J Comput Asst Tomogr. 1988;12:736-739.
126. Lee S.R., Sauches J., Mark A.S., et al. Posterior fossa hemangioblastomas: MR imaging. Radiology. 1989;171:463-468.
127. Reyns N., Assaker R., Louis E., et al. Leptomeningeal hemangioblastomatosis in a case of von Hippel-Lindau disease: case report. Neurosurgery. 2003;52(5):1212-1215.
128. Niemela M., Lim Y.J., Soderman M., et al. Gamma knife radiosurgery in 11 hemangioblastomas. J Neurosurg. 1996;85(4):591-596.
129. Guidetti B., Gagliardi F.M. Epidermoid and dermoid cysts: clinical evaluation and late surgical results. J Neurosurg. 1977;47:12-18.
130. Berger M.S., Wilson C.B. Epidermoid cyst of the posterior fossa. J Neurosurg. 1985;62:214-219.
131. Rosario M., Becker D.H., Conley F.K. Epidermoid tumors involving the fourth ventricle. Neurosurgery. 1981;9:9-13.
132. Goffin J., Plets C., Van Calenbergh F., et al. Posterior fossa dermoid cyst associated with dermal fistula: report of 2 cases and review of the literature. Childs Nerv Syst. 1993;9:179-181.
133. Logue V., Till K. Posterior fossa dermoid cysts with special reference to intracranial infection. J Neurol Neurosurg Psychiatry. 1952;15:1-12.
134. Akhaddar A., Jiddane M., Chakir N., et al. Cerebellar abscesses secondary to occipital dermoid cyst with dermal sinus: case report. Surg Neurol. 2002;58(3):266-270. (4)
135. Barloon T.J., Jacoby C.G., Schultz D.H. MR of fourth ventricular epidermoid tumors. Am J Neuroradiol. 1988;9:794-796.
136. Tancredi A., Fiume D., Gazzeri G. Epidermoid cysts of the fourth ventricle: very long follow up in 9 cases and review of the literature. Acta Neurochir (Wien). 2003;145(10):905-911.
137. Muller A., Buttner A., Weis S. Rare occurrence of intracerebellar colloid cyst. Case report. J Neurosurg. 1999;91(1):128-131.
138. Jan M., BaZozo V., Velut S. Colloid cyst of the fourth ventricle: diagnostic problems and pathogenic considerations. Neurosurgery. 1989;24:939-942.
139. Parkinson D., Childe A.E. Colloid cyst of the fourth ventricle. Report of a case of two colloid cysts of the fourth ventricle. J Neurosurg. 1952;9:404-409.
140. Bejjani G.K., Wright D.C., Schessel D., et al. Endodermal cysts of the posterior fossa. Report of three cases and review of the literature. J Neurosurg. 1998;89(2):326-335.
141. Harris C., Dias M., Brockmeyer D., et al. Neurenteric cysts of the posterior fossa: recognition, management, and embryogenesis. Neurosurgery. 1991;29:893-898.
142. Akimoto J., Sato Y., Tsutsumi M., et al. Fourth ventricular meningioma in an adult—case report. Neurol Med Chir (Tokyo). 2001;41(8):402-405.
143. Ceylan K., Ilbay K., Kuzeyli M., et al. Intraventricular meningioma of the fourth ventricle. Clin Neurol Neurosurg. 1992;94:181-184.
144. Chaskis C., Buisseret T., Michotte A., et al. Meningioma of the fourth ventricle presenting with intermittent behaviour disorders: a case report and review of the literature. J Clin Neurosci. 2001;8(suppl 1):59-62.
145. Iseda T., Goya T., Nakano S., et al. Magnetic resonance imaging and angiographic appearance of meningioma of the fourth ventricle—two case reports. Neurol Med Chir (Tokyo). 1997;37(1):36-40.
146. Cushing H., Eisenhardt L. Meningiomas: their classification, regional behavior, life history, and surgical end results. Springfield, IL: Charles C. Thomas; 1938. 139–140
147. Black P. Meningiomas. Neurosurgery. 1993;32:643-657.
148. Johnson M.D., Tulipan N., Whetsell W.O. Osteoblastic meningioma of the fourth ventricle. Neurosurgery. 1989;24(4):587-590.
149. Gokalp H.Z., Ozkal E., Erdogan A., et al. A giant meningioma of the fourth ventricle associated with Sturge-Weber disease. Acta Neurochir. 1981;57:115-120.
150. Ferrara M., Bizzozero L., D’Aliberti G., et al. Inflammatory meningioma of the fourth ventricle. Case report. J Neurosurg Sci. 1994;38(1):59-62.
151. Scheithauer B.W. Symptomatic subependymoma. Report of 21 cases with review of the literature. J Neurosurg. 1978;49:689-696.
152. Jooma R., Torrens M.J., Bradshaw J., et al. Subependymoma of the fourth ventricle. Surgical treatment in 12 cases. J Neurosurg. 1985;62:508-512.
153. Lombardi D., Scheithauer B.W., Meyer F.B., et al. Symptomatic subependymoma: a clinicopathological and flow cytometric study. J Neurosurg. 1991;75:583-588.
154. Piatt J.H.Jr., D’Agostino A. The Chiari II malformation: lesions discovered within the fourth ventricle. Pediatr Neurosurg. 1999;30(2):79-85.
155. Chiechi M.V., Smirniotopoulos J.G., Jones R.V. Intracranial subependymomas: CT and MR imaging features in 24 cases. AJR Am J Roentgenol. 1995;165(5):1245-1250.
156. Gandolfi A., Brizzi R.E., Tedeschi F., et al. Symptomatic subependymoma of the fourth ventricle. Case report. J Neurosurg. 1981;55:841-844.
157. Moss T.H. Observations on the nature of subependymoma: an electron microscopic study. Neuropathol Appl Neurobiol. 1984;10(1):63-75.
158. Nowak D.A., Trost H.A. Lhermitte-Duclos disease (dysplastic cerebellar gangliocytoma): a malformation, hamartoma or neoplasm? Acta Neurol Scand. 2002;105(3):137-145.
159. Hair L.S., Symmans F., Powers J.M., et al. Immunohistochemistry and proliferative activity in Lhermitte-Duclos disease. Acta Neuropathol. 1992;84:570-573.
160. Reznick M., Schoenen J. Lhermitte-Duclos disease. Acta Neuropathol. 1983;59:88-94.
161. Ambler M., Pogacar S., Sidman R. Lhermitte-Duclos disease (granule cell hypertrophy of the cerebellum): pathological analysis of the first familial case. J Neuropathol Exp Neurol. 1969;28:622-647.
162. Vinchon M., Blond S., Lejeune J.P., et al. Association of Lhermitte-Duclos and Cowden disease: report of a new case and review of the literature. J Neurol Neurosurg Psychiatry. 1994;57(6):699-704.
163. Padberg G.W., Schot J.D.L., Vielvoye G.J., et al. Lhermitte-Duclos disease and Cowden disease: a single phakomatosis. Ann Neurol. 1991;29:517-523.
164. Albrecht S., Haber R.M., Goodman J.C., et al. Cowden syndrome and Lhermitte-Duclos disease. Cancer. 1991;70:869-876.
165. King M.A., Coyne T.J., Spearritt D.J., et al. Lhermitte-Duclos disease and Cowden disease: a third case. Ann of Neurol. 1992;32(1):112-113.
166. Roessmann U., Wongmongkolrit T. Dysplastic gangliocytoma of the cerebellum in a newborn. J Neurosurg. 1984;60:845-847.
167. Vieco P.T., del Carpio-O’Donovan R., Melanson D., et al. Dysplastic gangliocytoma (Lhermitte-Duclos disease): CT and MR imaging. Pediatr Radiol. 1992;22:366-369.
168. Ashley D.G., Zee C.S., Chandrasoma P.T., et al. Case report. Lhermitte-Duclos disease: CT and MR findings. J Comput Assist Tomogr. 1990;14(6):984-987.
169. Smith R.R., Grossman R.I., Goldberg H.I., et al. MR imaging of Lhermitte-Duclos disease: a case report. AJNR Am J Neuroradiol. 1989;10:187-189.
170. Choudhury A.R. Short report. Preoperative magnetic resonance imaging in Lhermitte-Duclos disease. Br J Neurosurg. 1990;4:225-230.
171. Spaargaren L., Cras P., Bomhof M.A., et al. Contrast enhancement in Lhermitte-Duclos disease of the cerebellum: correlation of imaging with neuropathology in two cases. Neuroradiology. 2003;45(6):381-385.
172. Awwad E.E., Levy E., Martin D.S., et al. Atypical MR appearance of Lhermitte-Duclos disease with contrast enhancement. AJNR Am J Neuroradiol. 1995;16(8):1719-1720.
173. Carter J.E., Merren M.D., Swann K.W., et al. Preoperative diagnosis of Lhermitte-Duclos disease by magnetic resonance imaging. Case report. J Neurosurg. 1989;70:135-137.
174. Meltzer C.C., Smirniotopoulos J.G., Jones R.V., et al. The striated cerebellum: an MR imaging sign in Lhermitte-Duclos disease (dysplastic gangliocytoma). Radiology. 1995;194(3):699-703.
175. Wolansky L.J., Malantic G.P., Heary R., et al. Preoperative MRI diagnosis of Lhermitte-Duclos disease: case report with associated enlarged vessel and syrinx. Surg Neurol. 1996;45(5):470-475.
176. Faillot T., Sichez J.P., Brault J.L., et al. Lhermitte-Duclos disease (dysplastic gangliocytoma of the cerebellum). Report of a case and review of the literature. Acta Neurochir. 1990;105:44-49.
177. Stapleton S.R., Wilkins P.R., Bell B.A., et al. Short report. Recurrent dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) presenting with subarachnoid haemorrhage. Br J Neurosurg. 1992;6:153-156.
178. Hashimoto H., Iida J., Masui K., et al. Recurrent Lhermitte-Duclos disease—case report. Neurol Med Chir (Tokyo). 1997;37(9):692-696.
179. Williams D.W.3rd, Elster A.D., Ginsberg L.E., et al. Recurrent Lhermitte-Duclos disease: report of two cases and association with Cowden’s disease. AJNR Am J Neuroradiol. 1992;13(1):287-290.
180. Patchell R., Tibbs P., Walsh J., et al. A randomized trial of surgery in the treatment of single metastases to the brain. N Engl J Med. 1990;322:494-500.
181. Soffietti R., Ruda R., Mutani R. Management of brain metastases. J Neurol. 2002;249(10):1357-1369.
182. Potts D.G., Abbott G.F., von Sneidem J.V. National Cancer Institute Study: evaluation of computed tomography in the diagnosis of intracranial neoplasms. III. Metastatic tumors. Radiology. 1980;136:657-664.
183. Bisese J.H. MRI of cranial metastases. Semin Ultrasound CT. MRI. 1992;13:473-483.
184. Iwasaki I., Horie H., Yu T.J., et al. Intracranial teratoma with a prominent rhabdomyogenic element and germinoma in the fourth ventricle. Neurol Med Chir (Tokyo). 1984;24:51-55.
185. Desai K., Nadkarni T., Muzumdar D., et al. Midline posterior fossa teratoma—case report. Neurol Med Chir (Tokyo). 2000;41(2):94-96.
186. Drapkin A.J., Rose W.S., Pellmar M.B. Mature teratoma in the fourth ventricle of an adult: case report and review of the literature. Neurosurgery. 1987;21(3):404-410.
187. Aoyama I., Makita Y., Nabeshima S., et al. Intracranial double teratomas. Surg Neurol. 1982;17:383-387.
188. Harada K., Okamoto H., Fujioka Y., et al. Teratoma in the fourth ventricle of an elderly adult. Case report. Neurol Med Chir (Tokyo). 1984;24:499-503.
189. Lagares A., Gomez P.A., Lobato R.D., et al. Ganglioglioma of the brainstem: report of three cases and review of the literature. Surg Neurol. 2001;56(5):315-322.
190. Bills D.C., Hanieh A. Hemifacial spasm in an infant due to fourth ventricular ganglioglioma. Case report. J Neurosurg. 1991;75:134-137.
191. Chen M.N., Nakazawa S., Shimura T., et al. Myxofibroxanthoma of the fourth ventricle. Case report. Neurol Med Chir (Tokyo). 1985;25:564-567.
192. Kobayashi H., Kawano H., Ito H., et al. Hemangioma calcificans in the fourth ventricle. Neurosurgery. 1984;14(6):737-739.
193. Dubuc A.M., Northcott P.A., Mack S., et al. The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep. 2010;10:215-223.
194. Carlotti C.G., Smith C., Rutka J.T., et al. The molecular genetics of medulloblastoma: an assessment of new therapeutic targets. Neurosurg Rev.. 2008;31:359-369.
195. Onvani S., Etame A., Smith C.A., et al. Genetics of medulloblastoma: clues for novel therapies. Exp Rev. 2010;10(5):811-823.
196. Thompson M.C., Fuller C., Hogg T.L., et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924-1931.
197. Northcott P.A., Rutka J.T., Taylor M.D., et al. Genomics of medulloblastoma: from Giemsa-banding to next-generation sequencing in 20 years. Neurosurg Focus. 2010;28(1):E6.
198. Northcott P.A., Korshunov A., Witt H., et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408-1414.