[level-membership-for-critical-care-medicine-category]
20 Management of Shock
Introduction
Shock is an altered physiological state that affects the functioning of every cell and organ system in the body. It is a complex syndrome reflecting changing blood flow to body tissues with accompanying cellular dysfunction and eventual organ failure.2,3 Shock presents as a result of impaired nutrient delivery to the tissue:
While the cause of shock may be multifactorial, treatment focuses on optimising tissue perfusion and oxygen de-livery. Shock is often classified according to the primary underlying mechanism: a disruption of intravascular blood volume, impaired vasomotor tone or altered cardiac contractility.5 The shock syndrome is one of the most pervasive manifestations of critical illness present in intensive care patients.
Early detection and management of shock to reverse pathological processes improves patient outcomes.6 Although the traditional hallmark of shock is hypotension (SBP <90 mmHg) this can be a late or misleading sign and is considered a medical emergency.7 It is therefore critical that other signs and symptoms are identified early by frequent observations to detect a patient’s deteriorating state and respond before irreversible shock ensues.8 No one vital sign is adequate in determining the level or extent of shock6 nor is there a specific laboratory test which diagnoses the shock syndrome.
Pathophysiology
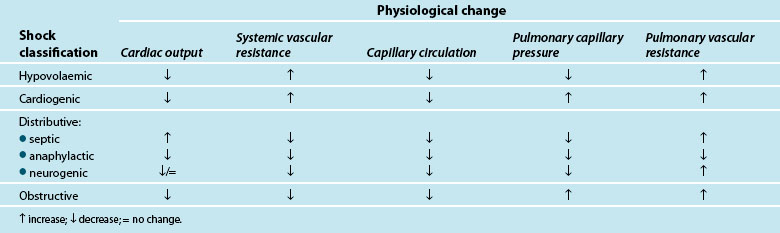
Traditionally, shock is classified by aetiology: hypovolaemic, cardiogenic and distributive.3,4,9 Each has a specific mechanism of action that leads to altered tissue perfusion and oxygen and nutrient uptake at the cellular level (see Table 20.1). In practice, it is common to find overlap between different shock types (e.g. in sepsis there may also be hypovolaemia and/or myocardial dysfunction).
| Shock type | Main characteristic |
|---|---|
| Hypovolaemic | a reduction in circulating blood volume through haemorrhage or dehydration or plasma fluid loss |
| Cardiogenic • obstructive shock |
pump failure (impaired cardiac contractility) usually as result of myocardial infarction a sub category of cardiogenic shock characterised by blockage of circulation to the tissues by impedance of outflow or filling in the heart (e.g. due to cardiac tamponade or pulmonary emboli) |
| Distributive shock | a maldistribution of circulation from sepsis, anaphylaxis or neurogenic injury |
Shock occurs when there is an inability of the body to meet metabolic demands of the tissues; hypoperfusion (decreased blood flow to the tissues) results in cellular dysfunction, as there is homeostatic imbalance between nutrient supply and demand,4,10 and adaptive responses can no longer accommodate circulatory changes. These adaptive responses are moderated via numerous ‘sensors’ throughout the thorax and large vessels in particular, which detect subtle changes in pressure (baroreceptors) or biochemical changes (chemoreceptors). These receptors feed back to the hypothalamus which regulates through the pituitary gland (for the release of a number of hormones such as antidiuretic hormone [ADH] and adrenocorticoid trophic hormone [ACTH] to target organs such as the kidney) and the cortex of the adrenal gland to respond and counter the developing effects of shock. Concurrently direct feedback stimulates the sympathetic nervous system to act on blood vessel tone, particularly the arterioles, and also target organs such as the adrenal gland and kidney to respond via the release of endogenous catecholamines (adrenaline and noradrenaline), mineral and glucocorticoids (aldosterone, cortisol), and the renin–angiotensin–aldosterone system (RAAS). RAAS activation results in synthesis of angiotensin II, a powerful vasoconstrictor that further potentiates the reduction in peripheral blood vessel capacity.
As adaptive responses fail, cardiac output becomes insufficient to provide adequate organ perfusion despite increasing tissue oxygen consumption (see Chapters 9 and 10). When oxygen is ‘supply dependent’, oxygen delivery is decreased and, to compensate, increased extraction occurs to enable continued tissue consumption. However, when oxygen delivery falls below a critical threshold, and extraction demand rises above the available blood oxygen levels, this compensation mechanism fails and oxygen debt results.6,11,12
Hypoperfusion may also exist despite a relatively normal cardiac output, and may not be immediately evident clinically.6 This maldistribution of bloodflow to some tissues while other areas receive more blood flow than needed,4,7,10,13,14 is often referred to as distributive shock, and is typical of the shock types that affect vasomotor tone (e.g. septic, neurogenic and anaphylactic shock). This maldistribution may leave some organ systems ischaemic for long periods leading to persistent organ dysfunction and failure.6 There is also evidence supporting the presence of cytopathic hypoxia as a result of excessive nitric oxide and tumour necrosis factor-alpha (TNFα) production (cellular proinflammatory mediators), where there is impaired mitochondrial (the powerhouse of the cell) oxygen utilisation which leads to depleted stores of adenosine tri-phosphate (ATP)4,11,13,15,16 and interferes with electron transport and metabolism16 (see Chapter 19). Nitric oxide is associated with vascular relaxation and is a major contributor to alterations in microvasculature and capillary leak in sepsis.17
Organ systems have varying responses in shock and are not measured directly. Often surrogate markers of global hypoperfusion are used to indicate the severity of shock.18–19 Lactate and acid–base disturbances, such as an increase in strong ion gap, have been suggested as early markers of mitochondrial dysfunction and cellular hypoperfusion.8,20 These ‘surrogate’ biochemical markers of hypoperfusion (pH, serum lactate and standard base excess) assess acidaemia and provide some insight into the degree of shock present.21 Lactate, a strong anion with normal production of 1500–4500 mmol/day, is a product of carbohydrate metabolism. Increased levels are present in tissue hypoxia, hypermetabolism, decreased lactate clearance, inhibition of pyruvate dehydrogenase and activation of inflammatory cells; all characteristics of developing shock (see Table 20.2). Increased lactate production is a warning sign of impending organ failure, as it is indicative of anaerobic metabolism. Blood lactate levels have been directly linked to deteriorating patient outcomes in shock.21,22
TABLE 20.2 Lactate production9
| Lactate production | |
| Product of carbohydrate metabolism (1400–4500 mmol/day) | Glucose, glycolysis; pyruvate, lactate |
| Rise in lactate levels | |
| Tissue hypoxia | |
As the shock state deteriorates and the body fails to compensate, organ systems begin to fail. This is complicated by a systemic inflammatory response (SIRS) which can be a direct cause of the shock state (see section on Distributive shock) or develop as a consequence of protracted shock. This results in ‘capillary leak’ or increased microvascular permeability which leads to interstitial oedema as a consequence of alterations to tissue endothelium. Many immune mediators including circulating cytokines, oxygen free-radicals and activated neutrophils alter the structure of the endothelial cells, creating space to allow larger intravascular molecules to cross into the extravascular space,23 with proteins and water moving from the intravascular space into the interstitium.24 This response mechanism improves the supply of nutrient-rich fluid to the site of local injury, however, systemically, fluid shifts lead to hypovolaemia, impaired organ function and development of acute organ injury such as acute lung injury (ALI) and acute kidney injury (AKI).24 This developing organ injury is the precedent to organ failure (more fully described in Chapter 21).
Patient Assessment
Critically ill patients often exhibit signs of tissue hypoxia as a result of cardiovascular disturbances.25 Table 20.3 provides an overview of the physiological changes in shock. Therapy is targeted to maintain oxygen delivery (DO2) to vital organs to prevent ischaemia and cell death.25,26 Ideally, organ systems and tissues should be monitored individually,25 however global measures such as perfusion pressure, cardiac output (CO) and DO2, are commonly used as surrogates to assist in treatment decision making.19 Patient assessment and haemodynamic monitoring, including calculation of CO, are used to differentiate shock states and assess progress in relation to treatment.26–28 CO is seen by many clinicians as an important assessment of shocked patients as it is a major determinant of DO2.25,26 Critically ill patients are frequently assessed clinically, although cardiac output estimations from physical examination are generally unreliable and patient status may change quickly.29 Therefore invasive techniques are most commonly used in critical care to measure CO (see also Chapter 9).
Non-Invasive Assessment
Perfusion status is determined clinically using gross organ function such as mental status, urine output and peripheral warmth and colour.6 Basic physical assessment of cardiovascular, central nervous system and renal function are essential when assessing a patient at risk of shock. Subtle changes in urine output, heart rate and capillary refill are all signs of physiological compensation in response to altered tissue perfusion associated with shock. Regular tracking of these vital signs and trend monitoring through careful documentation can alert clinicians to impending deterioration in the shock state. Level of consciousness may deteriorate; an early sign may be anxiety, and progress to restlessness, agitation or coma. Other assessment findings include cool, clammy skin, postural hypotension, tachycardia and decreased urine output.3 The reliability of these measures is questionable, particularly where multiple assessments by different clinicians are performed; in the ICU continuous ECG monitoring and invasive monitoring techniques are employed to assist in the objective assessment of changes in cardiovascular state.
Although CO estimations based on physical assessment findings are unreliable, physical examination using an estimation of vascular resistance has shown reasonable accuracy.30 Clinical assessment may determine CO using the rearranged equation of systemic vascular resistance (SVR = MAP − CVP/CO) where vascular resistance is measured through peripheral skin temperature changes.30 A reliable and accurate non-invasive clinical assessment technique of estimating cardiac output would be clinically useful27 allowing assessment of patients without invasive monitoring, or used to verify accuracy from invasive devices. While a number of non-invasive cardiac output measuring devices are available, further research and refinement is required before widespread application is considered in critical care.31
Invasive Assessment
The indicator dilution method using a thermal (thermodilution) signal (cold or hot) is the customary clinical standard for measuring CO26 in ICU. This is usually achieved by placement of a pulmonary artery catheter (PAC), or a central line in conjunction with a thermistor-tipped arterial cannula (transpulmonary aortic thermodilution). Other invasive techniques measure CO continuously using pulse contour or arterial pressure analysis and ultrasound doppler methods use an oesophageal probe. All methods have degrees of invasiveness, can be time-consuming to yield measurements of acceptable accuracy32, may be expensive and are not without risk of complications.27,33 The PAC is a controversial assessment tool26,28,33 due to the risk associated with the invasive line versus benefits for the measurement of CO34. This has led to increased interest in less or non-invasive measures of CO.
A further invasive assessment approach is the continuous estimation of mixed venous oxygen saturation using a light-emitting sensor in a PAC. As tissue oxygen delivery fails to meet demand and oxygen extraction rises, the residual oxygen content of blood returning to the lungs will fall; in effect a surrogate indicator of failure to meet body tissue oxygen demand. This technology was used in the landmark study by Rivers and colleagues.35 to monitor early deterioration of septic shock patients presenting to the ED in need of resuscitation and was part of a goal-directed approach to managing patients. This single-centre US study has been the subject of much interest for its claimed improvement in patient outcome, with this goal-directed approach being assessed in a major multicentre study in an effort to verify its findings within an international context and varying approach to critical care delivery.36
Management Principles
Managing a patient in shock focuses on treating the underlying cause, and restoration and optimisation of perfusion and oxygen delivery; this includes relevant activities using the acronym VIP37 (see Box 20.1). It is also suggested that giving critically ill patients a daily ‘FASTHUG’ improves the quality of care for patients in ICU.38 Specific management of patients with shock are discussed separately below depending on the cause.
Practice tip
Feeding (prevent malnutrition, promote adequate caloric intake)
Analgesia (reduce pain, improve physical and psychological wellbeing)
Sedation (titrate to the 3Cs – calm, cooperative, comfortable)
Thromboembolic prophylaxis (prevent DVT)
Head of bed elevated (up to 45° to reduce reflux and VAP)
Hypovolaemic Shock
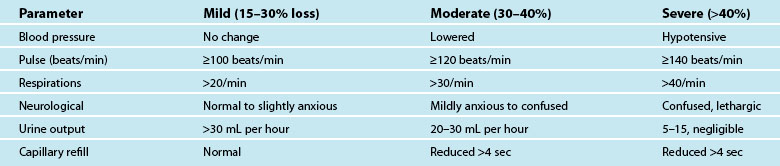
Hypovolaemia is a common primary cause of shock and also a factor in other shock states. Insufficient circulating blood volume is the underlying mechanism, leading to decreased cardiac output and altered perfusion.39,40 Death related to haemorrhage is most likely in the first few hours after injury.40 The most obvious cause is direct injury to vessels leading to haemorrhage, but there are more insidious causes such as dehydration from prolonged vomiting or diarrhoea, sepsis and burns.41 Hypovolaemic shock is classified as mild, moderate or severe, depending on the amount of volume loss (see Table 20.4). As the shock state worsens, associated compensatory mechanisms will be more pronounced,3 and hypovolaemic shock may deteriorate to Multi Organ Dysfunction Syndrome (MODS) if poor oxygen delivery is prolonged39 (see Chapter 21).
Clinical Manifestations
Symptoms of haemorrhage may not be present until more than 15–30% of blood volume is lost, and will deteriorate as the shock state worsens.3,41 Estimating blood or plasma loss is difficult and dilutional effects of resuscitation fluids may be evident when assessing haemoglobin and hematocrit.41 As the body compensates for the reduced circulating volume, widespread vasoconstriction occurs in most body systems apart from the heart and CNS; SVR rises markedly in an attempt to retain a viable circulatory system (this accounts for many of the signs and symptoms associated with circulatory compensation). However, as tissues are starved of oxygen and nutrients over a prolonged ischaemic time, local mediators are released as part of the inflammatory responses, leading to organ microvasculature vasodilation and capillaries re-open to maintain oxygen delivery and reduce hypoxia.41 This is a hallmark of developing MODS.
Nursing Practice
Clinical management of hypovolaemia centres on minimising fluid loss and rapid restoration of circulating blood volume41 once the airway and breathing are secure. More than one large-bore intravenous cannulae are usually inserted and lost circulating volume is replaced by colloids, isotonic crystalloids or blood products to achieve haemodynamic endpoints (e.g. MAP >65 mmHg). Body heat can be lost rapidly due to blood loss, the rapid infusion of room temperature fluids and exposure in the pre-hospital setting or during repeated physical examination. It is therefore important to institute measures to maintain patient temperature >35°C to avoid coagulopathies and loss of thermoregulation.42 The aim is to ameliorate the lethal triad of anaemia, coagulopathy and hypothermia.40–42
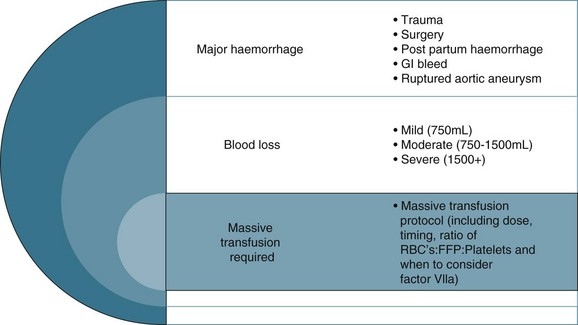
Debate surrounds early surgical intervention prior to aggressive fluid resuscitation.40 The premise is that allowing a lower perfusion pressure prior to achieving haemostasis with controlled or no fluid infusion results in less blood loss, due to the compensatory mechanisms described above.40 Use of medications such as Factor IVa and EPO also remains controversial in the setting of critical haemorrhage.42 Guidelines for ‘massive transfusion’ currently being finalised by the National Blood Authority (NBA) do not recommend use of Factor IVa beyond licensed indications, although there may be an indication when conventional therapy has failed to secure haemostasis following massive blood loss and transfusion of blood products. The current debate also includes dosage and thromboembolic complications associated with its use.42 The NBA is a statutory agency established in 2003 to improve and enhance the management of the Australian blood banks and plasma product sector. Current initiatives of the agency include development and promulgation of evidence based guidelines for both massive transfusion and intensive care.
Fluid resuscitation
Fluid resuscitation is a first-line treatment for hypovolaemic shock; providing fluid volume increases preload and therefore cardiac output (Starling’s law) and organ perfusion. A related principle is that the fluid infused should reflect fluid loss, e.g. plasma replacement in burns, fresh blood in massive haemorrhage. Giving a ‘fluid challenge’ is not always appropriate; the determining factors will be assessment of volume responsiveness, and whether the infusion will not be deleterious, causing overload, fluid shifts and perpetuating inflammatory responses.6 The fluid type, volume, rate and targeted endpoints is documented;41 often this is structured as a bolus dose in volume/kg to achieve a measured haemodynamic variable. When massive transfusion is required, attention should be given to product selection and hence a protocol can be employed.
Independent Practice
Critical care nurses must be efficient and practised at initial patient assessment to establish the degree of compensation occurring in a hypovolaemic patient. Figure 20.1 highlights clinical manifestations of haemorrhage. Careful consideration of a patient’s clinical picture will establish a hierarchy and priority of needs. Most hospitals have some level of track and trigger response that escalates care to appropriate levels (e.g. MET calling criteria), however nurses are in a position to establish first line management such as intravenous access where this is a required skill. There are also many examples of protocols and guidelines for nurses to initiate fluid resuscitation where a patient has indications of inadequate circulating blood volume; e.g. a fluid bolus up to 20 mL/kg of colloidor 30–40 mL/kg crystalloid may be recommended (depending on organisational guidelines).
Collaborative Management
Selection of the appropriate fluid indications for surgical management and ‘permissive hypotension’ (deliberate limiting or minimising resuscitation until after adequate surgical control of haemorrhage).40,42 will be assessed by the multidisciplinary team. Goal-directed therapy includes prevention of tissue hypoxia, typically through rigorous fluid resuscitation with either crystalloids or colloids to achieve specific haemodynamic endpoints (e.g. a CVP of 8–12 mmHg, MAP >70 mmHg, urine output >0.5 mL/kg/h). Vasopressor and inotrope therapy may be then added to maintain adequate perfusion pressure; noradrenaline is the vasopressor of choice because of vasoconstrictor effects.43
Preload management
The colloid versus crystalloid fluid resuscitation debate (use of albumin-based solutions or colloids) continues despite findings from the SAFE study conducted in Australasia; crystalloids (isotonic saline based solutions) were as effective as colloids for fluid resuscitation.44–46 The scientific rationale for using colloids over crystalloids is to preserve plasma oncotic pressure so as to retain intravascular fluid and minimise oedema. Colloids may also attenuate the inflammatory response.20 If moderate to severe hypovolaemia is suspected then blood is often used to improve oxygen-carrying capacity. Further dilution of blood by volume expanders increases hypoxia (otherwise known as isovolaemic anaemia) and red cells are usually needed. Use of isotonic saline as a volume expander is common, although resuscitation with large volumes of saline solutions can be associated with hyperchloraemic acidosis.40 Blood and blood components are usually considered necessary where patients exhibit signs of moderate to severe haemorrhage (see Figure 20.2). There is no perfect resuscitation fluid, and selection is guided by patient condition and the type of fluid lost.
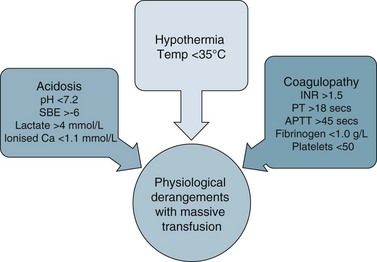
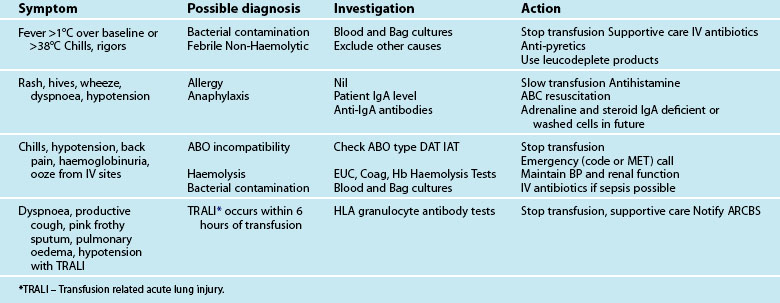
There are a number of factors to consider when administering blood products in massive volume. Massive transfusion is defined as replacement of a patient’s total blood volume in less than 24 hours (approximately 10 units of red cells);47,48 although the literature is inconsistent.48 A number of complications are evident (e.g. transfusion reactions, coagulopathies, hypothermia, sepsis)3 and is associated with high mortality.48 Patients receiving massive blood transfusions require careful monitoring for signs of metabolic derangements, hypothermia, citrate toxicity, hyperkalaemia and coagulopathies (due to depletion of clotting factors). Dilution and clotting factor consumption cause microvascular bleeding, often manifesting as oozing from multiple sites even after surgical correction.47,48 Massive transfusion of stored blood with high oxygen affinity adversely affects oxygen delivery to the tissues. It is therefore preferable to transfuse blood cells that are less than 1 week old; 2,3-diphosphoglycerate levels rise rapidly after transfusion, and normal oxygen affinity is usually restored within a few hours of transfusion.47
Each unit of blood contains approximately 3 g of citrate, which binds to ionised calcium. A healthy adult liver metabolises 3 g of citrate every 5 minutes. If blood is transfused rapidly or the liver is impaired, citrate toxicity and hypocalcaemia may develop. The patient should therefore be monitored for signs of tetany, hypotension and electrocardiographic evidence of hypocalcaemia.47 As stored blood ages, plasma potassium levels rise (possibly to over 30 mmol/L). Hypokalaemia may be more common as red cells begin active metabolism and intracellular uptake of potassium restarts with transfusion.47
Acid–base disturbances may also be evident due to the stored blood lactic acid levels and the citric acid. Citrate metabolises to bicarbonate, and a profound metabolic alkalosis may result from massive blood transfusion. As hypothermia causes reduced citrate and lactic acid metabolism, an increase in the affinity of haemoglobin to oxygen, platelet dysfunction and an increased tendency for cardiac dysrhythmias,47 the patient and the blood transfused should be warmed to avoid complications.
Leucocyte depletion occurs during donation in Australia and decreases up-regulation of the inflammatory immune response associated with transfusion. Current clinical practice guidelines for the administration of blood products and red cells to stable adult patients are listed in Tables 20.5 and 20.6. A new structure with multiple guidelines has been developed and the massive transfusion guideline is complete and will be followed by a number of other specialised guidelines. All guidelines will be available to download from the National Blood Authority website as they are completed (see Online resources).
TABLE 20.5 Clinical practice guidelines for red blood cell and platelet administration
| Appropriate Use of Blood Components For Stable Adults & Children >4 months (corrected) age Adapted from NHMRC/ASBT guidelines (www.anzsbt.org.au) Haemoglobin is NOT the sole deciding factor for transfusion – consider other patient factors e.g. signs of hypoxia and ongoing blood loss. |
|
| Red Cells | |
| Hb | Considerations |
| <70 g/L | Transfusion is often clinically useful unless early Hb recovery is expected. A threshold of <60 g/L may be appropriate for children. |
| 70–100 g/L | Likely to be appropriate during surgery with major blood loss or if there are signs or symptoms of impaired oxygen transport. |
| >80 g/L | May be appropriate to control anaemia-related symptoms in a patient on a chronic transfusion regimen or during marrow suppressive therapy. |
| >100 g/L | Not likely to be appropriate unless there are specific indications |
| WHAT DOSE? Red Cells (mL) = 0.4 × wt (kg) × (desired – actual) Hb (g/L) |
|
| Platelets | |
| Use of platelets is likely to be appropriate as prophylaxis for: | |
| Indication | Considerations |
| Bone Marrow Failure | At a platelet count of <10 Ö 109/L in the absence of risk factors and <20 Ö 109/L in the presence of risk factors (e.g. fever, antibiotics, evidence of haemostatic failure) |
| Surgery/Invasive | To maintain platelet count at >40 Ö 109/L. For surgical procedures with high risk of bleeding (e.g. ocular or neurosurgery) it may be procedure appropriate to maintain at 100 Ö 109/L |
| Platelet Function Disorders | May be appropriate in inherited or acquired disorders, depending on clinical features and setting. In this situation, platelet count is not a reliable indicator |
| Use of platelets is likely to be appropriate as therapy for: | |
| Bleeding | Any patient in whom thrombocytopenia is considered a major contributory factor. |
| Massive Bleeding/Transfusion | Confined to patients with thrombocytopenia and/or functional abnormalities who have significant bleeding. Often with platelet count <50 Ö 109/L (<100 Ö 109/L with diffuse microvascular bleeding). |
Cardiogenic Shock
Cardiogenic shock manifests as circulatory failure from cardiac dysfunction,49 and is reflected in a low cardiac output (CI <2.1 L/min/m2), hypotension (SBP <90 mmHg) and severe pulmonary congestion, high central vascular filling pressures (CVP; PAOP >18 mmHg).50 Additional invasive parameters are: intrathoracic blood volume index >850 mL/m2; global end-diastolic volume >700 mL/m2; and extravascular lung volume index >10 mL/kg.51,52 Cardiogenic shock is commonly associated with AMI and manifests when 40% or more of the left ventricle is ischaemic. It is also related to mechanical disorders (e.g. acute cardiac valvular dysfunction or septal defects), deteriorating cardiomyopathies or congestive cardiac failure,53,54 trauma and obstruction or inhibition of left ventricular ejection (referred to as obstructive shock e.g. pulmonary emboli, dissecting aneurysm, tamponade)37,53 (see Chapter 10). Myocardial depression from non-cardiac causes such as sepsis, acidosis, myocardial depressant factor, hypocalcaemia or drug impact55 may be so severe as to present as cardiogenic shock.
Incidence has been estimated at 3% of patients presenting with AMI, and mortality remains high (50–80%),56 given death from AMI overall is 7%. This is despite treatment advances including emergency revascularisation.57,58 Wider distribution of interventional cardiac revascularisation services has likely improved outcome for patients who present early in the course of their acute disease.
Clinical signs include poor peripheral perfusion, tachycardia and other signs of organ dysfunction such as confusion, agitation, oliguria, cool extremities, dyspnoea, many of which are present in hypovolaemic shock.49 Compensatory mechanisms are conflicting for a patient with cardiogenic shock, as cardiac workload is increased on an already-failing heart yet cardiac muscle oxygen delivery may be compromised.54 A careful but rapid assessment of the clinical history is helpful in differentiating the precipitant cause of this shock.
Clinical Manifestations
The clinical features of cardiogenic shock are reflective of congestive cardiac failure, although with greater severity:50,51,58
• low cardiac output and hypotension
• poor peripheral perfusion: pale, cool, clammy peripheries
• altered mentation, restlessness and anxiety
• pulmonary congestion with widespread inspiratory crackles and hypoxaemia (perhaps with frank pulmonary oedema)
• respiratory alkalosis (hyperventilation) or acidosis (respiratory fatigue)
In the absence of invasive monitoring, the profile of hypotension, peripheral hypoperfusion, and severe pulmonary and venous congestion are evident although this ‘classic’ profile is not universal. On initial examination, 30% of patients with shock of left ventricular aetiology had no pulmonary congestion and 9% had no hypoperfusion.59
Based on the underlying pathology of an acute left ventricular myocardial infarction, the structural or contractile abnormality impairs systolic performance resulting in incomplete left ventricular emptying.50 This results in subsequent progressive congestion of first the left atrium, then the pulmonary circulation, right ventricle, right atrium and finally the venous circulation.50,60,61 When invasive haemodynamic monitoring is available, sequence of changes exist as illustrated in Figure 20.3.
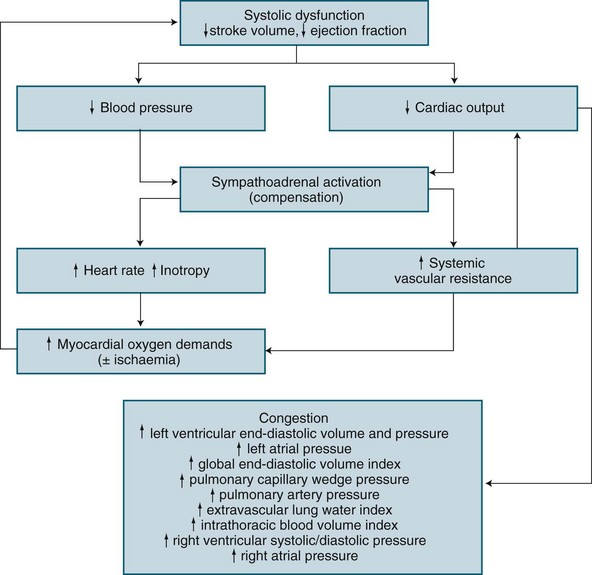
A patient with cardiogenic shock is also assessed and monitored for their oxygen delivery and tissue oxygen requirements (oxygen consumption). Systemic DO2 falls in proportion to a declining cardiac output, and is further worsened as hypoxaemia develops due to pulmonary oedema. Initially, VO2 may be sustained by an increase in tissue oxygen extraction ratio (O2ER).62 Normally 25% of delivered oxygen is extracted by tissues, but as delivery falls, tissues extract proportionally more oxygen to meet metabolic needs. Oxygen consumption can therefore be sustained until the severity of oxygen delivery deficit exceeds the ability to increase extraction. Maximal extraction is approximately 50%, and consumption falters when oxygen delivery falls to around 500–600 mL/min (cardiac index <2.2 L/min/m2).62–64 While use of a PAC is a well-described measure of severity in cardiogenic shock (as with hypovolaemic shock), evidence of improved patient outcome is unclear.28,65
Once oxygen consumption falls below tissue needs, resulting anaerobic metabolism causes lactate generation and the subsequent lactic acidosis.50,62 Progressive tissue ischaemia and injury ensues, along with worsening metabolic acidosis unless oxygen delivery can be restored. Myocardial contractile performance further worsens when myocardial ischaemia develops or when existing ischaemia or infarction is worsened, and a vicious cycle of ischaemia and dysfunction ensues.62
• Tachycardia offsets low stroke volume but increases myocardial oxygen consumption and decreases diastolic duration, reducing coronary perfusion time.
• Vasoconstriction limits the severity of hypotension but increases resistance to left ventricular emptying and may contribute to worsening of the cardiac output, in particular when cardiogenic shock is due to contractile dysfunction.
• An increase in cardiac workload to overcome the rise in systemic afterload increases myocardial oxygen demand, but cannot be met due to coronary artery occlusion.
• Developing pulmonary congestion is no longer contained within the pulmonary capillary and moves into the alveolar capillary space, creating pulmonary oedema, further impeding oxygen delivery to the circulation.
Nursing Practice
Independent Practice
Assessment
Frequent, thorough assessment of the patient’s status is essential, focusing on:
Optimising oxygen supply and demand
• positioning the patient upright to promote optimum ventilation by reducing venous return and lessening pulmonary oedema (but may contribute to worsening hypotension)
• administering oxygen, continuous positive airway pressure (CPAP) and bi-level positive airway pressure (BiPAP) support as required.66
Strategies to reduce oxygen demand include:
• implementing measures to reduce patient anxiety, including communication, explanation and analgesic and sedative medications (avoiding those that are cardio-depressive) where appropriate
• ensuring that visiting practices are appropriate for the patient (which may require facilitating lengthy visits by a loved one, limiting visiting time, or being selective with the visitors who remain with the patient).
Collaborative Management
Typical treatment regimens require preload reduction, augmentation of contractility with intravenous inotropes and afterload manipulation. These aspects are undertaken concurrently due to the potential severity of cardiogenic shock. Endotracheal intubation with mechanical ventilation is implemented if necessary (the need for mechanical ventilation is associated with an increase in mortality)67 (see Chapter 15).
Preload management
Preload reduction relieves pulmonary congestion, reduces myocardial workload and improves contractility, which is in part impaired by overstretched ventricles. Careful assessment of patient fluid status is necessary prior to either the administration of small aliquots of fluid to enhance deteriorating myocardial function or enhanced diuresis to reduce circulating blood volume. Any fluid offloading is balanced against the risk of excessive blood volume depletion and depression of cardiac output and blood pressure.68 Desired endpoints of therapy are a reduction in right atrial, pulmonary artery, and pulmonary artery wedge pressures, or in intrathoracic blood volume, global end-diastolic volume and extravascular lung water, depending on available monitoring equipment. Measures to reduce preload include:
• sitting a patient up with their legs either hanging over the side of the bed or in a dependent position
• IV diuretics (frusemide)68 given usually as intermittent boluses or if necessary as a continuous infusion
• venodilation (glyceryl trinitrate infusions at 10–200 µg/min titrated to blood pressure)69
• continuous haemofiltration (might be considered to rapidly reduce circulating volume)
• continuous positive airway pressure (indicated for pulmonary relief, with the additional benefit of reducing venous return).
Additional measures to reduce pulmonary hypertension may be employed. Morphine is useful to lessen the anxiety and oxygen demands during cardiogenic shock, and may offer additional benefits by reducing pulmonary artery pressure and pulmonary oedema.68 Other treatment options include correction of hypercapnoea if present, and nitric oxide by inhalation.
Inotropic therapy
Intravenous positive inotropes promote myocardial contractility to improve cardiac output and blood pressure. Currently available inotropes are not uniform in their beneficial effect on cardiac output and blood pressure because of additional vasoactive actions (either vasodilation or constriction) (see Table 20.7). Selection of an inotropic agent is therefore partly based on inotropic potency as well as the desired effect on vascular resistance:
• vasodilation in addition to inotropy (inodilator effect) favours cardiac output, but may compromise blood pressure70
• vasoconstriction in addition to inotropy (inoconstrictor effect) improves blood pressure, but may at times compromise left ventricular emptying and cardiac output.
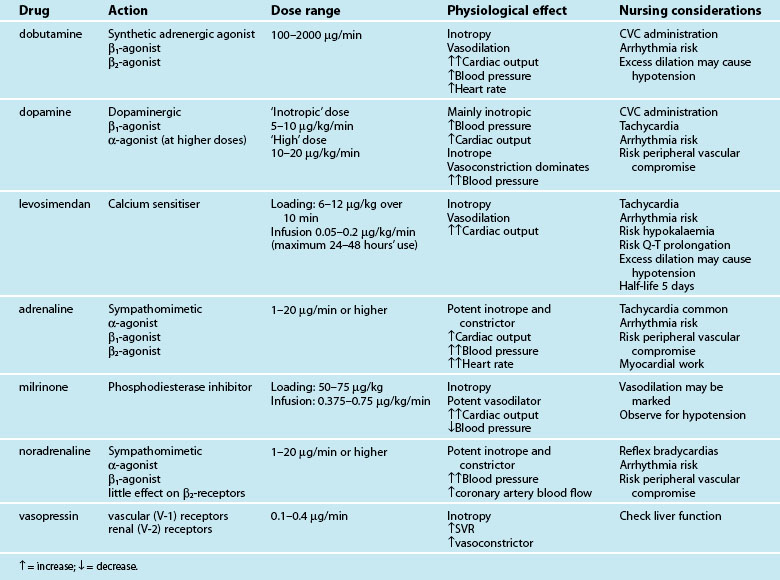
The vasodilation seen with inodilator agents may reduce both preload and afterload, leading to more effective myocardial pumping and an increased cardiac output. The effect on blood pressure is variable, as the opposing actions of increased contractility and vasodilation are not uniform in potency, and occur with differing effects between patients. Inodilators are generally selected when a patient has an elevated afterload and low cardiac output.70 By reducing afterload, left ventricular emptying is favoured with a reduction in cardiac contractility, reducing myocardial oxygen demand. Inodilators are therefore preferred in ischaemic cardiogenic shock.71–73
In contrast, inoconstrictors constrict the vasculature, resulting in increased preload and afterload while also increasing myocardial contractility.70 These increases, particularly in afterload, generally result in a raised blood pressure, but the impact on cardiac output is less predictable. An increase in cardiac output is often seen with these agents, but the increase in afterload may become limiting to left ventricular emptying when there is significant contractile impairment. Inoconstrictors are therefore generally selected when the afterload and resultant blood pressure are more severely compromised than the cardiac output. Vasoconstriction also further increases myocardial work and myocardial oxygen demand, and may worsen ischaemia.74
Dobutamine has traditionally been the inodilator of choice,75 although accumulating evidence for levosimendan, a calcium-sensitising agent, suggests improved outcomes.71,73 However, the slow onset of action time of levosimendan (hours) makes it a less suitable drug for acute resuscitation; other inotropes are therefore currently used initially and if required, levosimendan is then introduced. The long half-life (>5 days) of levosimendan confers a lasting impact on contractility after cessation of the infusion. Milrinone is also an effective inodilator,70 but excessive vasodilation may contribute to significant hypotension; in practice a concurrent vasoconstrictor (e.g. noradrenaline) may be administered. Close management of intravascular fluid volume is critical when using these agents.
Afterload control
Specific management of afterload, independent of contractility, is sometimes necessary, although caution is needed as the maintenance of blood pressure often provides little scope for further afterload reduction. Arteriodilators such as sodium nitroprusside reduce afterload and increase cardiac output, although with limitations due to hypotension.76 The introduction of oral angiotensin-converting enzyme (ACE) inhibitors as soon as possible after stabilisation of the patient with infarct-related cardiogenic shock is strongly recommended.77,78
Adjunctive therapies
Intra-aortic balloon pumping
Low cardiac output, pulmonary congestion, reduced MAP, and myocardial ischaemia from cardiogenic shock may all be improved by the introduction of intra-aortic balloon pump (IABP) therapy (see Chapter 12). Balloon inflation during diastole raises MAP and promotes coronary and systemic blood flow, while balloon deflation in advance of systole reduces afterload. This afterload reduction improves cardiac output and reduces left ventricular systolic pressure, lessening the oxygen demands of the ischaemic ventricle by reducing the necessary contractile force of the left ventricle.
Respiratory support
Varying degrees of pulmonary oedema accompany cardiogenic shock, causing hypoxaemia due to intrapulmonary shunt, decreased compliance and increased work of breathing (WOB). Hyperventilation with respiratory alkalosis may initially compensate for hypoxaemia and lactic acidosis, but fatigue during this increased WOB may cause patient progression to hypoventilation and respiratory acidosis. Oxygen is administered for hypoxaemia, but responses may be limited as the primary gas exchange defect is an intrapulmonary shunt. Non-invasive ventilatory approaches may be sufficient, but a wary eye for the need to intubate and mechanically ventilate should be maintained in the acute phase of treatment. CPAP at conventional levels of 5–15 cmH2O is well established as a support for the spontaneously breathing patient with pulmonary oedema.79 CPAP improves hypoxaemia, lessens WOB, reduces left ventricular afterload and provides additional benefit by impeding venous return, an effect that may lessen pulmonary congestion. These benefits are weighed against the potential for hypotension.
If hypoventilation and dyspnoea continue despite the use of CPAP, non-invasive bi-level positive airway pressure (BiPAP) is considered. Additional pressure support is applied during inspiration, above existing CPAP, improving inspiratory efficiency, with increased tidal volume and less work of breathing.66,80 Endotracheal intubation and ventilation should be undertaken when neither CPAP nor BiPAP result in improvement, or when the patient continues to deteriorate or tire. Many clinicians prefer to intubate and ventilate early, even in the absence of a specific respiratory need, to decrease the cardiovascular demands of the greater ventilatory effort. However this approach is controversial as mechanical ventilation is associated with poorer patient outcomes81 and disturbs cardiovascular balance as it exerts changes to intrathoracic pressures, particularly at inspiratory initiation.
Ventilation strategies largely reflect those for other compliance disorders (e.g. ARDS), and are described in more detail in Chapter 15. Initially, full mechanical ventilation with little or no contribution from the patient is appropriate to correct arterial blood gases and lessen the cardiovascular demands of the ventilatory burden. Subsequent reduction of ventilatory support, as the patient’s respiratory ability improves, follows conventional processes.
Biochemical normalisation
Frequent biochemistry measurement is necessary to detect and monitor the following aspects of care:
• arterial blood gases to identify the adequacy of ventilation and oxygenation and the presence of metabolic acidosis
• lactic acid measurement to assess the level of shock and changes in patient response to treatment
• hypokalaemia or hypomagnesaemia due to aggressive diuretic use
• hyperkalaemia due to severe acidosis, especially in the presence of renal failure
• hyperglycaemia due to the stress response to acute illness, and in response to sympathomimetic administration
• bicarbonate levels decline due to pH buffering, but replacement therapy is not routinely undertaken unless the arterial pH is life-threatening
• urea and creatinine to detect the onset of acute renal failure due to renal hypoperfusion.
Haemofiltration-based therapies (as slow continuous ultrafiltration, continuous veno-venous haemofiltration or haemodialysis) are used for fluid and electrolyte control when renal function suffers or as acute method for unloading fluid from the circulation (see Chapter 18).
Distributive Shock States
Sepsis and Septic Shock
Systemic Inflammatory Response Syndrome (SIRS) was a term developed to describe the clinical manifestations of many processes characterised by systemic inflammation including sepsis, burns, pancreatitis and trauma.82 This definition was however limited and problematic as it described general signs and was non-specific.83,84 Despite a revision in 2001,84 SIRS was viewed as a valid descriptor but not useful for clinical diagnosis in that form. It was however noted that the use of the SIRS definition in sepsis to aid in early identification was important. Signs and symptoms were subsequently added to SIRS in response to infection (sepsis): hyperglycaemia, altered mentation, generalised oedema, as well as a number of inflammatory, haemodynamic, organ dysfunction and tissue perfusion variables. A staging system (PIRO) was also introduced to profile the processes in septic patients85 (see Table 20.8).
| Predisposition | Factors that dispose certain patient groups to be more susceptible to infection and organ dysfunction, including genetic predisposition, age, and comorbidities like alcohol use and diabetes. |
| Infection | Type of infecting organism. How is it diagnosed? How severe is the infection? Is it local or general? What is the site of infection and the related outcomes? Hospital/ICU or community-acquired? |
| Response | Stratify severity, using biomarkers (e.g. IL-6 or procalcitonin) to gauge severity of the inflammatory/immune responses, and to predict how patients will respond and potential outcomes. Also assess ABGs, lactate levels, WBC, temperature, C reactive protein. |
| Organ dysfunction | Describe using either physiological levels or level of intervention. Use scoring systems to quantify level (mild, moderate, severe) and predict outcomes. |
Severe sepsis and septic shock is a leading cause of admission to ICU and has an associated high mortality. The terms ‘severe sepsis’ and ‘septic shock’ were defined and then refined during international consensus meetings that also described SIRS82,84 (see Table 20.9 and Chapter 21). The incidence of severe sepsis in Australia and New Zealand was 11.8% of ICU admissions, with median ICU and hospital stays of 6 days and 18 days respectively, and corresponding mortality rates of 32% at 28 days and an in-hospital mortality of 40%.86 A Victorian epidemiological study reflected similar results.87 More recent Australian data shows mortality remaining relatively high but in decline.36,88 The consequence of this high mortality focused attention on sepsis and its associated sequelae in the critical care literature, and led to a worldwide campaign in 2002 to reduce the mortality from sepsis.
TABLE 20.9 Sepsis, severe sepsis and MODS definitions82,84
| Term | Definition |
|---|---|
| Infection |
• A non-specific syndrome that results from a wide variety of severe clinical insults; present with two or more of the following:
• Other signs include: altered mental status, positive fluid balance or significant oedema, hyperglycaemia in the absence of diabetes, raised procalcitonin and/or C reactive protein, hypotension, hypoxaemia, acute oliguria, raised serum creatinine, coagulation abnormalities, ileus, thrombocytopenia, hyperbilirubinaemia, hyperlactaemia, decreased capillary refill/mottling.
• Systemic inflammatory response to infection. Manifestations of sepsis are the same as defined for SIRS. Determine if symptoms are a result of a direct systemic response to an infectious process and represent an acute alteration from baseline in the absence of other known causes for the abnormalities.
• A subset of severe sepsis; sepsis-induced hypotension (a systolic blood pressure <90 mmHg or a reduction of ≥40 mmHg from baseline) in the absence of other causes, despite adequate fluid resuscitation, and perfusion abnormalities (e.g. lactic acidosis, oliguria, acute alteration in mental status). Patients receiving vasopressor or inotropic agents may not be hypotensive by the time they manifest hypoperfusion abnormalities or organ dysfunction, but are still considered to have septic shock.
• Acute circulatory failure with persistent arterial hypotension unexplained by other causes and despite adequate fluid resuscitation (see also sepsis-induced hypotension).
MODS = multiple organ dysfunction syndrome; SIRS = systemic inflammatory response syndrome.
The Surviving Sepsis Campaign
The Surviving Sepsis campaign is an international collaborative formed after the Barcelona Declaration in 2002 to reduce the mortality of sepsis by 25% over a 5-year period, by increasing awareness and developing treatment guidelines for severe sepsis and shock, including a comprehensive list of graded recommendations.89,90
Various recommendations were combined to form ‘care bundles’ (‘a group of interventions related to a disease process that, when executed together, result in better outcomes than when implemented individually’)91, p.5 and promulgated through professional organisations (e.g. Institute of Healthcare Improvement [IHI]). Bundles have been introduced to change processes of care and as quality or benchmarking measures (see Chapter 3). Although the first version of the sepsis guidelines was supported by ANZICS, the subsequent and much expanded version was not,88 as many of the recommendations were based on research involving non-ICU and/or non-sepsis patients. Further research and evaluation is needed as mortality benefits of ‘care bundles’ may be a result of increased clinician awareness rather than the impact of treatment changes.92
An example of a refuted bundle relates to tight glycaemic control. The recommendation in the surviving sepsis guidelines supported tight glycaemic control and originated from research where the glycaemic control practice differed from Australia and New Zealand.93 The NICE-SUGAR study subsequently concluded that measures to maintain blood glucose level of ≤10 mmol/L increased mortality particularly in relation to severe hypoglycaemia.94 A recent meta analysis of 26 ICU related ‘tight glycaemic control’ studies, suggested that the practice could increase risk to ICU patients.95 The more pragmatic approach of maintaining blood glucose levels close to normal without inducing hypoglycaemia and other metabolic imbalances is therefore appropriate.96 The guideline was subsequently modified in 2009 to include findings from NICE-SUGAR.97
Clinical Manifestations
Septic shock results when infectious agents or infection-induced mediators in the blood stream produce haemodynamic compromise. Primarily a form of distributive shock, it is characterised by ineffective tissue oxygen delivery and extraction associated with inappropriate peripheral vasodilation, despite preserved or increased cardiac output.98 Hypovolaemia is also associated with septic shock due to the characteristic increased vasodilatation. This presents a clinical picture of a warm, pink and apparently well-perfused patient in early stages of septic shock with an elevated cardiac output, in contrast to that seen in hypovolaemic or cardiogenic shock patients.
Unchecked, cellular dysfunction in the presence of a failing compensatory process leads to cellular membrane damage, loss of ion gradients, leakage of lysosomal enzymes, proteolysis due to activation of cellular proteases and reductions in cellular energy stores which may result in cell death. Once enough cells from vital organs have reached this stage, shock becomes irreversible and death can occur despite eradication of the underlying septic focus. About half of the patients who succumb to septic shock die of failure of multiple organs.98
The effect of sepsis and septic shock on the cardiovascular system is profound; the haemodynamic hallmark is generalised arterial vasodilation with an associated decrease in systemic vascular resistance. Arterial vasodilation is mediated in part by cytokines that upregulate the expression of inducible nitric oxide synthase in the vasculature. Vascular response to the vasodilatory effect of nitric oxide and the activation of ATP-sensitive potassium channels combine to cause closure of the voltage-gated calcium channels in the cell membrane. As the vasoconstrictor effect of noradrenaline and angiotensin II depend on open calcium channels, lack of response to these pressor hormones that are central to compensatory mechanisms in shock can occur with the inevitable failure of delivery of oxygen to the functional mitochondria resulting in lactic acidosis in patients with sepsis.99 With high circulating levels of endogenous vasoactive hormones during sepsis, downregulation of their receptors occurs.
Nursing Practice and Collaborative Management
Initial Management: Fluid Resuscitation
Measuring surrogate markers of preload as an indicator of volume status is a contentious issue, as CVP as a measure of preload is not a good marker of volume responsiveness.32,100 While CVP was used in sepsis trials of early goal-directed therapy (EGDT) protocols35,101–103 and is an often documented endpoint of resuscitation, EGDT has been widely discussed and criticised in the literature. Australian data indicates that the incidence of patients meeting the criteria and mortality is lower than the treatment group in the original EGDT trial.36 This is currently the focus of a large trial by the ANZICS Clinical Trials Group (ARISE).36
Fluid resuscitation with crystalloid or colloid has long been controversial in the critical care literature. The landmark Saline versus Albumin Fluid Evaluation (SAFE) study44 demonstrated that in the adult intensive care patient population, albumin can be considered safe, without demonstrating any clear advantage over saline. In the study conducted in 14 Australian and 2 New Zealand ICUs, 6997 patients were randomised to receive either saline (n = 3500) or albumin (n = 3497). No significant differences were noted between the two treatment groups for 28-day all-cause mortality, days in intensive care, days in hospital, days on mechanical ventilation and days of renal replacement therapy.44 The Surviving Sepsis Campaign guidelines do not advocate one preferred resuscitation fluid.89 Irrespective of fluid selection, the disruption of the vascular bed in early septic shock through widespread vasodilatation results in increased capillary permeability and rapidly developing interstitial oedema. Large amounts of fluid can be administered without seemingly improving oxygen delivery whilst adding to developing generalised oedema which further impairs cellular delivery of oxygen and nutrients. Fluid resuscitation alone is therefore of limited value in septic shock and other measures must be considered.
Initial Management: Diagnosis, Source Control and Antimicrobial Therapy
Identifying and removing the source of infection and treating the infection with appropriate antimicrobial therapy are the mainstays of therapy for a patient with sepsis. Australian data indicate that in the ICU setting the most prevalent site of primary infection is pulmonary, followed by abdominal, together accounting for 70% of cases.86 Similar epidemiology is reported in international sepsis studies.104,105 In 2005, ICU pneumonia practices were studied in 14 ICUs and demonstrated a ventilator associated pneumonia (VAP) incidence of 28%.106 A further cohort study comparing Australian and Danish hospitals noted a lower incidence of VAP with a concomitant increase in broad spectrum antibiotics prescribed based on clinical signs and multiresistant organisms at the Australian site.107
To provide patients with appropriate antimicrobial treatment for targeting the infecting organism, obtaining appropriate samples prior to instigating antimicrobial therapy is the clinical standard, although any prescribed treatment should not be delayed as time to antibiotic administration is important in severe sepsis.108 In one large retrospective study, every additional hour to effective antimicrobial initiation in the first 6 hours after onset of hypotension was associated with >7% decrease in survival.108,109 Optimising dosage to achieve a therapeutic concentration is also important. Current practice is to continuously infuse glycopeptides to maintain a serum concentration above the minimum inhibitory concentration and therefore kill microbes more effectively. More recently there has been evidence that β-lactams should also be infused.110 Recently a paradigm shift has been suggested in relation to antimicrobial therapy; to get it right the first time with high doses, while limiting the duration of therapy and the potential to increase resistance.111
Ongoing Collaborative Management: Drug Therapy
Inotropes and vasopressors
A goal of maintaining MAP greater than 65 mmHg is common, with inotropes and vasopressors commenced when fluid resuscitation is considered adequate. Administration of these drugs requires continuous blood pressure monitoring and enables effective titration to meet the treatment goal. Australian practice preferences noradrenaline (for its specific alpha-receptor effects) and adrenaline as the vasopressors of choice. Dobutamine (2.5–10 mcg/kg) is often added to support patients with myocardial dysfunction to increase myocardial contractility and oxygen delivery to the tissues.90 Refractory hypotension, resistant to vasopressors, has been linked to downregulation of receptors. Vasopressin (0.4–0.6 units/hour) has been shown to reduce the requirements of other vasopressor agents.
Administration of arginine vasopressin in vasodilatory shock may help maintain blood pressure despite the relative ineffectiveness of other vasopressor hormones.99 Specifically, arginine vasopressin may inactivate the KATP channels and thereby lessen vascular resistance to noradrenaline and angiotensin II. It also decreases the synthesis of nitric oxide (as a result of a decrease in the expression of nitric oxide synthase) as well as cyclic guanosine monophosphate (cGMP) signalling by nitric oxide.99 The sites of major arterial vasodilation in sepsis – the splanchnic circulation, the muscles and the skin – are vascular beds that contain abundant arginine vasopressin receptors. In sepsis, vasopressin stores are quickly depleted. Administration of exogenous arginine vasopressin (0.04–0.06 units/min) can raise blood pressure by 25–50 mmHg by returning plasma concentrations of antidiuretic hormones to their earlier high levels.99
Steroids
The use of steroid therapy in severe sepsis remains controversial. At times, steroid replacement therapy may be used when patients display resistance to increasing doses of adrenergic agonists, i.e. adrenal insufficiency. Some research indicates that patients with septic shock that are unable to increase cortisol levels in response to a challenge may benefit from administration of low-dose corticosteroids112 (see Chapters 19 and 21 for further information).
Recombinant human activated protein C
• decreased inflammation through reduced levels of TNFα and NFKβ
• decreased thrombin production leading to anticoagulation
• profibrinolytic action through modulation of fibrinolysis inhibitors.
Drotrecogin alfa-activated (rhu), a recombinant form of activated protein C, was developed using DNA recombinant technology as a treatment for sepsis. Previous drug trials had targeted particular aspects of the host response to sepsis but had not yielded positive results. Preclinical studies devised dose-dependent reductions in the markers of fibrinolysis (d-dimer) and inflammation (Interleukin 6) leading to a Phase III clinical trial which resulted in drotrecogin alfa-activated rhu being the first drug approved for the treatment of severe sepsis. The published phase III trial was a large multi-centre, double-blind randomised controlled trial (PROWESS);113 this landmark study was the first drug trial to demonstrate a positive result in the treatment of severe sepsis. PROWESS was conducted in 11 countries with the hypothesis that administration of drotrecogin alfa at 24 mcg/kg/min for 96 hours would reduce 28-day all-cause mortality in patients with severe sepsis, with an acceptable safety profile.
Controversy followed publication of the PROWESS paper and licensing of drotrecogin alfa. A further study, ADDRESS, mandated by the Food and Drug Administration in the USA to investigate the effect of the drug on patients with a low risk of death, was stopped prematurely due to futility and an increased risk of significant bleeding. Studies on children were also stopped due to the unacceptable risk profile. An open label trial, ENHANCE,114 was then conducted to replicate the results of PROWESS; the Australian data indicated that depending on the selection criteria used, up to 8% of patients may be eligible for treatment with drotrecogin alfa (activated).115 Evidence for use remains equivocal. Use is currently rare and where used in severe sepsis, preparation and administration requires additional education and continual assessment and specific attention to signs of bleeding including cerebral haemorrhage.
Other adjuncts
Adequate nutritional support to offset high caloric and protein demands is relevant with enteral feeding preferred. Translocation of gut bacteria due to splanchnic hypoperfusion and increased permeability is a factor in secondary septic insults and stress ulceration.116 Equally important to patient-specific measures is institution of diligent infection control practices in ICU.117 For more information on organ support refer to the relevant chapters in Section II and Chapter 21.
Anaphylaxis
Anaphylaxis is the most severe, potentially life-threatening form of an allergic reaction,118–121 usually as a type I hypersensitivity classification (IgE-mediated hypersensitivity).121 Anaphylaxis appears rare,120 although data are sporadic in the literature; 0.01–0.02% of the general population is affected.121 Anaphylaxis appears more common in Western countries, but this may be related to more thorough reporting mechanisms.122 The prevalence of allergy with anaphylaxis has been documented as high as 7% in one Australian study of children, with insect stings, oral medications or food the most often cited causes. However, in this study, less than 1% of the population actually suffered an anaphylactic reaction manifesting with generalised multisystem allergic reaction, including evidence of airway involvement, rashes, GIT and cardiovascular dysfunction.123
This allergic response is via a host mast-cell reaction mediated by immunoglobulin E (IgE),118 and an antibody produced in response to the allergen that is attached to basophils (mast cells). Once sensitised to an allergen, subsequent exposure may lead to an anaphylactic reaction in affected individuals. The mechanism is that subsequent exposure leads to mast-cell–allergen complexes and the release of histamine.124 Reactions to an allergen cannot be predicted in anaphylaxis, with a subsequent exposure leading to an amplified or lesser response.119 There can be an initial reaction, which subsides with treatment over about 24 hours, but often described is a second or rebound reaction up to 8–10 hours after initial exposure to an allergen.118,122
Clinical Manifestations
Exposure to an allergen causes release of histamine and other mediators, with subsequent vasodilation and increased microvascular permeability – a distributive form of shock. Histamine acts, and is metabolised, rapidly while other mediators have a sustained effect.121 The antigen–antibody reaction may directly damage vascular walls, while release of vasoactive mediators such as histamine, serotonin, bradykinins and prostaglandins trigger a systemic response, resulting in vasodilation and increased capillary permeability, with widespread loss of fluid into the interstitial space and hypovolaemia. Blood pressure and cardiac output/index may fall with a compensatory rise in heart rate. Severe bronchospasm may also occur from mediator-induced bronchial oedema and pulmonary smooth muscle contraction.9 Abdominal pain is thought to be due to the inflammation of Peyer’s patches (clusters of lymphatic tissue containing B-lymphocytes, located in the mucosa and submucosa of the small intestine).124 A list of signs and symptoms for anaphylaxis appears in Table 20.10. Anaphylaxis should be considered when there are two or more organ systems involved.125
TABLE 20.10 Clinical manifestations of anaphylaxis119,122,123
| System | Clinical manifestations |
|---|---|
| Nervous | Syncope, dizziness, weakness, seizures, anxiety |
| Respiratory | Stridor, wheeze, cough, pharyngeal/laryngeal oedema, dyspnoea, bronchospasm, tachypnoea, cyanosis, use of accessory muscles |
| Cardiovascular | Tachycardia, hypotension, arrhythmias |
| Abdominal | Nausea, vomiting, cramps, pain, diarrhoea |
| Other | Flushed skin, pruritus, urticaria, angiooedema, erythema, rash, lacrimation, conjunctival injection, warmth, itching |
Of note is the high mortality in patients with asthma and those on beta-blocker or ACE inhibitor medications;119,126 these medications may limit the effectiveness of adrenaline therapy. Age and preexisting lung disease are the most important factors in relation to severity; older people and those with asthma or airways disease have a higher risk of a life-threatening reaction.124
Nursing Practice and Collaborative Care: Initial Management
Diagnosis of an anaphylactic reaction requires an appropriate assessment and history, including acute onset, history of allergic reaction and initial measures instituted to support airway, breathing and circulation (ABC). Removal of the causative agent (if possible) and early treatment (within 30 minutes of exposure to an allergen) results in improved outcomes. ABC measures are important considering the rapid impact of circulating mediators and potential decline in respiratory and cardiovascular function. Securing the airway is vital as most anaphylactic related deaths are due to asphyxiation.121 Adrenaline is recommended as first-line drug treatment119,121,122,124 often as an IM injection.
Nursing Practice and Collaborative Care: Airway Management
Early elective intubation is recommended for patients with airway oedema, stridor, or any oropharyngeal swelling. Patients with airway swelling and/or angiooedema are at high risk for rapid deterioration and respiratory compromise.125 Late presentation to hospital or delayed intubation when airway swelling is present may mean that intubation and other emergency airway procedures may be extremely difficult. Multiple attempts at intubation increase laryngeal oedema or cause trauma to the airway. Early recognition of the potentially difficult airway allows planning for alternative airway management by experts in difficult airways.125
Nursing Practice and Collaborative Care: Adjunctive Support
Adjunctive drugs include H2-antagonists, antihistamines, corticosteroids and other beta2-agonists for airway symptoms. The H2-antagonists are competitive antagonists of histamine at the parietal cell H2 receptor. Blocking both H1 and H2 receptors is an advantage with urticaria present. Corticosteroids may be beneficial for persistent bronchospasm, asthma and severe cutaneous reactions but not in acute management. Glucagon and noradrenaline may be required for patients on beta-blockers who may have resistant severe hypotension and bradycardia.127 Glucagon exerts positive inotropic and chronotropic effects, independently of catecholamines, while atropine may reverse bradycardia. Vasopressin is also suggested where shock is refractory to adrenaline.121 Given that a second reaction may occur after the initial allergic response, monitoring should continue for up to 48 hours.121
Neurogenic/Spinal Shock
Neurogenic shock is a form of distributive shock caused by loss of vasomotor (sympathetic) tone from disruption to or inhibition of neural output. Characteristics include SBP <90–100 mmHg and a HR <80 bpm without other obvious causes.128 Note that the HR is within otherwise accepted normal limits. Most often it is described as a triad of hypotension, bradycardia and hypothermia. The primary cause is a spinal cord injury above T6, secondary to disruption of sympathetic outflow from T1–L2 and to unopposed vagal tone, leading to decreased vascular resistance and associated vascular dilation.129 It may also develop after anaesthesia, particularly spinal, cerebral medullary ischaemia or when there is spinal cord complete or partial injury above the midthoracic region (thoracic outflow tract).
Spinal shock is a subclass of neurogenic shock, with a transient physiological (rather than anatomical) reflex depression of cord function below the level of injury and associated loss of sensorimotor functions. Incidence has been reported at 14% of patients presenting to the ED within 2 hours of injury and predominantly affects patients with cervical damage.128 Spinal shock can also occur with a spinal cord laceration or contusion, and is associated with varying degrees of motor and sensory deficit (see also Chapters 17 and 23). Trauma is frequently the reason for primary injury and simultaneous injuries may also be responsible for haemodynamic compromise.128 Haemorrhagic shock in combination with neurogenic shock has a poor outcome.
Clinical Manifestations
Inhibited sympathetic outflow results in dominance of the parasympathetic nervous system, with a reduction in systemic vascular resistance and lowered blood pressure. Preload to the right heart is reduced, which lowers stroke volume and subsequent cardiac output/index. The usual response to reduction in cardiac output (a raised heart rate) does not occur due to the parasympathetic nervous system and blockage of sympathetic compensatory responses, and the patient may be bradycardic and hypotensive,129 with their skin warm and dry.
In spinal shock there may be an initial rise in blood pressure due to release of catecholamines, followed by hypotension.129 Flaccid paralysis, including that of the bladder and bowel, is observed and sustained priapism may develop. Symptoms may last hours to days, until the reflex arcs below the level of injury begin to regain function. This is a result of damage to the spinal cord, and results in pale, cold skin above the site of injury, and warm, pink skin below the site of injury. Anhidrosis (absence of sweating) may be present. Heart rate may be slow, requiring intervention.
Nursing Practice and Collaborative Management
After neck and torso stabilisation, a patient is placed in a position that supports spinal precautions (neutral neck positioning) with the spinal boards removed within 20 minutes if possible. Caution for spinal instability remains despite medical imaging clearance, due to the potential for spinal ligament damage. The patient is positioned supine, with their legs in alignment with the torso. Elevation of the head may cause pooling of blood in the lower limbs, exacerbating hypotension,130 and makes the patient sensitive to sudden position changes.
Loss of sympathetic outflow requires close cardiac and haemodynamic monitoring for bradycardia and hypotension. Symptomatic bradycardia is treated and may require cardiac pacing if unresponsive to atropine. Therapies include fluid resuscitation with the addition of inotropes if necessary to improve vasomotor tone to increase preload and maintain a MAP >80–85 mmHg129 to restore spinal cord perfusion and to prevent secondary neuronal hypoperfusion.131 A higher (supranormal) MAP may be targeted to improve recovery and prevent secondary injuries.131 Volume expansion with colloids and crystalloids or blood products will vary depending on patient situation, however subgroup analysis in the SAFE trial indicated that colloids and hypotonic solutions may not be the best options.44
Respiratory function is closely monitored to prevent or minimise atelectasis, pneumonia131 and secretion retention. The level of injury is indicative of the potential for respiratory muscle weakness (see Table 20.11). The diaphragm is innervated by the phrenic nerve (originating at C3–C5); any injury above C3 leads to complete respiratory muscle paralysis and patients will require ventilatory support.131 Incomplete injuries between C3 and C5 may also require ventilation initially but subsequently recover some respiratory function.
| Cord level innervation | Accessory muscle |
|---|---|
| C3–C5 (mostly C4) | Diaphragm |
| C6 | Serratus anteriorLatissimus dorsiPectoralis |
| T1–11 | Intercostals |
| T6–L1 | Abdominals |
Paralytic ileus is a concern in the acute phase of injuries above T5, where disruption of integrative innervation pathways leads to unmodulated colonic functioning132 and peristaltic hypomotility. Ileus may lead to respiratory compromise and should be managed. The patient should remain ‘nil by mouth’ and treatment includes gastric decompression, adequate IV hydration and electrolyte balance. Drug therapy with prokinetics, probiotics, aperients and IV neostigmine or lignocaine has been reported to be useful.
Summary
Research vignette
Critique
Not surprisingly, early signs, when combined with late signs, were more strongly predictive than early signs alone of risk of death. Having noted this, many of the early signs listed did not result in death so any system of response based on the early signs alone could be expensive in terms of demand with limited benefit, at least in reduction in mortality. So whilst this study further indentifies both early and late signs of deterioration that will allow for more refined calling criteria, it does fail to deliver the definitive set of calling criteria for clinical emergency response systems. Weaknesses include the retrospective nature of the review and the reliance on charted records. The original sample is now over 10 years old and care practices may have moved on since then. It does acknowledge the MERIT study,134 the only large-scale prospective assessment of the MET system in Australia but does not discuss in detail why this research failed to show a difference where a MET system was in place.
Although there are more signs included in the SOCCER study than would be available on a standard bedside observation chart,133 research such as this has led to initiatives to standardise observation charts and highlight appropriate calling criteria and escalation procedures. Standardisation in this way supports organisations to provide equitable service to patients. In NSW there has been statewide implementation of the ‘Between the Flags’ program135 which includes a colour-coded chart, escalation procedure and indepth online training modules. This program enables clinicians to respond appropriately and communicate effectively when patients deteriorate. The Australian Commission on Safety and Quality in Healthcare has also developed a program to support organisations in increasing structures for hospital patients to receive comprehensive care regardless of location and time of day.136 These are important initiatives to combat avoidable in-hospital complications and deaths.
Learning activities
1. What assessments are important to obtain appropriate information for a patient presenting with signs of hypoperfusion?
2. What systems are in place where you work to ensure adequate processes are in place to assess patients presenting in shock?
3. How could the patient in the case study have been managed differently?
4. What are the clinical escalation processes in the facilities you have worked in?
American Heart Association. http://www.heart.org/HEARTORG/.
Anaphylaxis Australia www.allergyfacts.org.au/
Australian Commission on Safety and Quality in Healthcare. http://www.safetyandquality.gov.au/internet/safety/publishing.nsf/Content/home.
Cardiac Society of Australia and New Zealand. http://www.csanz.edu.au/Home/tabid/62/Default.aspx.
Clinical Excellence Commission: Between the flags. http://www.cec.health.nsw.gov.au/programs/between-the-flags.html.
National Blood Authority Australia. www.nba.gov.au.
Sepsis. http://www.ihi.org/ihi/topics/criticalcare/sepsis.
Spinal cord injury network. https://spinalnetwork.org.au/.
Surviving sepsis. www.survivingsepsis.org/.
World Allergy Organisation. http://www.worldallergy.org/index.php.
Manji RA, Wood KE, Kumar A. The history and evolution of circulatory shock. Critical Care Clinics. 2009;25(1):1–29.
Australian Commission on Safety and Quality in Healthcare. Recognising and responding to clinical deterioration: Background Paper. http://www.safetyandquality.gov.au/internet/safety/publishing.nsf/Content/AB9325A491E10CF1CA257483000C9AC4/$File/BackgroundPaper-2009.pdf, June 2008. Available from
1 Adams F. The book of prognostics, by Hippocrates. In: eBooks@Adelaide University of Adelaide Library; 2007. Available from http://ebooks.adelaide.edu.au/h/hippocrates/prognostics/
2 Rice TW, Bernard BG. Drotrecogin alfa (activated) for the treatment of severe sepsis and septic shock. Am J Med Sci. 2004;328(4):205–214.
3 Kelley D. Hypovolemic shock: an overview. Crit Care Nurs Q. 2005;28(1):2–19.
4 Bridges EJ, Duke S. Cardiovascular aspects of septic shock: pathophysiology, monitoring, and treatment. Crit Care Nurs. 2005;25(2):14–24.
5 Manji RA, Wood KE, Kumar A. The history and evolution of circulatory shock. Critical Care Clinics. 2009;25(1):1–29.
6 Barbee R, Reynolds P, Ward K. Assessing shock resuscitation strategies by oxygen debit repayment. Shock. 2010;33(2):113–122.
7 Kellum JA. Use of vasopressor agents in critically ill patients. Curr Opin Crit Care. 2002;8(3):236–241.
8 Strehlow MC. Early identification of shock in critically ill patients. Emerg Med Clin N Am. 2010;28(1):57–66.
9 Kinney M, Dunbar S, Brooks-Brunn J, Molter N, Vitello-Cicciu J. AACN’s clinical reference for critical care nursing. St Louis: Mosby; 1998.
10 Vallet B, Weil E, Lebuffe G. Resuscitation from circulatory shock: an approach based on oxygen-derived parameters. Berlin: Springer-Verlag; 2005.
11 Hameed SM, Aird WC, Cohn SM. Oxygen delivery. Crit Care Med. 2003;31(12Suppl):5658–5667.
12 ACCCN. National Advanced Life Support Education Package. Melbourne: Cambridge Press; 2004.
13 Brealey D, Brand M, Hargreaves I, Heales S, Land J, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219–223.
14 Trager K, DeBacker D, Radermacher P. Metabolic alterations in sepsis and vasoactive drug-related metabolic effects. Curr Opin Crit Care. 2003;9(4):271–278.
15 Hubbard WJ, Bland KI, Chaudry IH. The role of the mitochondrion in trauma and shock. Shock. 2004;22(5):395–402.
16 Al-Khafaji AH, Sharma S, Eschun G, Franklin C, Talavera F, et al. Multisystem organ failure of sepsis. http://www.emedicine.com/med/topic3372.htm, 2004. [Cited Aug 2004]. Available from
17 Fortin C, McDonald P, Fulop T, Lesur O. Sepsis, leukocytes, and nitric oxide (NO): An intricate affair. Shock. 2010;33(4):344–352.
18 Cheatham M. The Holy Grail of shock resuscitation. Crit Care Med. 2005;33(11):2691–2692.
19 Casserly B, Read R, Levy M. Hemodynamic monitoring in sepsis. Crit Care Clinics. 2009;25(4):803–823.
20 Wagner F, Baumgart K, Simkova V, Georgieff M, Radermacher P, Calzia E. Year in review 2007: Critical Care: shock. Critical Care. 2008;12(5):227.
21 Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med. 2009;37(5):1670–1677.
22 Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med. 2004;32(8):1637–1642.
23 Fishel RS, Are C, Barbul A. Vessel injury and capillary leak. Crit Care Med. 2003;31(8Suppl):S502–S511.
24 Sherwood ER, Toliver-Kinsky T. Mechanisms of the inflammatory response. Best Pract Res Clin Anaesth. 2004;18(3):385–405.
25 Leach R, Treacher D. Oxygen transport-2. Tissue hypoxia. BMJ. 1998;317(7169):1370–1373.
26 Adams K. Hemodynamic assessment: the physiologic basis for turning data into clinical information. AACN Clinical Issues: Advanced Practice in Acute and Critical Care. 2004;15(4):534–546.
27 Moshkovitz Y, Kaluski E, Milo O, Vered Z, Cotter G. Recent developments in cardiac output determination by bioimpedance: comparison with invasive cardiac output and potential cardiovascular applications. Curr Opin Cardiol. 2004;19(3):229–237.
28 Levin PD, Sprung CL. Another point of view: no swan song for the pulmonary artery catheter. Crit Care Med. 2005;33(5):1123–1124.
29 Boettger S, Pavlovic D, Grundling M, Wendt M, Hung O, et al. Comparison of arterial pressure cardiac output monitoring with transpulmonary thermodilution in septic patients. Med Sci Monitor. 2010;16(3):PR1–PR7.
30 Treacher D, Harvey C, Bradley R. Can cardiac output be assessed clinically with sufficient accuracy to be of value in patient management? Comparison with thermo-dilution in Intensive Care Unit patients. London: The Intensive Care Unit, UMDS, St Thomas’ Hospital; 2006.
31 Boyle M, Steel L, Flynn G, Murgo M, Nicholson L, et al. Assessment of the clinical utility of an ultrasonic monitor of cardiac output (the USCOM) and agreement with thermodilution measurement. Crit Care Resusc. 2009;11(3):198–203.
32 Godje O, Hoke K, Goetz AE, Felbinger TW, Reuter DA, et al. Reliability of a new algorithm for continuous cardiac output determination by pulse-contour analysis during hemodynamic instability. Crit Care Med. 2002;30(1):52–58.
33 Shah MR, Hasselblad V, Stevenson LW, et al. Impact of the pulmonary artery catheter in critically ill patients: meta-analysis of randomized clinical trials. JAMA. 2005;294(13):1664–1670.
34 Harvey S, Welch C, Harrison D, Rowan K, Singer M. Post hoc insights from PAC-Man: the UK pulmonary artery catheter trial. Crit Care Med. 2008;36(6):1714–1721.
35 Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Eng J Med. 2001;345(19):1368–1377.
36 Ho BC, Bellomo R, McGain F, et al. The incidence and outcome of septic shock patients in the absence of early-goal directed therapy. Critical Care. 2006;10(3):R80.
37 Weil M. Personal commentary on the diagnosis and treatment of circulatory shock states. Curr Opin Crit Care. 2004;10(4):246–249.
38 Vincent J-L. Give your patient a fast hug (at least) once a day. Crit Care Med. 2005;33(6):1225–1229.
39 Stennett A, Gainer J. TSC for hemorrhagic shock: effects on cytokines and blood pressure. Shock. 2004;22(6):569–574.
40 Santry HP, Alam HB. Fluid resuscitation: past, present, and the future. Shock. 2010;33(3):229–241.
41 Gutierrez G, Reines HD, Wulf-Gutierrez ME. Clinical review: hemorrhagic shock. Critical Care. 2004;8(5):373–381.
42 Beekley A. Damage control resuscitation: a sensible approach to the exsanguinating surgical patient. Crit Care Med. 2008;36(7S):S267–S274.
43 Bauer M. Multiple organ failure: update on pathophysiology and treatment strategies. Euroanesthesia: European Society of Anaesthesiology,conference proceedings, Vienna, Austria; 2005. p. 203–6.
44 Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. New Engl J Med. 2004;350(22):2247–2256.
45 Alderson P, Bunn F, Lefebvre C, Li W, Li L, et al. Human albumin solution for resuscitation and volume expansion in critically ill patients. Cochrane Database of Systematic Reviews 2004; CD001208.
46 Lighthall GK, Pearl RG. Volume resuscitation in the critically ill: choosing the best solution: how do crystalloid solutions compare with colloids? J Crit Illness. 2003;18:252–260.
47 Lopez-Plaza I. Massive blood transfusion [Transfusion medicine update. Institute for transfusion medicine, Available from] http://www.itxm.org/tmu/tmu1998/tmu4-98.htm.
48 Criddle L, Eldridge D, Walker J. Variables predicting trauma patient survival following massive transfusion. J Emerg Nurs. 2005;31:236–242.
49 Lim N, Dubois M, De Backer D, Vincent J-L. Do all nonsurvivors of cardiogenic shock die with a low cardiac index? Chest. 2003;124(5):1885–1891.
50 Stacy K. Shock. In: Urden L, Stacy K, Lough ME. Thelan’s critical care nursing: diagnosis and management. St Louis: Mosby, 2002.
51 Hofer CK, Furrer L, Matter-Ensner S, Maloigne M, Klaghofer R, et al. Volumetric preload measurement by thermodilution: a comparison with transoesophageal echocardiography. Br J Anaes. 2005;94(6):748–755.
52 Agricola E, Bove T, Oppizzi M, Marino G, Zangrillo A, et al. Ultrasound comet-tail images: a marker of pulmonary edema: a comparative study with wedge pressure and extravascular lung water. Chest. 2005;127(5):1690–1695.
53 Worthley L. Shock: a review of pathophysiology and management, Part 1. Crit Care Resusc. 2000;2:55–65.
54 Lenneman A, Ooi HH. Cardiogenic shock. http://www.emedicine.com/MED/topic285.htm, 2005. [Cited 2005]. Medscape. Available from
55 Di Marco JP, Gersh B, Opie LH. Antiarrhythmic drugs and strategies. Opie LH, Gersch B. Drugs for the heart, 6th edn, Philadelphia: Elsevier Saunders, 2005.
56 Santoro GM, Buonamici P. Reperfusion therapy in cardiogenic shock complicating acute myocardial infarction. Am Heart J. 1999;138(2Pt2):S126–S138.
57 Blanton C, Thompson P. Cardiogenic shock and myocardial infarction in 19 Australian teaching hospitals. Cardiac Society of Australia and New Zealand conference proceedings, 49th ASM; 2001.
58 Carnendran L, Abboud R, Sleeper LA, Gurunathan R, Webb JG, et al. Trends in cardiogenic shock: report from the SHOCK Study. The SHould we emergently revascularize Occluded Coronaries for cardiogenic shocK? Eur Heart J. 2001;22(6):472–478.
59 Menon V, White H, LeJemtel T, Webb JG, Sleeper LA, Hochman JS. The clinical profile of patients with suspected cardiogenic shock due to predominant left ventricular failure: a report from the SHOCK Trial Registry. SHould we emergently revascularize Occluded Coronaries in cardiogenic shocK? J Am Coll Cardiol. 2000;36(3SupplA):1071–1076.
60 Hollenberg SM, Kavinsky CJ, Parrillo JE. Cardiogenic shock. Ann Intern Med. 1999;131(1):47–59.
61 Rahimtoola S. Acute rheumatic fever. In: Fuster V, Alexander R, O’Rourke RA. Hurst’s the heart. New York: McGraw-Hill, 2004.
62 Leach RM, Treacher DF. The pulmonary physician in critical care. Number 2: oxygen delivery and consumption in the critically ill. Thorax. 2002;57(2):170–177.
63 Leach RM, Treacher D. The relationship between oxygen delivery and consumption. Disease-Month. 1994;40:301–368.
64 Shoemaker WC, Appel P, Kram HB, Waxman K, Lee TS, et al. Prospective trial of supranormal values of survivors as therapeutic goals in high-risk surgical patients. Chest. 1988;94:1176–1186.
65 Harvey SE, Welch CA, Harrison DA, Rowan KM, Singer M. Post hoc insights from PAC-Man – the UK pulmonary artery catheter trial. Crit Care Med. 2008;36(6):1714–1721.
66 Park M, Sangean M, Volpe MdeS, Feltrim MI, Nozqwa F, et al. Randomized, prospective trial of oxygen, continuous positive airway pressure, and bilevel positive airway pressure by face mask in acute cardiogenic pulmonary edema. Crit Care Med. 2004;32(12):2546–2548.
67 Lesage A, Ramakers M, Daubin C, Verrier V, Beynier D, et al. Complicated acute myocardial infarction requiring mechanical ventilation in the intensive care unit: prognostic factors of clinical outcome in a series of 157 patients. Crit Care Med. 2004;32(1):100–105.
68 Opie LH, Kaplan NM. Diuretics. Opie LH, Gersch BJ. Drugs for the heart, 6th edn, Philadelphia: Elsevier, 2005.
69 Jugbutt B. Nitrates in myocardial infarction. Cardiovasc Drugs Ther. 1994;8:635–646.
70 Poole-Wilson PA, Opie LH. Digitalis, acute inotropes and inotropic dilators: acute and chronic heart failure. Opie LH, Gersch BJ. Drugs for the heart, 6th edn, Philadelphia: Elsevier, 2005.
71 Follath F, Cleland JGF, Just H, Papp JG, Scholz H, et al. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): a randomised double-blind trial. Lancet. 2002;360(9328):196–202.
72 Mebazaa A, Barraud D, Welschbillig S. Randomized clinical trials with levosimendan. Am J Cardiol. 2005;96(6A):G74–G79.
73 Moiseyev VS, Poder P, Andrejevs N, Ruda MV, Golikov AP, et al. Safety and efficacy of a novel calcium sensitizer, levosimendan, in patients with left ventricular failure due to an acute myocardial infarction: a randomized, placebo-controlled, double-blind study (RUSSLAN). (RUSSLAN Study Investigators). Eur Heart J. 2002;23(18):1422–1432.
74 Rosano G, Fini M, Caminiti G, Barbaro G. Cardiac metabolism in myocardial ischemia. Current Pharmaceutical Design. 2008;14:2551–2562.
75 O’Connor CM, Gattis WA, Uretsky BF, Adams KF, Jr., McNutty SE, et al. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: insights from the Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1999;138(1Pt1):78–86.
76 Gersh BJ. Which therapy for which condition?. Opie LH, Gersch BJ. Drugs for the heart, 6th edn, Philadelphia: Elsevier, 2005.
77 Annane D, Bellissant E, Pussard E, Asmar R, Lacombe F, et al. Placebo-controlled, randomized, double-blind study of intravenous enalaprilat efficacy and safety in acute cardiogenic pulmonary edema. Circulation. 1996;94(6):1316–1324.
78 AIRE Study Investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure: the Acute Infarction Ramipril Efficacy (AIRE) Study. Lancet. 1993;342(8875):821–828.
79 Murray S. Bi-level positive airway pressure (BiPAP) and acute cardiogenic pulmonary oedema (ACPO) in the emergency department. Aust Crit Care. 2002;15(2):51–63.
80 Agarwal R, Agarwal A, Gupta D, Jindal SK, et al. Non-invasive ventilation in acute cardiogenic pulmonary oedema. Postgrad Med J. 2005;81(960):637–643.
81 Tallman T, Peacock W, Emerman C, et al. Noninvasive ventilation outcomes in 2,430 acute decompensated heart failure patients: an ADHERE Registry Analysis. Academ Emerg Med. 2008;15(4):355–362.
82 Bone R, Balk R, Cerra F, Dellinger R, Fein A, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. ACCP/SCCM Consensus Conference Committee American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655.
83 Angus D, Wax R. Epidemiology of sepsis: an update. Crit Care Med. 2001;29(7Suppl):S109–S116.
84 Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intens Care Med. 2003;29(4):530–538.
85 Angus D, Burgner D, Wunderink R, et al. The PIRO concept: P is for predisposition. Crit Care. 2003;7(3):248–251.
86 Finfer S, Bellomo R, Lipman J, French C, Dobb G, Myburgh J. Adult-population incidence of severe sepsis in Australian and New Zealand intensive care units. Intens Care Med. 2004;30(4):589–596.
87 Sundararajan V, Macisaac CM, Presneill JJ, Cade JF, Visvanathan K. Epidemiology of sepsis in Victoria, Australia. Crit Care Med. 2005;33(1):71–80.
88 Hicks P, Cooper DJ, Webb S, et al. The Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. An assessment by the Australian and New Zealand intensive care society. Anaesthesia & Intensive Care. 2008;36(2):149–151.
89 Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med. 2004;32(3):858–873.
90 Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(4):296–327.
91 Joint Commission on Accreditation of Healthcare Organizations. Raising the bar with bundles treating patients with an all-or-nothing standard. Joint Commission Perspectives on Patient Safety. 2006;6(4):5–6.
92 Finfer S. The Surviving Sepsis Campaign: robust evaluation and high-quality primary research is still needed. Intens Care Med. 2010;36(2):187–189.
93 van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. New Eng J Med. 2001;345(19):1359–1367.
94 Nice-Sugar Study InvestigatorsFinfer S, Chittock DR, Su SY, Blair D, Foster D, et al. Intensive versus conventional glucose control in critically ill patients. New Eng J Med. 2009;360(13):1283–1297.
95 Griesdale DE, de Souza RJ, van Dam RM, et al. Intensive insulin therapy and mortality among critically ill patients: a meta-analysis including NICE-SUGAR study data. CMAJ. 2009;180(8):821–827.
96 Van den Berghe G, Schetz M, Vlasselaers D, et al. Clinical review: Intensive insulin therapy in critically ill patients: NICE-SUGAR or Leuven blood glucose target? J Clin Endocrinol Metabolism. 2009;94(9):3163–3170.
97 Surviving Sepsis Campaign. Statement on Glucose Control in Severe Sepsis 2009. [Cited January 2011]. Available from http://www.survivingsepsis.org/About_the_Campaign/Documents/SSC%20Statement%20on%20Glucose%20Control%20in%20Severe%20Sepsis.pdf
98 Hollenberg SM, Ahrens TS, Annane D, Astiz ME, Chalfin DB, et al. Practice parameters for hemodynamic support of sepsis in adult patients: 2004 update. Crit Care Med. 2004;32(9):1928–1948.
99 Schrier RW, Wang W. Acute renal failure and sepsis. New Engl J Med. 2004;351:159–169.
100 Marik PE, Varon J. Early goal-directed therapy: on terminal life support? Am J Emerg Med. 2010;28(2):243–245.
101 Puskarich MA, Marchick MR, Kline JA, Steuerwald MT, Jones AE. One year mortality of patients treated with an emergency department based early goal directed therapy protocol for severe sepsis and septic shock: a before and after study. Critical Care. 2009;13(5):R167.
102 Focht A, Jones AE, Lowe TJ. Early goal-directed therapy: improving mortality and morbidity of sepsis in the emergency department. Joint Commiss J Qual Patient Safety. 2009;35(4):186–191.
103 Rivers EP, Coba V, Whitmill M. Early goal-directed therapy in severe sepsis and septic shock: a contemporary review of the literature. Curr Opin Anaesthesiol. 2008;21(2):128–140.
104 Padkin A, Goldfrad C, Brady AR, Young D, Black N, Rowan K. Epidemiology of severe sepsis occurring in the first 24 hrs in intensive care units in England, Wales, and Northern Ireland. Crit Care Med. 2003;31(9):2332–2338.
105 Brun-Buisson C, Meshaka P, Pinton P, Vallet B, Group ES. EPISEPSIS: a reappraisal of the epidemiology and outcome of severe sepsis in French intensive care units. Intens Care Med. 2004;30(4):580–588.
106 Boots RJ, Lipman J, Bellomo R, Stephens D, Heller RE. The spectrum of practice in the diagnosis and management of pneumonia in patients requiring mechanical ventilation. Australian and New Zealand practice in intensive care (ANZPIC II). Anaesthes Intens Care. 2005;33(1):87–100.
107 Sogaard OS, Lemoh C, Spelman D, et al. A binational cohort study of ventilator-associated pneumonia in Denmark and Australia. Scand J Infect Dis. 2006;38(4):256–264.
108 Kumar A. Optimizing antimicrobial therapy in sepsis and septic shock. Criti Care Clinics. 2009;25:733–751.
109 Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006;34(6):1589–1596.
110 Taccone FS, Laterre P-F, Dugernier T, et al. Insufficient beta-lactam concentrations in the early phase of severe sepsis and septic shock. Crit Care. 2010;14(4):R126.
111 Lipman J, Boots R. A new paradigm for treating infections: “go hard and go home. Crit Care Resusc. 2009;11(4):276–281.
112 Lipiner-Friedman D, Sprung CL, Laterre PF, et al. Adrenal function in sepsis: the retrospective Corticus cohort study. Crit Care Med. 2007;35(4):1012–1018.
113 Bernard G, Macias W, Joyce D, Williams M, Bailey J, Vincent J. Safety assessment of drotrecogin alfa (activated) in the treatment of adult patients with severe sepsis. Critical Care. 2003;7(2):155–163.
114 Vincent JL, Bernard GR, Beale R, et al. Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE: further evidence for survival and safety and implications for early treatment. Crit Care Med. 2005;33(10):2266–2277.
115 Finfer S, Felton T, Blundell A, Lipman J, ANZICS Clinical Trials Group Sepsis Investigators. Estimate of the number of patients eligible for treatment with drotrecogin alfa (activated) based on differing international indications: post-hoc analysis of an inception cohort study in Australia and New Zealand. Anaesthes Intens Care. 2006;34(2):184–190.
116 Magnotti L, Deitch E. Burns, bacterial translocation, gut barrier function, and failure. J Burn Care Rehab. 2005;26:383–391.
117 Maragakis L. Recognition and prevention of multidrug-resistant Gram-negative bacteria in the intensive care unit. Crit Care Med. 2010;38(8Suppl):S345–S351.
118 Ellis AK, Day J. Diagnosis and management of anaphylaxis. Can Med Assoc J. 2003;169:307–311.
119 McLean-Tooke AP, Bethune C, Fay AC, Spickett GP. Adrenaline in the treatment of anaphylaxis: what is the evidence? BMJ. 2003;327(7427):1332–1335.
120 Gold M. EpiPen epidemic or good clinical practice? J Paediatr Child Health. 2003;39:376–377.
121 Kanji S, Chant C. Allergic and hypersensitivity reactions in the intensive care unit. Crit Care Med. 2010;38(6Suppl):S162–S168.
122 World Allergy website. Anaphylaxis. http://www.worldallergy.org/public/allergic_diseases_center/anaphylaxis/anaphylaxis.shtml, 2004. [Cited Jun 2004]. Available from
123 Boros CA, Kay D, Gold MS. Parent reported allergy and anaphylaxis in 4173 South Australian children. J Paediatr Child Health. 2000;36:36–40.
124 Brown S. Clinical features and severity grading of anaphylaxis. J Allergy Clin Immunol. 2004;114:371–376.
125 American Heart Association. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 10.6: Anaphylaxis. Circulation. 2005;112(IV):143–145.
126 Pumphrey R. Anaphylaxis: can we tell who is at risk of a fatal reaction? Curr Opin Allergy Clin Immunol. 2004;4:285–290.
127 Tang A. A practical guide to anaphylaxis. Am Fam Physician. 2003;68:1325–1332.
128 Guly HR, Bouamra O, Lecky FE, Trauma Audit, Research N. The incidence of neurogenic shock in patients with isolated spinal cord injury in the emergency department. Resuscitation. 2008;76(1):57–62.
129 Dawodu S. Spinal cord injury: definition, epidemiology, pathophysiology. Medscape [Cited 2005]. Available from http://www.emedicine.com/PMR/topic182.htm
130 Mattera C. Spinal trauma: new guidelines for assessment and management in the out-of-hospital environment. J Emerg Nurs. 1998;24:523–534.
131 Miko I, Gould R, Wolf S, Afifi S. Acute spinal cord injury. Int Anesthesiol Clin. 2009;47(1):37–54.
132 Baumann A, Audibert G, Klein O, Mertes P. Continuous intravenous lidocaine in the treatment of paralytic ileus due to severe spinal cord injury. Acta Anaesthesiologica Scandinavica. 2009;53(1):128–130.
133 Harrison GA, Jacques T, McLaws ML, Kilborn G. Combinations of early signs of critical illness predict in-hospital death-The SOCCER Study (signs of critical conditions and emergency responses). Resuscitation. 2006;71(3):327–334.
134 Hillman K, Chen J, Cretikos M, Bellomo R, Brown D, et al. Introduction of the medical emergency team (MET) system: a cluster-randomised controlled trial.]. Lancet. 2005;365(9477):2091–2097. Erratum appears in Lancet 2005; 366(9492): 1164
135 Clinical Excellence Commission. Between the Flags: Keeping patients safe. NSW Government. http://www.cec.health.nsw.gov.au/programs/between-the-flags.html, 2010. [Cited January 2011]. Available from
136 Australian Commission on Safety and Quality in Healthcare. Recognising and responding to clinical deterioration. http://www.safetyandquality.gov.au/internet/safety/publishing.nsf/Content/prog-patientsrisk-lp, 2009. [Cited January 2011]. Available from
137 Barochia AV, Cui X, Vitberg D, Suffredini AF, O’Grady NP, et al. Bundled care for septic shock: an analysis of clinical trials. Critical Care Medicine. 2010;38(2):668–678.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
20 Management of Shock
Introduction
Shock is an altered physiological state that affects the functioning of every cell and organ system in the body. It is a complex syndrome reflecting changing blood flow to body tissues with accompanying cellular dysfunction and eventual organ failure.2,3 Shock presents as a result of impaired nutrient delivery to the tissue:
While the cause of shock may be multifactorial, treatment focuses on optimising tissue perfusion and oxygen de-livery. Shock is often classified according to the primary underlying mechanism: a disruption of intravascular blood volume, impaired vasomotor tone or altered cardiac contractility.5 The shock syndrome is one of the most pervasive manifestations of critical illness present in intensive care patients.
Early detection and management of shock to reverse pathological processes improves patient outcomes.6 Although the traditional hallmark of shock is hypotension (SBP <90 mmHg) this can be a late or misleading sign and is considered a medical emergency.7 It is therefore critical that other signs and symptoms are identified early by frequent observations to detect a patient’s deteriorating state and respond before irreversible shock ensues.8 No one vital sign is adequate in determining the level or extent of shock6 nor is there a specific laboratory test which diagnoses the shock syndrome.
Pathophysiology
Traditionally, shock is classified by aetiology: hypovolaemic, cardiogenic and distributive.3,4,9 Each has a specific mechanism of action that leads to altered tissue perfusion and oxygen and nutrient uptake at the cellular level (see Table 20.1). In practice, it is common to find overlap between different shock types (e.g. in sepsis there may also be hypovolaemia and/or myocardial dysfunction).
| Shock type | Main characteristic |
|---|---|
| Hypovolaemic | a reduction in circulating blood volume through haemorrhage or dehydration or plasma fluid loss |
| Cardiogenic • obstructive shock |
pump failure (impaired cardiac contractility) usually as result of myocardial infarction a sub category of cardiogenic shock characterised by blockage of circulation to the tissues by impedance of outflow or filling in the heart (e.g. due to cardiac tamponade or pulmonary emboli) |
| Distributive shock | a maldistribution of circulation from sepsis, anaphylaxis or neurogenic injury |
Shock occurs when there is an inability of the body to meet metabolic demands of the tissues; hypoperfusion (decreased blood flow to the tissues) results in cellular dysfunction, as there is homeostatic imbalance between nutrient supply and demand,4,10 and adaptive responses can no longer accommodate circulatory changes. These adaptive responses are moderated via numerous ‘sensors’ throughout the thorax and large vessels in particular, which detect subtle changes in pressure (baroreceptors) or biochemical changes (chemoreceptors). These receptors feed back to the hypothalamus which regulates through the pituitary gland (for the release of a number of hormones such as antidiuretic hormone [ADH] and adrenocorticoid trophic hormone [ACTH] to target organs such as the kidney) and the cortex of the adrenal gland to respond and counter the developing effects of shock. Concurrently direct feedback stimulates the sympathetic nervous system to act on blood vessel tone, particularly the arterioles, and also target organs such as the adrenal gland and kidney to respond via the release of endogenous catecholamines (adrenaline and noradrenaline), mineral and glucocorticoids (aldosterone, cortisol), and the renin–angiotensin–aldosterone system (RAAS). RAAS activation results in synthesis of angiotensin II, a powerful vasoconstrictor that further potentiates the reduction in peripheral blood vessel capacity.
As adaptive responses fail, cardiac output becomes insufficient to provide adequate organ perfusion despite increasing tissue oxygen consumption (see Chapters 9 and 10). When oxygen is ‘supply dependent’, oxygen delivery is decreased and, to compensate, increased extraction occurs to enable continued tissue consumption. However, when oxygen delivery falls below a critical threshold, and extraction demand rises above the available blood oxygen levels, this compensation mechanism fails and oxygen debt results.6,11,12
Hypoperfusion may also exist despite a relatively normal cardiac output, and may not be immediately evident clinically.6 This maldistribution of bloodflow to some tissues while other areas receive more blood flow than needed,4,7,10,13,14 is often referred to as distributive shock, and is typical of the shock types that affect vasomotor tone (e.g. septic, neurogenic and anaphylactic shock). This maldistribution may leave some organ systems ischaemic for long periods leading to persistent organ dysfunction and failure.6 There is also evidence supporting the presence of cytopathic hypoxia as a result of excessive nitric oxide and tumour necrosis factor-alpha (TNFα) production (cellular proinflammatory mediators), where there is impaired mitochondrial (the powerhouse of the cell) oxygen utilisation which leads to depleted stores of adenosine tri-phosphate (ATP)4,11,13,15,16 and interferes with electron transport and metabolism16 (see Chapter 19). Nitric oxide is associated with vascular relaxation and is a major contributor to alterations in microvasculature and capillary leak in sepsis.17
Organ systems have varying responses in shock and are not measured directly. Often surrogate markers of global hypoperfusion are used to indicate the severity of shock.18–19 Lactate and acid–base disturbances, such as an increase in strong ion gap, have been suggested as early markers of mitochondrial dysfunction and cellular hypoperfusion.8,20 These ‘surrogate’ biochemical markers of hypoperfusion (pH, serum lactate and standard base excess) assess acidaemia and provide some insight into the degree of shock present.21 Lactate, a strong anion with normal production of 1500–4500 mmol/day, is a product of carbohydrate metabolism. Increased levels are present in tissue hypoxia, hypermetabolism, decreased lactate clearance, inhibition of pyruvate dehydrogenase and activation of inflammatory cells; all characteristics of developing shock (see Table 20.2). Increased lactate production is a warning sign of impending organ failure, as it is indicative of anaerobic metabolism. Blood lactate levels have been directly linked to deteriorating patient outcomes in shock.21,22
TABLE 20.2 Lactate production9
| Lactate production | |
| Product of carbohydrate metabolism (1400–4500 mmol/day) | Glucose, glycolysis; pyruvate, lactate |
| Rise in lactate levels | |
| Tissue hypoxia | |
As the shock state deteriorates and the body fails to compensate, organ systems begin to fail. This is complicated by a systemic inflammatory response (SIRS) which can be a direct cause of the shock state (see section on Distributive shock) or develop as a consequence of protracted shock. This results in ‘capillary leak’ or increased microvascular permeability which leads to interstitial oedema as a consequence of alterations to tissue endothelium. Many immune mediators including circulating cytokines, oxygen free-radicals and activated neutrophils alter the structure of the endothelial cells, creating space to allow larger intravascular molecules to cross into the extravascular space,23 with proteins and water moving from the intravascular space into the interstitium.24 This response mechanism improves the supply of nutrient-rich fluid to the site of local injury, however, systemically, fluid shifts lead to hypovolaemia, impaired organ function and development of acute organ injury such as acute lung injury (ALI) and acute kidney injury (AKI).24 This developing organ injury is the precedent to organ failure (more fully described in Chapter 21).
Patient Assessment
Critically ill patients often exhibit signs of tissue hypoxia as a result of cardiovascular disturbances.25 Table 20.3 provides an overview of the physiological changes in shock. Therapy is targeted to maintain oxygen delivery (DO2) to vital organs to prevent ischaemia and cell death.25,26 Ideally, organ systems and tissues should be monitored individually,25 however global measures such as perfusion pressure, cardiac output (CO) and DO2, are commonly used as surrogates to assist in treatment decision making.19 Patient assessment and haemodynamic monitoring, including calculation of CO, are used to differentiate shock states and assess progress in relation to treatment.26–28 CO is seen by many clinicians as an important assessment of shocked patients as it is a major determinant of DO2.25,26 Critically ill patients are frequently assessed clinically, although cardiac output estimations from physical examination are generally unreliable and patient status may change quickly.29 Therefore invasive techniques are most commonly used in critical care to measure CO (see also Chapter 9).
Non-Invasive Assessment
Perfusion status is determined clinically using gross organ function such as mental status, urine output and peripheral warmth and colour.6 Basic physical assessment of cardiovascular, central nervous system and renal function are essential when assessing a patient at risk of shock. Subtle changes in urine output, heart rate and capillary refill are all signs of physiological compensation in response to altered tissue perfusion associated with shock. Regular tracking of these vital signs and trend monitoring through careful documentation can alert clinicians to impending deterioration in the shock state. Level of consciousness may deteriorate; an early sign may be anxiety, and progress to restlessness, agitation or coma. Other assessment findings include cool, clammy skin, postural hypotension, tachycardia and decreased urine output.3 The reliability of these measures is questionable, particularly where multiple assessments by different clinicians are performed; in the ICU continuous ECG monitoring and invasive monitoring techniques are employed to assist in the objective assessment of changes in cardiovascular state.
Although CO estimations based on physical assessment findings are unreliable, physical examination using an estimation of vascular resistance has shown reasonable accuracy.30 Clinical assessment may determine CO using the rearranged equation of systemic vascular resistance (SVR = MAP − CVP/CO) where vascular resistance is measured through peripheral skin temperature changes.30 A reliable and accurate non-invasive clinical assessment technique of estimating cardiac output would be clinically useful27 allowing assessment of patients without invasive monitoring, or used to verify accuracy from invasive devices. While a number of non-invasive cardiac output measuring devices are available, further research and refinement is required before widespread application is considered in critical care.31
Invasive Assessment
The indicator dilution method using a thermal (thermodilution) signal (cold or hot) is the customary clinical standard for measuring CO26 in ICU. This is usually achieved by placement of a pulmonary artery catheter (PAC), or a central line in conjunction with a thermistor-tipped arterial cannula (transpulmonary aortic thermodilution). Other invasive techniques measure CO continuously using pulse contour or arterial pressure analysis and ultrasound doppler methods use an oesophageal probe. All methods have degrees of invasiveness, can be time-consuming to yield measurements of acceptable accuracy32, may be expensive and are not without risk of complications.27,33 The PAC is a controversial assessment tool26,28,33 due to the risk associated with the invasive line versus benefits for the measurement of CO34. This has led to increased interest in less or non-invasive measures of CO.
A further invasive assessment approach is the continuous estimation of mixed venous oxygen saturation using a light-emitting sensor in a PAC. As tissue oxygen delivery fails to meet demand and oxygen extraction rises, the residual oxygen content of blood returning to the lungs will fall; in effect a surrogate indicator of failure to meet body tissue oxygen demand. This technology was used in the landmark study by Rivers and colleagues.35 to monitor early deterioration of septic shock patients presenting to the ED in need of resuscitation and was part of a goal-directed approach to managing patients. This single-centre US study has been the subject of much interest for its claimed improvement in patient outcome, with this goal-directed approach being assessed in a major multicentre study in an effort to verify its findings within an international context and varying approach to critical care delivery.36
Management Principles
Managing a patient in shock focuses on treating the underlying cause, and restoration and optimisation of perfusion and oxygen delivery; this includes relevant activities using the acronym VIP37 (see Box 20.1). It is also suggested that giving critically ill patients a daily ‘FASTHUG’ improves the quality of care for patients in ICU.38 Specific management of patients with shock are discussed separately below depending on the cause.
Practice tip
Feeding (prevent malnutrition, promote adequate caloric intake)
Analgesia (reduce pain, improve physical and psychological wellbeing)
Sedation (titrate to the 3Cs – calm, cooperative, comfortable)
Thromboembolic prophylaxis (prevent DVT)
Head of bed elevated (up to 45° to reduce reflux and VAP)
Hypovolaemic Shock
Hypovolaemia is a common primary cause of shock and also a factor in other shock states. Insufficient circulating blood volume is the underlying mechanism, leading to decreased cardiac output and altered perfusion.39,40 Death related to haemorrhage is most likely in the first few hours after injury.40 The most obvious cause is direct injury to vessels leading to haemorrhage, but there are more insidious causes such as dehydration from prolonged vomiting or diarrhoea, sepsis and burns.41 Hypovolaemic shock is classified as mild, moderate or severe, depending on the amount of volume loss (see Table 20.4). As the shock state worsens, associated compensatory mechanisms will be more pronounced,3 and hypovolaemic shock may deteriorate to Multi Organ Dysfunction Syndrome (MODS) if poor oxygen delivery is prolonged39 (see Chapter 21).
Clinical Manifestations
Symptoms of haemorrhage may not be present until more than 15–30% of blood volume is lost, and will deteriorate as the shock state worsens.3,41 Estimating blood or plasma loss is difficult and dilutional effects of resuscitation fluids may be evident when assessing haemoglobin and hematocrit.41 As the body compensates for the reduced circulating volume, widespread vasoconstriction occurs in most body systems apart from the heart and CNS; SVR rises markedly in an attempt to retain a viable circulatory system (this accounts for many of the signs and symptoms associated with circulatory compensation). However, as tissues are starved of oxygen and nutrients over a prolonged ischaemic time, local mediators are released as part of the inflammatory responses, leading to organ microvasculature vasodilation and capillaries re-open to maintain oxygen delivery and reduce hypoxia.41 This is a hallmark of developing MODS.
Nursing Practice
Clinical management of hypovolaemia centres on minimising fluid loss and rapid restoration of circulating blood volume41 once the airway and breathing are secure. More than one large-bore intravenous cannulae are usually inserted and lost circulating volume is replaced by colloids, isotonic crystalloids or blood products to achieve haemodynamic endpoints (e.g. MAP >65 mmHg). Body heat can be lost rapidly due to blood loss, the rapid infusion of room temperature fluids and exposure in the pre-hospital setting or during repeated physical examination. It is therefore important to institute measures to maintain patient temperature >35°C to avoid coagulopathies and loss of thermoregulation.42 The aim is to ameliorate the lethal triad of anaemia, coagulopathy and hypothermia.40–42
Debate surrounds early surgical intervention prior to aggressive fluid resuscitation.40 The premise is that allowing a lower perfusion pressure prior to achieving haemostasis with controlled or no fluid infusion results in less blood loss, due to the compensatory mechanisms described above.40 Use of medications such as Factor IVa and EPO also remains controversial in the setting of critical haemorrhage.42 Guidelines for ‘massive transfusion’ currently being finalised by the National Blood Authority (NBA) do not recommend use of Factor IVa beyond licensed indications, although there may be an indication when conventional therapy has failed to secure haemostasis following massive blood loss and transfusion of blood products. The current debate also includes dosage and thromboembolic complications associated with its use.42 The NBA is a statutory agency established in 2003 to improve and enhance the management of the Australian blood banks and plasma product sector. Current initiatives of the agency include development and promulgation of evidence based guidelines for both massive transfusion and intensive care.
Fluid resuscitation
Fluid resuscitation is a first-line treatment for hypovolaemic shock; providing fluid volume increases preload and therefore cardiac output (Starling’s law) and organ perfusion. A related principle is that the fluid infused should reflect fluid loss, e.g. plasma replacement in burns, fresh blood in massive haemorrhage. Giving a ‘fluid challenge’ is not always appropriate; the determining factors will be assessment of volume responsiveness, and whether the infusion will not be deleterious, causing overload, fluid shifts and perpetuating inflammatory responses.6 The fluid type, volume, rate and targeted endpoints is documented;41 often this is structured as a bolus dose in volume/kg to achieve a measured haemodynamic variable. When massive transfusion is required, attention should be given to product selection and hence a protocol can be employed.
Independent Practice
Critical care nurses must be efficient and practised at initial patient assessment to establish the degree of compensation occurring in a hypovolaemic patient. Figure 20.1 highlights clinical manifestations of haemorrhage. Careful consideration of a patient’s clinical picture will establish a hierarchy and priority of needs. Most hospitals have some level of track and trigger response that escalates care to appropriate levels (e.g. MET calling criteria), however nurses are in a position to establish first line management such as intravenous access where this is a required skill. There are also many examples of protocols and guidelines for nurses to initiate fluid resuscitation where a patient has indications of inadequate circulating blood volume; e.g. a fluid bolus up to 20 mL/kg of colloidor 30–40 mL/kg crystalloid may be recommended (depending on organisational guidelines).
Collaborative Management
Selection of the appropriate fluid indications for surgical management and ‘permissive hypotension’ (deliberate limiting or minimising resuscitation until after adequate surgical control of haemorrhage).40,42 will be assessed by the multidisciplinary team. Goal-directed therapy includes prevention of tissue hypoxia, typically through rigorous fluid resuscitation with either crystalloids or colloids to achieve specific haemodynamic endpoints (e.g. a CVP of 8–12 mmHg, MAP >70 mmHg, urine output >0.5 mL/kg/h). Vasopressor and inotrope therapy may be then added to maintain adequate perfusion pressure; noradrenaline is the vasopressor of choice because of vasoconstrictor effects.43
Preload management
The colloid versus crystalloid fluid resuscitation debate (use of albumin-based solutions or colloids) continues despite findings from the SAFE study conducted in Australasia; crystalloids (isotonic saline based solutions) were as effective as colloids for fluid resuscitation.44–46 The scientific rationale for using colloids over crystalloids is to preserve plasma oncotic pressure so as to retain intravascular fluid and minimise oedema. Colloids may also attenuate the inflammatory response.20 If moderate to severe hypovolaemia is suspected then blood is often used to improve oxygen-carrying capacity. Further dilution of blood by volume expanders increases hypoxia (otherwise known as isovolaemic anaemia) and red cells are usually needed. Use of isotonic saline as a volume expander is common, although resuscitation with large volumes of saline solutions can be associated with hyperchloraemic acidosis.40 Blood and blood components are usually considered necessary where patients exhibit signs of moderate to severe haemorrhage (see Figure 20.2). There is no perfect resuscitation fluid, and selection is guided by patient condition and the type of fluid lost.
There are a number of factors to consider when administering blood products in massive volume. Massive transfusion is defined as replacement of a patient’s total blood volume in less than 24 hours (approximately 10 units of red cells);47,48 although the literature is inconsistent.48 A number of complications are evident (e.g. transfusion reactions, coagulopathies, hypothermia, sepsis)3 and is associated with high mortality.48 Patients receiving massive blood transfusions require careful monitoring for signs of metabolic derangements, hypothermia, citrate toxicity, hyperkalaemia and coagulopathies (due to depletion of clotting factors). Dilution and clotting factor consumption cause microvascular bleeding, often manifesting as oozing from multiple sites even after surgical correction.47,48 Massive transfusion of stored blood with high oxygen affinity adversely affects oxygen delivery to the tissues. It is therefore preferable to transfuse blood cells that are less than 1 week old; 2,3-diphosphoglycerate levels rise rapidly after transfusion, and normal oxygen affinity is usually restored within a few hours of transfusion.47
Each unit of blood contains approximately 3 g of citrate, which binds to ionised calcium. A healthy adult liver metabolises 3 g of citrate every 5 minutes. If blood is transfused rapidly or the liver is impaired, citrate toxicity and hypocalcaemia may develop. The patient should therefore be monitored for signs of tetany, hypotension and electrocardiographic evidence of hypocalcaemia.47 As stored blood ages, plasma potassium levels rise (possibly to over 30 mmol/L). Hypokalaemia may be more common as red cells begin active metabolism and intracellular uptake of potassium restarts with transfusion.47
Acid–base disturbances may also be evident due to the stored blood lactic acid levels and the citric acid. Citrate metabolises to bicarbonate, and a profound metabolic alkalosis may result from massive blood transfusion. As hypothermia causes reduced citrate and lactic acid metabolism, an increase in the affinity of haemoglobin to oxygen, platelet dysfunction and an increased tendency for cardiac dysrhythmias,47 the patient and the blood transfused should be warmed to avoid complications.
Leucocyte depletion occurs during donation in Australia and decreases up-regulation of the inflammatory immune response associated with transfusion. Current clinical practice guidelines for the administration of blood products and red cells to stable adult patients are listed in Tables 20.5 and 20.6. A new structure with multiple guidelines has been developed and the massive transfusion guideline is complete and will be followed by a number of other specialised guidelines. All guidelines will be available to download from the National Blood Authority website as they are completed (see Online resources).
TABLE 20.5 Clinical practice guidelines for red blood cell and platelet administration
| Appropriate Use of Blood Components For Stable Adults & Children >4 months (corrected) age Adapted from NHMRC/ASBT guidelines (www.anzsbt.org.au) Haemoglobin is NOT the sole deciding factor for transfusion – consider other patient factors e.g. signs of hypoxia and ongoing blood loss. |
|
| Red Cells | |
| Hb | Considerations |
| <70 g/L | Transfusion is often clinically useful unless early Hb recovery is expected. A threshold of <60 g/L may be appropriate for children. |
| 70–100 g/L | Likely to be appropriate during surgery with major blood loss or if there are signs or symptoms of impaired oxygen transport. |
| >80 g/L | May be appropriate to control anaemia-related symptoms in a patient on a chronic transfusion regimen or during marrow suppressive therapy. |
| >100 g/L | Not likely to be appropriate unless there are specific indications |
| WHAT DOSE? Red Cells (mL) = 0.4 × wt (kg) × (desired – actual) Hb (g/L) |
|
| Platelets | |
| Use of platelets is likely to be appropriate as prophylaxis for: | |
| Indication | Considerations |
| Bone Marrow Failure | At a platelet count of <10 Ö 109/L in the absence of risk factors and <20 Ö 109/L in the presence of risk factors (e.g. fever, antibiotics, evidence of haemostatic failure) |
| Surgery/Invasive | To maintain platelet count at >40 Ö 109/L. For surgical procedures with high risk of bleeding (e.g. ocular or neurosurgery) it may be procedure appropriate to maintain at 100 Ö 109/L |
| Platelet Function Disorders | May be appropriate in inherited or acquired disorders, depending on clinical features and setting. In this situation, platelet count is not a reliable indicator |
| Use of platelets is likely to be appropriate as therapy for: | |
| Bleeding | Any patient in whom thrombocytopenia is considered a major contributory factor. |
| Massive Bleeding/Transfusion | Confined to patients with thrombocytopenia and/or functional abnormalities who have significant bleeding. Often with platelet count <50 Ö 109/L (<100 Ö 109/L with diffuse microvascular bleeding). |
Cardiogenic Shock
Cardiogenic shock manifests as circulatory failure from cardiac dysfunction,49 and is reflected in a low cardiac output (CI <2.1 L/min/m2), hypotension (SBP <90 mmHg) and severe pulmonary congestion, high central vascular filling pressures (CVP; PAOP >18 mmHg).50 Additional invasive parameters are: intrathoracic blood volume index >850 mL/m2; global end-diastolic volume >700 mL/m2; and extravascular lung volume index >10 mL/kg.51,52 Cardiogenic shock is commonly associated with AMI and manifests when 40% or more of the left ventricle is ischaemic. It is also related to mechanical disorders (e.g. acute cardiac valvular dysfunction or septal defects), deteriorating cardiomyopathies or congestive cardiac failure,53,54 trauma and obstruction or inhibition of left ventricular ejection (referred to as obstructive shock e.g. pulmonary emboli, dissecting aneurysm, tamponade)37,53 (see Chapter 10). Myocardial depression from non-cardiac causes such as sepsis, acidosis, myocardial depressant factor, hypocalcaemia or drug impact55 may be so severe as to present as cardiogenic shock.
Incidence has been estimated at 3% of patients presenting with AMI, and mortality remains high (50–80%),56 given death from AMI overall is 7%. This is despite treatment advances including emergency revascularisation.57,58 Wider distribution of interventional cardiac revascularisation services has likely improved outcome for patients who present early in the course of their acute disease.
Clinical signs include poor peripheral perfusion, tachycardia and other signs of organ dysfunction such as confusion, agitation, oliguria, cool extremities, dyspnoea, many of which are present in hypovolaemic shock.49 Compensatory mechanisms are conflicting for a patient with cardiogenic shock, as cardiac workload is increased on an already-failing heart yet cardiac muscle oxygen delivery may be compromised.54 A careful but rapid assessment of the clinical history is helpful in differentiating the precipitant cause of this shock.
Clinical Manifestations
The clinical features of cardiogenic shock are reflective of congestive cardiac failure, although with greater severity:50,51,58
• low cardiac output and hypotension
• poor peripheral perfusion: pale, cool, clammy peripheries
• altered mentation, restlessness and anxiety
• pulmonary congestion with widespread inspiratory crackles and hypoxaemia (perhaps with frank pulmonary oedema)
• respiratory alkalosis (hyperventilation) or acidosis (respiratory fatigue)
In the absence of invasive monitoring, the profile of hypotension, peripheral hypoperfusion, and severe pulmonary and venous congestion are evident although this ‘classic’ profile is not universal. On initial examination, 30% of patients with shock of left ventricular aetiology had no pulmonary congestion and 9% had no hypoperfusion.59
Based on the underlying pathology of an acute left ventricular myocardial infarction, the structural or contractile abnormality impairs systolic performance resulting in incomplete left ventricular emptying.50 This results in subsequent progressive congestion of first the left atrium, then the pulmonary circulation, right ventricle, right atrium and finally the venous circulation.50,60,61 When invasive haemodynamic monitoring is available, sequence of changes exist as illustrated in Figure 20.3.
A patient with cardiogenic shock is also assessed and monitored for their oxygen delivery and tissue oxygen requirements (oxygen consumption). Systemic DO2 falls in proportion to a declining cardiac output, and is further worsened as hypoxaemia develops due to pulmonary oedema. Initially, VO2 may be sustained by an increase in tissue oxygen extraction ratio (O2ER).62 Normally 25% of delivered oxygen is extracted by tissues, but as delivery falls, tissues extract proportionally more oxygen to meet metabolic needs. Oxygen consumption can therefore be sustained until the severity of oxygen delivery deficit exceeds the ability to increase extraction. Maximal extraction is approximately 50%, and consumption falters when oxygen delivery falls to around 500–600 mL/min (cardiac index <2.2 L/min/m2).62–64 While use of a PAC is a well-described measure of severity in cardiogenic shock (as with hypovolaemic shock), evidence of improved patient outcome is unclear.28,65
Once oxygen consumption falls below tissue needs, resulting anaerobic metabolism causes lactate generation and the subsequent lactic acidosis.50,62 Progressive tissue ischaemia and injury ensues, along with worsening metabolic acidosis unless oxygen delivery can be restored. Myocardial contractile performance further worsens when myocardial ischaemia develops or when existing ischaemia or infarction is worsened, and a vicious cycle of ischaemia and dysfunction ensues.62
• Tachycardia offsets low stroke volume but increases myocardial oxygen consumption and decreases diastolic duration, reducing coronary perfusion time.
• Vasoconstriction limits the severity of hypotension but increases resistance to left ventricular emptying and may contribute to worsening of the cardiac output, in particular when cardiogenic shock is due to contractile dysfunction.
• An increase in cardiac workload to overcome the rise in systemic afterload increases myocardial oxygen demand, but cannot be met due to coronary artery occlusion.
• Developing pulmonary congestion is no longer contained within the pulmonary capillary and moves into the alveolar capillary space, creating pulmonary oedema, further impeding oxygen delivery to the circulation.
Nursing Practice
Independent Practice
Assessment
Frequent, thorough assessment of the patient’s status is essential, focusing on:
1. identification of patients at risk of clinical deterioration;
2. assessment of the severity of shock and identification of organ or system dysfunction;
[/not-level-membership-for-critical-care-medicine-category]
