CHAPTER 54 MANAGEMENT OF STATUS EPILEPTICUS
Known case reports of epilepsy date as far back as the 16th century, but the term status epilepticus was first noted in the English language in 1868 in Bazire’s translation of Trousseau’s Lecture on Clinical Medicine1,2: “The status epilepticus is characterized, not by a single attack, but by a series of attacks … the stupor which succeeds the convulsions lasts from ten minutes to three-quarters of an hour at most. But before the stupor has passed away another attack, exactly similar to the first, supervenes, and is confounded with it. Now, as the third stage of an epileptic fit is not usually regarded as distinct from the convulsive stage, the patient seems to be still in a fit, although his comatose condition is only an effect of the fit. He has not therefore got over the disturbances caused by the first attack before a second occurs, then a third, a fourth, a fifth; and in proportion to the recurrence of the fits the cerebral congestion increases, the apoplectic coma is prolonged, and extends over a period varying from two to twenty-four hours, and after a time the patient does not recover his senses at all …” Although the basic definition of status epilepticus has not changed significantly over time, the understanding of the pathophysiology, etiology, and particularly treatment since the initial advent of the bromides has dramatically evolved. This chapter focuses on these aforementioned concepts, with particular emphasis on the advances in treatment of status epilepticus.
DEFINITION
Status epilepticus is defined as continuous seizure activity lasting more than 30 minutes or two or more sequential seizures without full recovery of consciousness between seizures.3 However, most physicians agree that treatment should not be withheld if the manifestation does not conform to this 30-minute definition. Thus, new operational definitions, such as those by Lowenstein and colleagues, define generalized convulsive status epilepticus (GCSE) in adults and older children (>5 years old) as 5 minutes or more of continuous seizures or two or more discrete seizures between which there is incomplete recovery of consciousness.4
CLASSIFICATION
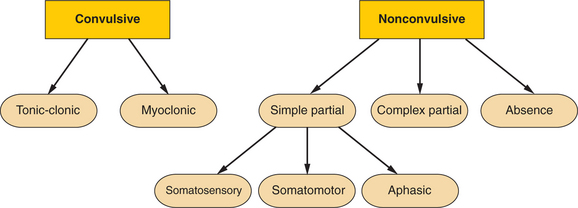
Many different complicated classification schemes for status epilepticus have been proposed. In general, status epilepticus can be defined on the basis of electrographic onset—specifically, partial versus generalized—or on clinical grounds (convulsive versus nonconvulsive) (Fig. 54-1). All forms of partial status epilepticus, such as complex partial status epilepticus, are classified clinically as nonconvulsive.
The most common form of status epilepticus is GCSE and consists of tonic and/or clonic motor activity. This motor activity may be symmetrical or asymmetrical and clinically overt or subtle. There is associated alteration of consciousness and bihemispherical, generalized, ictal discharges (which maybe asymmetrical) on the electroencephalogram (EEG).5 If GCSE is allowed to progress, convulsive activity may become less apparent and clinical manifestations more subtle. Thus, the term subtle generalized convulsive status epilepticus has been used to denote twitching movements of the face, eyelids, trunk, and distal extremities or nystagmoid movements of the eyes in association with bilateral ictal discharges on EEG.6
Complex partial status epilepticus (Fig. 54-2) has the most varied clinical manifestation. Although the prevailing constant is the definite impairment of consciousness and very localized electrographic seizures, the extent of the impairment can be variable, ranging from mild alteration of consciousness with or without automatisms, waxing and waning confusion, and agitation with near-psychotic behavior to complete stupor.7–10
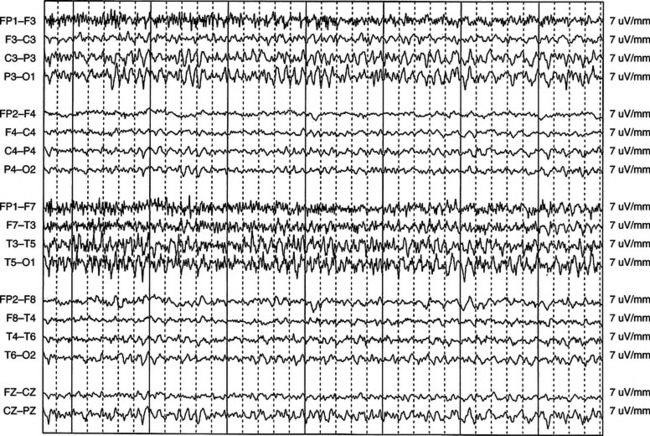
Absence status epilepticus is considered a nonconvulsive form of the condition and is often categorized as either typical or atypical absence status epilepticus. Both types are associated with paroxysms or loss of consciousness of abrupt onset and offset. However, typical absence status epilepticus is characterized by the hallmark 3-Hz spike-and-wave discharges on EEG (Fig. 54-3), a normal interictal background, and normal neurological examination findings. Atypical absence status epilepticus usually occurs in patients with underlying neurological dysfunction (Lennox-Gastaut syndrome) and is characterized by diffuse slow spike-and-wave discharges (<3 Hz) and an abnormal interictal background.11 Whatever the form, absence status epilepticus typically occurs more often in children and adolescents than in adults.
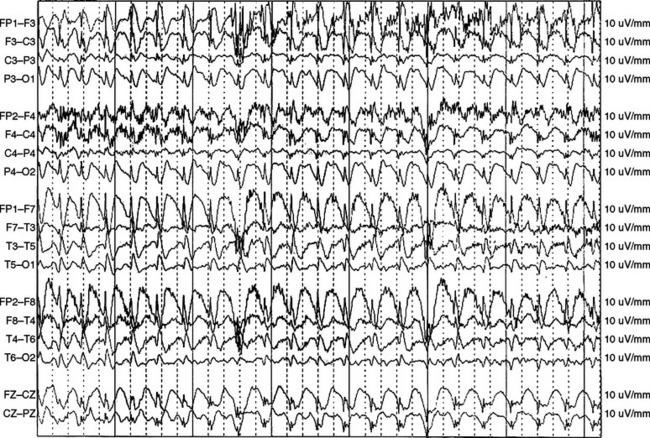
Myoclonic status epilepticus as defined by Gastaut11 is divided into two subgroups. The first group includes primary generalized epilepsy characterized by massive bilateral myoclonic jerks that occur at irregular intervals and by preservation of consciousness. This is a rare form in comparison with the second group, which occurs in secondary (symptomatic) generalized epilepsy in children and is associated with alteration of consciousness. Examples of this second group include myoclonic-astatic epilepsy, Lennox-Gastaut syndrome, and epilepsy with myoclonic absences.12 In addition, there is a separate category of myoclonic status epilepticus that occurs in nonepileptic patients with acute or subacute encephalopathies.11 This category includes degenerative encephalopathies such as dyssynergia cerebellaris myoclonica of Ramsay Hunt, progressive myoclonic epilepsy, Lafora’s disease, and various toxic and metabolic (liver, renal insufficiency) encephalopathies.12
Myoclonic status epilepticus caused by anoxic encephalopathy has not been regarded as status epilepticus in the strictest sense11; however, some authorities have argued5 that this symptomatic myoclonic syndrome should instead be considered a subtle form of GCSE.12
Simple partial status epilepticus includes somatomotor, somatosensory, and aphasic status epilepticus. Somatomotor status epilepticus can occur during or at the onset of a somatomotor partial epileptic seizure (somatomotor simple partial status epilepticus in the strictest sense)11,13 or herald the presence of a metastatic embolus or asymptomatic sylvian or suprasylvian infarction in elderly persons.11,14 Ictal discharges are usually more pronounced in the rolandic area with definite preservation of consciousness.11 A well-known type of somatomotor status epilepticus is epilepsia partialis continua (Kojewnikoff’s epilepsy), characterized by segmental myoclonus between seizures. Lastly, somatomotor simple partial status epilepticus can accompany severe brain lesions in nonepileptic persons and is characterized by proximal limb or truncal myoclonus repeated approximately once per second in conjunction with periodic lateralized epileptiform discharges (PLEDs) on EEG.8 This syndrome occurs almost exclusively in the elderly population and most often represents a watershed infarct or, less frequently, metastatic tumor or brain contusion.11,15,16
Somatosensory status epilepticus is very rare and may in fact be residual activity after surgical resection of the primary epileptogenic area in medically refractory epilepsy.13
Aphasic or dysphasic status epilepticus is less frequent than somatomotor status epilepticus, but it is well documented in the literature.17–22 This syndrome is characterized by aphasia (either receptive or expressive), sometimes in association with alexia and agraphia, lasting from seconds to days.
One special situation that deserves mention is simple partial status epilepticus in the context of hyperosmolar nonketotic hyperglycemia. Simple partial motor seizures can accompany medical symptoms such as polydipsia and loss of appetite. Osmolar values can be as high as 320 mmol, and in this situation, the use of phenytoin is contraindicated because of its inherent ability to exacerbate hyperglycemia.13
INCIDENCE, ETIOLOGY, AND MORTALITY
Multiple case series and large hospital-based retrospective studies of GCSE are noted in the literature.23–35 However, the true incidence of status epilepticus is not well defined, for multiple reasons, including clinically missed cases, variability in reporting, and inherent differences between populations. In initial hospital-based reports, Hauser31 estimated an annual incidence of 50,000 to 60,000 cases of status epilepticus in the United States. However, in a more recent prospective population-based study of status epilepticus,36 a higher U.S. incidence was estimated, approximately 100,000 to 150,000 patients with at least one episode of status epilepticus per year, according to extrapolation from community population data in Richmond, Virginia. Several retrospective and fewer prospective studies have been performed, and although methods of reporting and subpopulations of case studies are variable, a few generalizations can be drawn.
First, the incidence of status epilepticus appears to be highest at the extremes of age: the young (1 to 12 months of age) and the elderly (>60 years of age).36,37 Recurrence, however, appears most common in the pediatric population.36 The Richmond study36 revealed a recurrence rate of 35% in the pediatric population versus 7% and 10% in the adult and elderly populations, respectively. Second, the major etiology for status epilepticus in the pediatric population overall appears to be systemic non–central nervous system infections.36 In adults, low antiepileptic drug levels, temporally remote symptomatic lesions (particularly cerebrovascular accident), and acute stroke accounted for the majority of cases.38 Thus, stroke, whether acute or subacute/chronic, accounted for 61% of cases of status epilepticus in the elderly population in the Richmond study.36 Third, the overall rate of mortality from status epilepticus appears to be approximately 15% to 22% in adults25,27,28,36 but much lower in children (5% to 11%).24,26,29 Fourth, death from status epilepticus is intricately linked with etiology, age, and duration of status epilepticus. Anoxia and hypoxia were much more common causes of status epilepticus in adults than in the pediatric population and were associated with the highest rates of mortality.36 The mortality rate is higher for status epilepticus lasting longer than 60 minutes (nearly 20%) than for status epilepticus lasting 30 to 59 minutes (<5% respectively).28
PHYSIOLOGICAL CHANGES IN GENERALIZED CONVULSIVE STATUS EPILEPTICUS
Acute Systemic Complications of Generalized Convulsive Status Epilepticus
Acute systemic effects of GCSE include cardiovascular alteration, acidosis, respiratory distress, hyperthermia, and renal failure secondary to rhabdomyolysis (Table 54-1). These physiological alterations occur in essentially two phases.39 Initially, the early phase of GCSE is characterized by sympathetic overdrive,40 but after 30 minutes, failure of homeostatic mechanisms causes initial physiological alterations to normalize or even move in the exact opposite direction.39
TABLE 54-1 Systemic Physiological Changes in Generalized Convulsive Status Epilepticus
The most significant and most life-threatening changes occur within the cardiac system. Initially systemic hypertension predominates, along with tachycardia.41 Mean arterial blood pressure is elevated as a result of increased vascular resistance. However, later on, relative hypotension occurs, secondary to failure of splanchnic and renal constriction.42–44 Mortality is associated with the development of life-threatening arrhythmias in nearly 60% of patients with GCSE.45
Hyperthermia seen early on in GCSE in animal studies is not only associated with significant cerebellar damage but is also responsible for ischemic injury in outer cortical layers and hippocampus.46,47 Studies by Liu and associates48 in experimental rat models (kainic acid–induced seizures) demonstrated that decreasing temperature to 28° C decreased status epilepticus by 50%, and further reduction to 23° C abolished status epilepticus entirely. Increasing temperatures to 42° C uniformly resulted in death within 2 hours. Of most importance, decreasing temperature to 28° C prevented hippocampal cell loss, emphasizing the need to treat hyperthermia aggressively at the onset of status epilepticus.48
Derangements in blood chemistry are also seen. The natural occurrence of lactic acidosis is secondary to anaerobic metabolism in the setting of repetitive muscle activity from ongoing convulsions. Hyperglycemia occurs in the setting of increased levels of circulating catecholamines and exacerbates existing acidosis. Thus, supplemental glucose should be given only if true hypoglycemia exists.42,59
Respiratory changes include central apnea and pulmonary edema secondary to increased intravascular pressures within the pulmonary vasculature.43
Other systemic changes that may occur in GCSE include peripheral leukocytosis and possible rhabdomyolysis secondary to muscle injury from repetitive convulsions, producing subsequent acute tubular necrosis and possible renal failure.50
Central Nervous System Physiological Changes
Essentially, the central nervous system changes reflect the eventual difficulty of the brain in keeping up with the ongoing metabolic demands induced by GCSE. Initially, cerebral blood flow increases profoundly to supply the brain with the necessary glucose and oxygen.51 However, late in status epilepticus (hours after onset of seizure activity), breakdown of the body’s homeostatic mechanisms (cerebrovascular autoregulation) occurs.46 In addition, mean arterial pressure starts to fall, which causes further drop in cerebral blood flow.51,52 An eventual mismatch between elevated cerebral metabolism and insufficient cerebral blood flow (supply/demand mismatch) occurs. As lactate accumulates, brain glucose levels decline47 and, in combination with decreased oxygenation at later stages of status epilepticus, contribute to ongoing hypoxia and anoxia.53
PATHOPHYSIOLOGY OF NEURONAL INJURY
The aforementioned systemic alterations can contribute to neuronal injury, but the primary mechanisms leading to ongoing seizures and neuronal injury are at the cellular level. Although an eventual cerebral supply/demand mismatch occurs in the later stages of GCSE, neuronal injury occurs within 60 minutes.46,52 The combination of decreased γ-aminobutyric acid (GABA) neurotransmitter–mediated inhibition and abundance of glutamate excitation can lead to neuronal injury. Normal excitatory synaptic neurotransmission is essentially altered. Under normal circumstances, glutamate binds to postsynaptic non–N-methyl-D-aspartate (NMDA)–type glutamate receptors after its release from the axon terminal. This promotes sodium flow into the cell and produces depolarization, which results in further propagation down the axon. However, in the case of status epilepticus, when glutamate binds to the postsynaptic NMDA receptor, the usual self-limited depolarization does not occur. Instead, prolonged depolarization occurs, because the magnesium ion that generally blocks the NMDA channel pore is displaced.42,53 Thus, calcium ions flow into the cell, causing intracellular accumulation of calcium, which leads to cell necrosis and apoptosis.42 Because NMDA receptors are located primarily in the limbic system, neuronal cell loss thus results in hippocampal injury.42
MANAGEMENT AND TREATMENT OF STATUS EPILEPTICUS
The goal with any acute medical emergency is the timely diagnosis, management, and treatment of the problem. The longer the duration of status epilepticus, the more subtle the motor manifestations,54 the later the electroencephalographic stage (impairing adequate detection of the syndrome),55 and the more refractory to pharmacological treatment the seizures will be54,56 (Fig. 54-4).
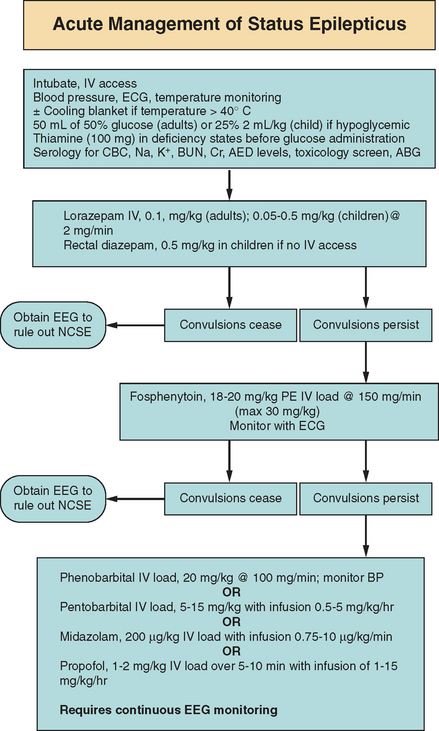
Management is essentially divided into two sections: immediate general concerns to prevent the ongoing systemic complications described previously and specific pharmacotherapy to abort ongoing seizures. In general, the ABCs (airway, breathing, and circulation) of any acute medical problem should be addressed. A clear airway for the adequate passage of air is needed with availability of immediate endotracheal intubation if required. If ongoing convulsive motor activity causes hypoventilation, a paralytic agent may be needed temporarily to support respiration and prevent the patient from injuring himself or herself. Peripheral access also needs to be addressed immediately in order to initiate intravenous medication and provide fluid resuscitation if needed. Two large-gauge intravenous catheters, or even a central catheter if peripheral access cannot be obtained, should be in place. Blood pressure, the electrocardiogram, and temperature should be monitored. Hypotension should be avoided, and agents to support blood pressure may be required temporarily. If significant hyperthermia develops (>40° C), a cooling blanket may be necessary to stabilize body temperature.57 Once the airway, breathing, and circulation are secure, blood chemistry profiles should be drawn for potentially reversible causes or exacerbating factors of status epilepticus. Fingerstick blood glucose should be tested, and 50 mL of 50% glucose bolus injection for adults and 2 mL/kg of 25% glucose for children should be administered if hypoglycemia exists. A glucose bolus should also be administered if hypoglycemia is suspected and a blood glucose level cannot be established in a timely manner. Thiamine (100-mg bolus) should be administered in individuals suspected of thiamine deficiency (e.g., alcoholic patients) before or simultaneously with glucose administration to prevent precipitating acute Wernicke’s encephalopathy. Blood samples should be analyzed for complete blood cell count, metabolic profile (sodium, potassium, blood urea nitrogen, creatinine, calcium, magnesium), antiepileptic drug levels if applicable, urine/serum toxicological levels, and arterial blood gases.
The benzodiazepines, phenytoin, and phenobarbital have been used as first-line agents for aborting status epilepticus. The rationale for use of these agents for first-line therapy is the direct result mainly of four pivotal studies.58–61
Leppik and colleagues61 compared lorazepam with diazepam for the initial treatment of status epilepticus; 78 patients received either 10 mg of diazepam or 4 mg of lorazepam. No significant difference in terminating status epilepticus (58% with diazepam versus 78% with lorazepam) or latency of action (2 minutes for diazepam and 3 minutes for lorazepam) was noted.61 In 1988, Shaner and associates,59 in a randomized clinical trial, compared the efficacy of the combination of diazepam and phenytoin with phenobarbital alone in 36 patients with GCSE. Phenytoin was added to the phenobarbital regimen if seizures persisted for 10 minutes or more after therapy was initiated. Although phenobarbital aborted status epilepticus a little more quickly than did the combination, there was no statistical significance in the success frequency between the two treatments.59 However, these two studies paved the way for the Veterans Affairs cooperative trial (randomized, double-blind study)60 in 1998 in which four different treatment protocols were compared in 384 patients with overt (presumably early) status epilepticus and 134 patients with subtle (presumably late) status epilepticus. The four treatment regimens were as follows: (1) lorazepam, 0.1 mg/kg; (2) diazepam, 0.15 mg/kg, followed by 18 mg/kg of phenytoin; (3) phenytoin alone, 18 mg/kg; and (4) phenobarbital, 15 mg/kg. In the patients with overt status epilepticus, the best response (electroencephalographic and clinical termination of seizures within 20 minutes, and no seizure recurrence within 60 minutes, of therapy initiation) occurred in 64.9% of patients who received lorazepam alone, 58.2% of those who received phenobarbital alone, 55.8% of those who received diazepam with phenytoin, and 43.6% of those who received phenytoin alone. In the patients with subtle status epilepticus, overall response was much lower than in the patients with overt status epilepticus. The response rate was highest in the patients who received phenobarbital (24.2%), followed by those who received lorazepam (17.9%) and those who received diazepam and phenytoin (8.3%), and lowest in those who received phenytoin alone (7.7%). No statistically significant difference between recurrence during the 12-hour study, incidence of adverse effects, or 30-day outcome was noted. The authors concluded that lorazepam was more effective than phenytoin as initial intravenous treatment for overt GCSE. Although lorazepam was not found to be more efficacious than phenobarbital alone or diazepam and phenytoin combined, it was found to be easier to use.60 In the most recent randomized, double-blind trial, Alldredge and colleagues58 in 2001 evaluated intravenous benzodiazepine administration by paramedics for the treatment of out-of-hospital status epilepticus. In this trial, 205 patients were randomly assigned to receive intravenous diazepam (5 mg), lorazepam (2 mg), or placebo. Status epilepticus was aborted in 59.1% of patients receiving lorazepam who arrived in the emergency department, in comparison with 42.6% of the diazepam recipients and 21.1% of the placebo recipients. The duration of status epilepticus was also shorter in the lorazepam recipients.
PHARMACOTHERAPY
Benzodiazepines
The benzodiazepines commonly used in the United States to treat status epilepticus include lorazepam (Ativan), diazepam (Valium), and midazolam (Versed). The agents in this class enhance GABA-mediated inhibition by binding to the benzodiazepine-binding site on the GABA receptor complex. Diazepam enters the brain rapidly because of its lipophilic quality; however, nearly 20 minutes later, it is rapidly distributed to other fatty tissues, which causes a fall of serum concentrations.58,62 Thus, duration of action is approximately 15 to 30 minutes.63 Side effects include respiratory suppression, hypotension, and sedation. Lorazepam is less lipid soluble but is distributed to other tissues over a longer period of time than is diazepam. Thus, lorazepam has a longer duration of effect (12 to 24 hours) but a slightly longer delay in reaching peak concentration (6 to 10 minutes) than does diazepam (1 to 3 minutes).63 Side effects are similar to those of diazepam. The recommended dosage of lorazepam in status epilepticus is 0.1 mg/kg in adults and 0.05 to 0.5 mg/kg in children (maximal rate, 2 mg/minute).63,64 The recommended dosage of diazepam is 0.15 to 0.25 mg/kg in adults as an intravenous push and 0.1 to 1.0 mg/kg in children (maximal rate, 5 mg/minute).63,64 In children, in whom intravenous access may be difficult initially, rectal diazepam (Diastat) may be given at a dosage of 0.5 mg/kg.3 Midazolam is used mainly in Europe as a first-line agent, but it is used as an intravenous drip in refractory status epilepticus in the United States and hence is discussed elsewhere in this chapter (section on “Refractory Status Epilepticus”).
Phenytoin/Fosphenytoin
Phenytoin has been shown to be effective in terminating generalized status epilepticus in 41% to 90% of patients in five uncontrolled retrospective studies.65–69 Phenytoin’s main anticonvulsant mechanism of action involves blocking voltage-sensitive, use-dependent sodium channels.70 However, the main disadvantages of this drug involve the limitation of rapid administration because of phenytoin itself and the propylene glycol diluent. The maximum rate of infusion is 50 mg/minute, which in a 70-kg individual would take nearly 30 minutes to administer. In addition, significant hypotension and arrhythmias can occur in elderly patients and in patients with known cardiac disease.70 Phlebitis and local irritation can also occur with intravenous administration, which further limits timely use in status epilepticus. In 1996, the U.S. Food and Drug Administration approved fosphenytoin for the treatment of status epilepticus. Fosphenytoin is a water-soluble prodrug of phenytoin that converts to phenytoin in 8 minutes.71–73 Approximately 1.5 mg of fosphenytoin is equivalent to 1 mg of phenytoin, and the dosage is expressed as phenytoin equivalents. Fosphenytoin can be infused at 150 mg phenytoin equivalents per minute. However, therapeutic concentrations of phenytoin (≥1 mg/L unbound) are attained within 10 minutes when either fosphenytoin or phenytoin is administered at maximal infusion rates. The recommended initial loading dosage of phenytoin is 15 to 20 mg/kg (no faster than 50 mg/minute) to a total maximum of 30 mg/kg. Fosphenytoin loading dosage is 18 to 20 mg/kg phenytoin equivalents administered at 150 mg/minute.64 The dosages are the same in children. In some patients, up to 30 mg/kg may be necessary to stop the seizures, but additional phenytoin/fosphenytoin has not proved helpful and in fact may be epileptogenic.74 Thus, either agent phenytoin or fosphenytoin may have a similar time to onset of effect.73 However, the significant advantage of fosphenytoin is the fewer local side effects (pain at injection site, phlebitis) that result in fewer infusion rate decreases, interruptions, or change of site.72 In addition, infusion of fosphenytoin at 150 mg/minute resulted in some decline in blood pressure, but this was not judged to be clinically significant, and so no adjustment of infusion rates was required. In addition, no significant cardiac arrhythmias have been associated with fosphenytoin.73 Rate of administration, however, should be slowed down regardless of which agent is used if hypotension, arrhythmias, or prolongation of the QT interval on electrocardiogram should occur.
Phenobarbital
Although many actions at the cellular level have been reported for phenobarbital, evidence suggests that the main anticonvulsant effect is enhancement of GABA-mediated inhibition.75 Phenobarbital can be given at a loading dosage of 20 mg/kg in adults and children at a maximal rate of 100 mg/minute.63,64 However, respiratory suppression, along with hypotension, can occur with phenobarbital infusion, particularly if benzodiazepines were administered previously. Thus, intravenous phenobarbital loading requires careful observation for hypotension and usually should be performed with the patient already intubated for airway support.
REFRACTORY STATUS EPILEPTICUS
There is no general consensus regarding the definition of what constitutes refractory status epilepticus. There is discrepancy between duration of seizure activity and number of failed medications. As a general guideline, status epilepticus that is not responsive to the measures described previously (benzodiazepine, phenytoin/fosphenytoin, phenobarbital) should be considered refractory. To date, there are no prospective randomized trials comparing medication choices for refractory status epilepticus. However, in the Veterans Affairs study, nearly 55% of patients who had overt status epilepticus did not respond to first-line therapeutic agents (benzodiazepines, phenytoin, phenobarbital); the cumulative rate of response to a second first-line agent was 7%, and that to a third first-line agent, 2.3%. Addition of phenobarbital for individuals who did not respond to lorazepam and phenytoin therapy was effective in only 5%.60,76 Thus, a case can be made to start early intravenous anesthetic drips in patients who have not responded to initial benzodiazepine and phenytoin/fosphenytoin therapy. Pentobarbital is probably the best-studied intravenous drip used for refractory status epilepticus.77–84 Pentobarbital (sodium-5-ethyl-5,1-methylbutyl barbiturate) binds to the GABAA receptor and thus augments GABAergic transmission, resulting in anticonvulsant and sedative-hypnotic effects.80 Pentobarbital has an onset of action of 15 to 20 minutes and a half-life of 20 to 60 hours.80 Pentobarbital-induced coma has proved effective in treating refractory status epilepticus,78 although depression of cardiac contractility and vascular tone, leading to profound hypotension (necessitating pressor support), are major side effects.80 The protocol adopted by clinicians for pentobarbital-induced coma is based on the standardized protocol of Lowenstein and colleagues.83 Pentobarbital is initially administered at a loading intravenous dose of 5 to 15 mg/kg over an hour, followed by a maintenance infusion of 0.5 to 5 mg/kg/hour, titrated to burst-suppression. To ensure adequate respiration, the patient should be electively intubated before barbiturate induction of coma. Infusion should be slowed down if profound hypotension occurs, with addition of pressor agents if needed.
Midazolam is a water-soluble benzodiazepine with a relatively short half-life of 4 to 6 hours.85 Midazolam has been used in the intensive care unit for sedation and also in the operating room for preoperative induction of anesthesia in patients. Possible anticonvulsant effects were seen in animal studies initially,86–88 and since the early 1990s, a number of small case series and reports have been published about the use of midazolam infusion for refractory status epilepticus.86,89–92 A major disadvantage with midazolam infusion is tachyphylaxis. After 1 to 2 days, the dosage of the infusion must be increased substantially to maintain seizure control.92 With prolonged infusion, the drug accumulates as a result of an increase in elimination half-life, potentially leading to increasing difficulty in returning patients back to alertness.93 Initial studies suggest that midazolam may have fewer hypotensive effects than those observed with high-dose pentobarbital-induced coma.86,90,94 A protocol commonly used to treat refractory status epilepticus with midazolam was developed at the University of California, San Francisco.86 As in pentobarbital infusion, the patient should be intubated and continuous electroencephalographic monitoring should be in place. The initial loading dose is 200 μg/kg as a slow intravenous bolus, followed by 0.75 to 10 μg/kg/minute infusion. Titration of the infusion is based on electroencephalographic monitoring, the primary endpoint being suppression of electrographic seizures. The infusion should be discontinued after 12 to 24 hours of electroencephalographic monitoring and reinstated if seizures recur.86 However, because of the small number of patient studies in case series and reports, and because there have been no randomized prospective clinical trials to date, definite conclusions about dosing and efficacy cannot be drawn. In addition, electrographic endpoints differ among studies (complete suppression on EEG versus suppression of clinical/electrographic seizures) and not all of the investigators used continuous EEGs to monitor patients (and thus potentially missed electrographic seizures).
Propofol (2,6-diisopropylphenol), like midazolam, has been used for the induction and maintenance of anesthesia and also sedation in intensive care units. Propofol activates the GABAA receptor but is also a global central nervous system depressant.95 Propofol has an even shorter duration of action than midazolam because of its highly lipophilic nature and large volume of distribution. Thus, propofol is rapidly taken up by the brain and in turn rapidly eliminated. In contrast to midazolam, there is no accumulation with prolonged infusion. A rare complication that deserves mention is the “propofol infusion syndrome” seen in pediatric patients. This syndrome consists of metabolic acidosis, rhabdomyolysis, and cardiovascular collapse.96 Hence, propofol is contraindicated in children. Stecker and associates97 proposed an initial loading dose of 1 to 2 mg/kg over 5 to 10 minutes, followed by a maintenance infusion of 1 to 15 mg/kg/hour, titrated to induce electrographic burst suppression. It is recommended to slowly taper propofol infusions by 5% of the maintenance infusion per hour to prevent rebound of seizures on discontinuation.97 Multiple small case series advocating propofol’s efficacy in treating refractory status epilepticus have also been cited in the literature.97–105 In one small open-label, randomized study, investigators compared the outcome of patients treated with propofol versus pentobarbital infusion.97 It appeared that the time until attainment of control of refractory status epilepticus was longer (12 minutes) with pentobarbital than with propofol (2.6 minutes). However, propofol was not significantly better than pentobarbital in treatment of refractory status epilepticus in this study.97 One small (14-patient) retrospective comparison of propofol and midazolam infusion for refractory status epilepticus did not reveal differences in clinical and electrographic seizure control.106 Unfortunately, the same problems in drawing conclusions about midazolam’s efficacy also apply to propofol.
As indicated, more work is needed in three areas. First, depth of suppression on EEG and outcome with each of these three medications must be studied. A retrospective chart study by Krishnamurthy and Drislane107 suggested that deeper suppression correlated with better continued seizure control and survival. Second, the optimal electrographic endpoint of treatment also needs to be addressed. There is no consensus on how aggressively to treat persistent epileptiform discharges in those on anesthetic intravenous infusions for refractory status epilepticus. Most epileptologists would not consider PLEDs by themselves to be a manifestation of clinical seizures or status epilepticus. PLEDs have been seen in acute and subacute lesions (herpes simplex virus–related encephalitis, stroke, tumor, head trauma),108 as well as chronically.109 However, whether periodic epileptiform discharges occurring at a rate of 0.5 to 2 per second should be considered an ictal equivalent is still an area of debate. Some clinicians consider PLEDs or periodic epileptiform discharges that are present after a clear-cut clinical or electrographic seizure to be an ictal equivalent and treat them as such. Third, controlled, randomized prospective trials with head-to-head comparison of anesthetic infusions need to be conducted with similar subpopulations of patients.
NEW PHARMACOLOGICAL DEVELOPMENTS
The intravenous formulation of valproic acid has generated much attention as a possible intravenous alternative agent for treatment of refractory status epilepticus. The exact mechanism of action of valproate is unknown. Valproate has been shown to increase brain GABA levels, reduce repetitive high-frequency firing by blocking voltage-sensitive sodium channels, activate calcium-dependent potassium clearance, and possibly decrease levels of excitatory amino acids such as aspartate.110 In reality, the contribution of each of these actions to valproate’s anticonvulsant effect is unknown.
Very little literature exists about the use of intravenous sodium valproate (Depacon) for status epilepticus in adults and children. Depacon was approved in 1996 by the U.S. Food and Drug Administration for treatment of epilepsy with a recommended infusion rate of 20 mg/minute over 60 minutes, with a total maximum daily dose of 60 mg/kg/day. However, several studies have shown good tolerance by patients and relatively infrequent hypotension with more rapid infusion rates of up to 6 mg/kg/minute.111–116 In addition, loading doses achieving therapeutic concentrations with intravenous valproate sodium are about 25 to 30 mg/kg,113 and reports of better efficacy with dosages as high as 30 to 40 mg/kg in children have been noted.117
In one series reported by Uberall and associates,117 41 children with refractory status epilepticus (failure of benzodiazepines, phenytoin, barbiturates) were given an initial loading dose of 20 to 40 mg/kg of intravenous valproate sodium over 1 to 5 minutes, with repetition after 10 to 15 minutes if necessary, followed by continuous intravenous infusion at 5 mg/kg/hour. After a 12-hour seizure free period, infusion was decreased to 1 mg/kg every 2 hours. Uberall and associates noted a response to intravenous valproate sodium (cessation of clinical and electrographic seizures) within 2 to 6 minutes by 65.9% of patients, response within 3 to 10 minutes after the second bolus by another 9.8%, and a response by one patient during the infusion. Overall, 32 (78%) of 41 patients responded to treatment. Success was highest in generalized tonic-clonic status epilepticus and for initial loading dosages between 30 and 40 mg/kg.117 In a retrospective study by Yu and colleagues,118 18 patients with status epilepticus were given an initial loading dose of 25 mg/kg of intravenous valproate at a rate of approximately 3 mg/kg/minute with cessation of seizures within 20 minutes of completion of the infusion. Patients tolerated the infusion well; one patient reported transient tremor afterward. However, this study was limited by design (retrospective), lack of stratification into seizure subtypes, and lack of electrographic confirmation of seizure cessation. A more recent retrospective study in adults of 63 patients revealed an overall efficacy of 63% when valproate was used as the first antiepileptic drug in 14 patients, the second in 20 patients, the third in 19 patients, and the fourth or fifth in 8 patients.119 Of the 63 patients, 3 experienced hypotension with intravenous infusion rates up to 500 mg/minute.
Since 2003, anecdotal case reports of topiramate administered in suspension form (via nasogastric tube) for refractory status epilepticus have been published.120–122 Dosages ranged from 300 to 1600 mg/day.120–122 However, currently no definite conclusions can be made about efficacy. In addition, lack of an intravenous form poses a difficulty for acute administration.
Other experimental medications that have been tried for refractory status epilepticus have included ketamine,123 inhalational anesthetics (desflurane, isoflurane),124–128 and lidocaine.129,130
Ketamine is an intravenous agent with a short half-life of 2 to 3 hours that in animal studies has shown anticonvulsant properties by causing NMDA receptor blockade.131 Isoflurane’s and desflurane’s antiepileptic effects may result from potentiation of GABAA inhibition. Both of these agents necessitate continuous electroencephalographic monitoring for dosage titration. Lidocaine has been tried mainly in Scandinavia and has an anticonvulsant effect primarily through blocking sodium-dependent channels, although the exact mechanism of action is not known.129 However, potential serious cardiac effects and proconvulsive action at higher doses pose serious risk in using this agent for refractory status epilepticus.
One final point in treatment of GCSE involves electrographic assessment of the patient’s condition with medically stabilization (when convulsive activity has ceased). Ongoing NCSE has been observed in patients after GTCS (generalized tonic-clonic seizures) have ceased.132 In a study by DeLorenzo and associates,132 both continuous and intermittent ictal activity in NCSE were picked up within the first hour after GCSE was controlled.132 Therefore, although continuous electroencephalographic monitoring may not be feasible in all centers, a continuous 1-hour “follow-up” EEG, after GCSE has ceased and after initial pharmacotherapy has been initiated, should be obtained.
NONCONVULSIVE STATUS EPILEPTICUS
The discussion up to this point has focused mainly on treatment of GCSE. Overall consensus is to treat GCSE aggressively because of associated high rates of morbidity and mortality. However, the incidence of and rate of mortality from NCSE are not so clear, and thus aggressiveness of therapy has not been uniform. In addition, diagnosis of NCSE itself poses a problem. Underrecognition of the problem can occur because diagnosis is based largely on electrographic data. Investigators have classified comatose patients in intensive care units with incidental findings of electrographic status epilepticus as having NCSE.133 Thus, in an effort to assess the morbidity and mortality in NCSE, Shneker and colleagues134 retrospectively reviewed a computerized database at their institution, identifying 100 consecutive cases of NCSE by the following characteristics: mental status, etiology, presence of complications, and electroencephalographic findings. Patients’ conditions were stratified into three categories according to etiology: acute medical, epilepsy, and cryptogenic. Overall, the rate of mortality was similar to that for GCSE (18%). The authors found the highest rate of mortality to be in the group with acute medical conditions (27%), in contrast to only 3% in the patients with known epilepsy (status epilepticus in patients with epilepsy).134 Again, as with GCSE, etiology was also closely linked to increased rates of mortality.36 In addition, patients with severe mental status impairment and acute complications also had increased mortality rates.134 Thus, the authors concluded that patients with epilepsy as the only cause of NCSE should probably not be routinely treated aggressively with anesthetic coma.134 Other authors have concluded that typical absence status epilepticus and complex partial status epilepticus should be treated with benzodiazepines or an intravenous agent, but anesthetic coma is not recommended. This is in contrast with NCSE in patients with coma, for which a more aggressive approach has been recommended.135 Ultimately, treatment of NCSE needs to be individually tailored to the underlying etiology, associated comorbidity, and individual response to initial first- and second-line therapy.
Arzimanaglou A, Hirsch E, Nehlig A, et al. Epilepsy and neuroprotection: an illustrated review. Epilept Disord. 2002;4:173-182.
Husain AM, Mebust KA, Radtke RA, et al. Generalized periodic epileptiform discharges: etiologies, relationship to status epilepticus, and prognosis. J Clin Neurophysiol. 1999;16:51-58.
Logroscino G, Hesdorffer DC, Cascino GD, et al. Long-term mortality after a first episode of status epilepticus. Neurology. 2002;58:537-541.
Shorvon S. Definition, classification, and frequency of status epilepticus. In: Shorvon S, editor. Status Epilepticus: Its Clinical Features and Treatment in Children and Adults. Cambridge, UK: Cambridge University Press; 1994:21-33.
Treiman DM, Walton NY, Kendrick C. A progressive sequence of electroencephalographic changes during generalized convulsive status epilepticus. Epilepsy Res. 1990;5:49-60.
1 Hunter RA. Status epilepticus: history, incidence and problems. Epilepsia. 1959;60:162-188.
2 Trousseau A. Lecture of clinical medicine. Bazire PV, editor. Lectures on Clinical Medicine Delivered at the Hôtel-Dieu, Paris. vol 1. London: New Sydenham Society; 1868:53-54.
3 Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. JAMA. 1993;270:854-859.
4 Lowenstein DH, Bleck TP, Macdonald RL. It’s time to revise the definition of status epilepticus. Epilepsia. 1999;40:120-122.
5 Treiman DM. Generalized convulsive status epilepticus in the adult. Epilepsia. 1993;34(Suppl 1):S2-S11.
6 Treiman DM, DeGiorgio CM, Salisbury SM, et al. Subtle generalized convulsive status epilepticus. Epilepsia. 1984;25:653.
7 Williamson P. Complex partial status epilepticus. In: Engel J, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven; 1997:681-699.
8 Lowenstein DH, Aminoff MJ. Clinical and EEG features of status epilepticus in comatose patients. Neurology. 1992;42:100-104.
9 Tomson T, Lindom U, Nilsson BY. Nonconvulsive status epilepticus in adults: thirty-two consecutive patients from a general hospital population. Epilepsia. 1992;33:829-835.
10 Tomson T, Svanborg E, Wedlund JE. Nonconvulsive status epilepticus: high incidence of complex partial status. Epilepsia. 1986;27:276-285.
11 Gastaut H. Classification of status epilepticus. Adv Neurol. 1983;34:15-35.
12 Ohtahara S, Ohtsuka Y. Myoclonic status epilepticus. In: Engel J, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven; 1997:725-729.
13 Wieser H-G. Simple partial status epilepticus. In: Engel J, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven; 1997:709-723.
14 Gastaut H, Boudouresques G, Gastaut JL, Michel B, et al. Jacksonian epileptic seizures as inaugural manifestations of sylvian infarctus. In: Boulay G, Moseley IF, editors. Computerized Axial Tomography in Clinical Practice. New York: Springer Verlag; 1977:237-245.
15 Chatrian G, Shaw C, Leffman H. The significance of periodic lateralized epileptiform discharges on EEG: an electrographic, clinical, and pathologic study. Electroencephalogr Clin Neurophysiol. 1964;17:177-193.
16 Naquet R, Franck G, Vigouroux R. Decharge paroxystique du carrefour parieto-temporo-occipital [Paroxysmal discharge from the parieto-temporo-occipital junction]. Zentralbl Neurochir. 1915;4/5:153-180.
17 De Pasquet E, Gaudin E, Bianchi A, et al. Prolonged and monosymptomatic dysphasic status epilepticus. Neurology. 1976;26:244-247.
18 Gastaut H. Aphasia. The sole manifestation of focal status epilepticus [Letter]. Neurology. 1979;29:1638.
19 Hamilton N, Matthews T. Aphasia, the sole manifestation of focal status epilepticus. Neurology. 1979;29:745-748.
20 Vernea J. Partial status epilepticus with speech arrest. Proc Aust Assoc Neurol. 1974;11:223-228.
21 Racy A, Osborn MA, Vern BA, et al. Epileptic aphasia: first onset of prolonged monosymptomatic status epilepticus in adults. Arch Neurol. 1980;37:419-422.
22 Wells CR, Labar DR, Solomon GE. Aphasia as the sole manifestation of simple partial status epilepticus. Epilepsia. 1992;33:84-87.
23 Celesia GG. Prognosis in convulsive status epilepticus. In: Delgado-Escueta A, Wasterlain C, Treiman D, et al, editors. Status Epilepticus: Mechanisms of Brain Damage and Treatment. New York: Raven Press; 1983:55-59.
24 Aicardi J, Chevrie JJ. Convulsive status epilepticus in infants and children: a study of 239 cases. Epilepsia. 1970;11:187-197.
25 Aminoff NJ, Simon RP. Status epilepticus: causes, clinical features and consequences in 98 patients. Am J Med. 1980;69:657-666.
26 Maytal J, Shinnar S, Moshe SL, et al. Low morbidity and mortality of status epilepticus in children. Pediatrics. 1989;83:323-331.
27 DeLorenzo R, Towne AR, Pellock JM, et al. Status epilepticus in children, adults, and the elderly. Epilepsia. 1992;33(Suppl 4):515-525.
28 Towne AR, Pellock JM, Ko D, et al. Determinants of mortality in status epilepticus. Epilepsia. 1994;5:27-36.
29 Dunn DW. Status epilepticus in children: etiology, clinical features, and outcome. J Child Neurol. 1988;3:167-173.
30 Leppik IE. Status epilepticus. Clin Ther. 1985;7:272-278.
31 Hauser WA. Status epilepticus: epidemiology considerations. Neurology. 1990;40(Suppl 2):9-12.
32 Hauser WA. Status epilepticus: frequency, etiology, and neurologic sequelae. In: Delgado-Escueta A, Wasterlain C, Treiman D, et al, editors. Status Epilepticus: Mechanisms of Brain Damage and Treatment. New York: Raven Press; 1983:3-14.
33 Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980′s. Neurology. 1992;43:483-488.
34 Janz D. Conditions and causes of status epilepticus. Epilepsia. 1961;2:170-177.
35 Oxbury JM, Whitty CWM. Causes and consequences of status epilepticus in adults: a study of 86 cases. Brain. 1971;94:733-744.
36 De Lorenzo R, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316-325.
37 Hesdorffer DC, Logroscino G, Cascino G, et al. Incidence of status epilepticus in Rochester, Minnesota, 1965–1984. Neurology. 1998;50:735-741.
38 Shinnnar S, Pellock JM, Moshe S, et al. In whom does status epilepticus occur: Age-related differences in children. Epilepsia. 1997;38:907-914.
39 Lothman EW. Pathophysiology of status epilepticus. J Clin Neurophysiol. 1990;40(Suppl 2):13-23.
40 Simon RP, Aminoff MJ, Benowitz NL. Changes in plasma catecholamines after tonic-clonic seizures. Neurology. 1984;34:255-257.
41 White PT, Grant P, Mosier J, et al. Changes in cerebral dynamics associated with seizures. Neurology. 1961;11:345-361.
42 Fountain NB, Lothman EW. Pathophysiology of status epilepticus. J Clin Neurophysiol. 1995;12:326-342.
43 Bayne LL, Simon RP. Systemic and pulmonary vascular pressures during generalized seizures in sheep. Ann Neurol. 1997;42:588-594.
44 Kreisman NR, Gauthier-Lewis ML, Conklin SG, et al. Cardiac output and regional hemodynamics during recurrent seizures in rats. Brain Res. 1993;626:295-302.
45 Boggs JG, Painter JA, DeLorenzo RJ. Analysis of electrocardiographic changes in status epilepticus. Epilepsy Res. 1993;14:87-94.
46 Meldrum BS, Brierly JB. Prolonged epileptic seizures in primates. Arch Neurol. 1973;28:10-17.
47 Blennow G, Brierley JB, Meldrum BS, et al. Epileptic brain damage: the role of systemic factors that modify cerebral energy metabolism. Brain. 1978;101:687-700.
48 Liu Z, Gatt A, Mikati M, et al. Effect of temperature on kainic acid–induced seizures. Brain Res. 1993;631:51-58.
49 Tomlinson FH, Anderson RE, Meyer FB. Effect of arterial blood pressure and serum glucose on brain intracellular pH, cerebral and cortical blood flow during status epilepticus in the white New Zealand rabbit. Epilepsy Res. 1993;14:123-137.
50 Singhal PC, Chugh KS, Gulatie DR. Myoglobinuria and renal failure after status epilepticus. Neurology. 1978;28:200-201.
51 Seisjo BK, Ingvar M, Folbergrova J, et al. Local cerebral circulation and metabolism in bicuculline-induced status epilepticus: relevance for development of cell damage. In: Delgado-Escueta AV, Wasterlain CG, Treiman DM, et al, editors. Status Epilepticus. New York: Raven Press; 1983:217-230.
52 Meldrum BS. Metabolic factors during prolonged seizures and their relation to cell death. Adv Neurol. 1983;34:261-275.
53 Simon R, Pellock JM, DeLorenzo RJ. Acute morbidity and mortality of status epilepticus. In: Engel J, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven; 1997:741-753.
54 Treiman DM, Meyers PD, Walton NY. DVA Status Epilepticus Cooperative Study Group. Duration of generalized convulsive status epilepticus: relationship to clinical symptomatology and response to treatment. Epilepsia. 1992;33(Suppl 3):66.
55 Treiman DM, Walton NY, Kendrick C. A progressive sequence of electroencephalographic changes during generalized convulsive status epilepticus. Epilepsy Res. 1990;5:49-60.
56 Treiman DM, Meyers PD. VA Epilepsy Cooperative Study Group. Utility of the EEG pattern as a predictor of success in the treatment of generalized convulsive status epilepticus. Epilepsia. 1991;32(Suppl 3):93.
57 Bleck TP. Management approaches to prolonged seizures and status epilepticus. Epilepsia. 1999;40(Suppl 2):S59-S63.
58 Alldredge BK, Gelb AM, Isaacs SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med. 2001;345:631-637.
59 Shaner DM, McCurdy SA, Herring Mo, et al. Treatment of status epilepticus: a prospective comparison of diazepam and phenytoin versus phenobarbital and optional phenytoin. Neurology. 1988;38:202-207.
60 Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792-798.
61 Leppik IE, Derivan AT, Homan RW, et al. Double-blind study of lorazepam and diazepam in status epilepticus. JAMA. 1983;249:1452-1454.
62 Ramsay RE, Hammond EJ, Perchalski RJ, et al. Brain uptake of phenytoin, phenobarbital, and diazepam. Arch Neurol. 1979;36:535-539.
63 Treiman DM. Status epilepticus. In: Resor J, Kutt H, editors. The Medical Treatment of Epilepsy. New York: Marcel Dekker; 1992:183-193.
64 Treiman DM. Current treatment strategies is selected situations in epilepsy. Epilepsia. 1993;34(Suppl 5):S17-S23.
65 McWilliam PKA, Leeds MB. Intravenous phenytoin sodium in continuous convulsions in children. Lancet. 1958;2:1147-1149.
66 Wallis W, Kutt H, McDowell F. Intravenous diphenylhydantoin in treatment of acute repetitive seizures. Neurology. 1968;18:513-525.
67 Wilder BJ, Ramsay RE, Willmore LJ, et al. Efficacy of intravenous phenytoin in the treatment of status epilepticus: kinetics of central nervous system penetration. Ann Neurol. 1977;1:511-518.
68 Cranford RE, Leppik IE, Patrick B, et al. Intravenous phenytoin in acute treatment of seizures. Neurology. 1979;29:1474-1479.
69 Carter CH. Use of parenteral diphenylhydantoin sodium in control of status epilepticus. AMA Arch Neurol Psychiatry. 1958;79:136-137.
70 Glauser T, Graves N. Phenytoin and fosphenytoin. In: Wyllie E, editor. The Treatment of Epilepsy. Philadelphia: Lippincott Williams & Wilkins; 2001:853-868.
71 Allen FH, Runge RW, Legarda S. Safety, tolerance and pharmacokinetics of intravenous fosphenytoin in status epilepticus. Epilepsia. 1995;36(Suppl 4):90-94.
72 Browne TR, Kugler AR, Eldon MA. Pharmacology and pharmacokinetics of fosphenytoin. Neurology. 1996;46(Suppl 1):S3-S7.
73 Ramsey RE, DeToledo J. Intravenous administration of fosphenytoin: options for the management of seizures. Neurology. 1996;46(Suppl 1):S17-S19.
74 Osorio I, Reed RC. Treatment of refractory generalized tonic-clonic status epilepticus with pentobarbital anesthesia after high-dose phenytoin. Epilepsia. 1989;30:464-471.
75 Prichard J, Ransom B. Phenobarbital: mechanism of action. In: Levy R, Mattson R, Meldrum B, editors. Antiepileptic Drugs. New York: Raven Press; 1995:359-369.
76 Treiman DM, Walton NY, Collins JF, et al. Treatment of status epilepticus if first drug fails [Abstract]. Epilepsia. 1999;40(Suppl 7):243.
77 Van Ness PC. Pentobarbital and EEG burst suppression in treatment of status epilepticus refractory to benzodiazepines and phenytoin. Epilepsia. 1990;31:61-67.
78 Yaffe K, Lowenstein DH. Prognostic factors of pentobarbital therapy for refractory generalized status epilepticus. Neurology. 1993;43:890-895.
79 Orlowski JP, Erenberg G, Lueders H, et al. Hypothermia and barbiturate coma for refractory status epilepticus. Crit Care Med. 1984;12:367-372.
80 Rashkin MC, Youngs C, Penovich P. Pentobarbital treatment of refractory status epilepticus. Neurology. 1987;37:500-503.
81 Partinen M, Kovanen J, Nilsson E. Status epilepticus treated by barbiturate anesthesia with continuous monitoring of cerebral function. BMJ (Clin Res Ed). 1981;282:520-521.
82 Young GB, Blume WT, Bolton CF, et al. Anesthetic barbiturates in refractory status epilepticus. Can J Neurol Sci. 1980;7:291-292.
83 Lowenstein DH, Aminoff MJ, Simon RP. Barbiturate anesthesia in the treatment of status epilepticus. Neurology. 1988;38:395-400.
84 Mirski MA, Williams MA, Hanley DF. Prolonged pentobarbital and phenobarbital coma for refractory generalized status epilepticus. Crit Care Med. 1995;23:400-404.
85 Reves JG, Fragen RJ, Vinik R, et al. Midazolam: pharmacology and uses. Anesthesiology. 1985;62:310-324.
86 Parent JM, Lowenstein DH. Treatment of refractory generalized status epilepticus with continuous infusion of midazolam. Neurology. 1994;44:1837-1840.
87 Domino EF. Comparative seizure inducing properties of various cholinesterase inhibitors: antagonism by diazepam and midazolam. Neurotoxicology. 1987;8:113-122.
88 Raines A, Henderson TR, Swinyard EA, et al. Comparison of midazolam and diazepam by the intramuscular route for the control of seizures in a mouse model of status epilepticus. Epilepsia. 1990;31:313-317.
89 Crisp CB, Gannon R, Knauft F. Continuous infusion of midazolam hydrochloride to control status epilepticus. Clin Pharmacol. 1988;7:322-324.
90 Kumar A, Bleck TP. Intravenous midazolam for the treatment of refractory status epilepticus. Crit Care Med. 1992;20:483-488.
91 Cortina J, Ancillo P, Duarte J, et al. Intravenous midazolam suppression of complex partial status refractory to intravenous phenytoin and diazepam. Clin Neuropharmacol. 1993;16:468-470.
92 Noritoku DK, Sinha S. Prolongation of midazolam half-life after sustained infusion for status epilepticus. Neurology. 2000;54:1366-1368.
93 Malacrida R, Fritz ME, Suter PM, et al. Pharmacokinetics of midazolam administered by continuous venous infusion to intensive care patients. Crit Care Med. 1992;20:1123-1126.
94 Rivera R, Segnini M, Baltodano A, et al. Midazolam in the treatment of status epilepticus in children. Crit Care Med. 1993;21:991-994.
95 Borgeat A, Wilder-Smith OHG, Suter PM. The non-hypnotic therapeutic applications of propofol. Anesthesiology. 1994;80:642-656.
96 Vasile B, Rasulo F, Candiani A, et al. The pathophysiology of propofol infusion syndrome: a simple name for a complex syndrome. Intensive Care Med. 2003;29:1417-1425.
97 Stecker MM, Kramer TH, Raps EC, et al. Treatment of refractory status epilepticus with propofol: clinical and pharmacokinetic findings. Epilepsia. 1998;39:18-26.
98 Yanny HF, Christmas D. Propofol infusions for status epilepticus [Letter]. Anesthesia. 1988;43:514.
99 Wood PR, Browne GPR, Pugh S. Propofol infusion for the treatment of status epilepticus. Lancet. 1988;1:480-481.
100 Chilvers CR, Laurie PS. Successful use of propofol in status epilepticus. Anesthesia. 1990;45:995-996.
101 Mackenzie SJ, Kapadia F, Grant IS. Propofol infusion for control of status epilepticus. Anesthesia. 1990;45:1043-1045.
102 Alia G, Natale E, Mattaliano A, et al. On two cases of status epilepticus treated with propofol [Abstract]. Epilepsia. 1991;32(Suppl 1):77.
103 Borgeat A, Wilder-Smith OH, Jallon P, et al. Propofol in the management of status epilepticus: a case report. Intensive Care Med. 1994;20:148-149.
104 Pitt-Miller PL, Elcock BJ, Maharaj M. The management of status epilepticus with a continuous propofol infusion. Anesth Analg. 1994;78:1193-1194.
105 Hantson P, VanBrandt N, Verbeeck R, et al. Propofol for refractory status epilepticus. Intensive Care Med. 1994;20:611-612.
106 Prasad A, Worrall B, Bertram E, et al. Propofol and midazolam in the treatment of refractory status epilepticus. Epilepsia. 2001;42:380-386.
107 Krishnamurthy K, Drislane F. Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory status epilepticus. Epilepsia. 1999;40:759-762.
108 Chatrian GE, Shaw CM, Leffman H. The significance of periodic, lateralizing epileptiform discharges in EEG: an electrographic, clinical, and pathological study. Electroencephalogr Clin Neurophysiol. 1964;17:177-193.
109 Westmoreland BF, Klass DW, Sharbrough FW. Chronic periodic lateralized epileptiform discharges. Arch Neurol. 1986;43:494-496.
110 Bourgeois B. Valproate. In: Wyllie E, editor. The Treatment of Epilepsy. Philadelphia: Lippincott Williams & Wilkins; 2001:843-851.
111 Ramsey RE, Cantrell D, Collins SD, et al. Safety and tolerance of rapidly infused Depacon. A randomized trial in subjects with epilepsy. Epilepsy Res. 2003;52:189-201.
112 Sinha S, Naritoku DK. Intravenous valproate is well tolerated in unstable patients with status epilepticus. Neurology. 2000;55:722-724.
113 Venkataraman V, Wheless JW. Safety of rapid intravenous infusion of valproate loading in epilepsy patients. Epilepsy Res. 1999;35:147-153.
114 Wheless J, Venkataraman V. Safety of high intravenous valproate loading doses in epilepsy patients. J Epilepsy. 1998;11:319-324.
115 Naritoku DK, Mueed S. Intravenous loading of valproate for epilepsy. Clin Neuropharmacol. 1999;22:102-106.
116 Wheless JW, Vazquez BR, Kanner AM, et al. Rapid infusion with valproate sodium is well tolerated in patients with epilepsy. Neurology. 2004;63:1507-1508.
117 Uberall M, Trollman R, Wunsiedler U, et al. Intravenous valproate in pediatric epilepsy patients with refractory status epilepticus. Neurology. 2000;54:2188-2189.
118 Yu K, Mills S, Thompson N, et al. Safety and efficacy of intravenous valproate in pediatric status epilepticus and acute repetitive seizures. Epilepsia. 2003;44:724-726.
119 Limdi NA, Shimpi AV, Faught E, et al. Efficacy of rapid IV administration of valproic acid for status epilepticus. Neurology. 2005;64:353-355.
120 Kahriman M, Minecan D, Kutluay E, et al. Efficacy of topiramate in children with refractory status epilepticus. Epilepsia. 2003;44:1353-1356.
121 Towne AR, Garnett LK, Waterhouse EJ, et al. The use of topiramate in refractory status epilepticus. Neurology. 2003;60:332-334.
122 Bensalem MK, Fakhoury TA. Topiramate and status epilepticus: report of three cases. Epilepsy Behav. 2003;4(6):757-760.
123 Sheth R, Gidal B. Refractory status epilepticus: response to ketamine. Neurology. 1998;51:1765-1766.
124 Mirsattari S, Sharpe M, Young B. Treatment of refractory status epilepticus with inhalational anesthetic agents isoflurane and desflurane. Arch Neurol. 2004;61:1254-1259.
125 Kofke WA, Young RSK, Davis P, et al. Isoflurane for refractory status epilepticus: a clinical series. Anesthesiology. 1989;71:653-659.
126 Kofke WA, Bloom MJ, Van Cott A, et al. Electrographic tachyphylaxis to etomidate and ketamine used for refractory status epilepticus controlled with isoflurane. J Neurosurg Anesthesiol. 1997;9:269-272.
127 Hughes DR, Sharpe MD, McLachlan RS. Control of epilepsia partialis continua and secondary generalized status epilepticus with isoflurane. J Neurol Neurosurg Psychiatry. 1992;55:739-740.
128 Sharpe MD, Young GB, Mirsattari S, et al. Prolonged desflurane administration for refractory status epilepticus. Anesthesiology. 2002;97:261-264.
129 De Giorgio C, Altman K, Hamilton-Byrd E, et al. Lidocaine in refractory status epilepticus: confirmation of efficacy with continuous EEG monitoring. Epilepsia. 1992;33:913-916.
130 Pascual J, Sedano M, Polo J, et al. Intravenous lidocaine for status epilepticus. Epilepsia. 1988;29:584-589.
131 Lodge D, Johnson KM. Noncompetitive excitatory amino acid receptor antagonists. Trends Pharmacol Sci. 1990;11:81-86.
132 DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia. 1998;39:833-840.
133 Towne AR, Waterhouse EJ, Boggs JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology. 2000;54:340-345.
134 Shneker B, Fountain N. Assessment of acute morbidity and mortality in nonconvulsive status epilepticus. Neurology. 2003;61:1066-1073.
135 Walker MC. Diagnosis and treatment of nonconvulsive status epilepticus. CNS Drugs. 2001;15:931-939.