[level-membership-for-surgery-category]
Chapter 22 Management of Pituitary, Adrenal, and Thyroid Disease
PITUITARY DISORDERS
Anatomy
The optic chiasma may be in front of (15%), above (80%), or behind the sella (5%). Within this optic chiasma, nerve fibers from the nasal half of the retina cross over to the opposite optic tract while those from the temporal half remain uncrossed. The close association of the pituitary gland with the optic chiasma explains the visual symptoms associated with expanding masses in this region.1
The median eminence is an intensely vascular component at the baseline of the hypothalamus that forms the floor of the third ventricle. The pituitary stalk arises from the median eminence. The hypothalamus extends anteriorly to the optic chiasma and posteriorly to the mammillary bodies. It was not until the mid-1960s that hypothalamic releasing hormones were isolated and identified (Table 22-1).
Table 22-1 Pituitary Hormones, Hypothalamic Hormones, and Other Regulatory Factors
| Pituitary Hormones | Hypothalamic Hormones | Other Regulatory Factors |
|---|---|---|
| Thyrotropin | TRH | T4, T3, dopamine, Pit-1 |
| Corticotropin | CRH | ADH, adrenaline, cortisol |
| Luteinizing hormone | LH-RH | Estrogen, progesterone, testosterone |
| Follicle-stimulating hormone | LH-RH | Activin, estrogen, inhibin, follistatin, testosterone |
| Growth hormone | GH-RH | Somatostatin, estrogens, T4, Pit-1 |
| Prolactin | PRF | Dopamine, TRH, Pit-1, estrogen, serotonin, vasoactive intestinal peptide, GnRH-associated peptide |
TRH, thyrotropin-releasing hormone; CRH, corticotropin-releasing hormone;
LH-RH, luteinizing hormone-releasing hormone; GH-RH, growth hormone-releasing hormone; PRF, prolactin-releasing factor; Pit-1, pituitary-specific transcription factor; ADH, antidiuretic hormone.
Pituitary Tumors
Pituitary tumors may present with either hypofunction or hyperfunction, as well as symptoms directly related to the mass effect of the tumor (Table 22-2). Since the advent of computed tomography (CT), microadenomas have been arbitrarily designated as equal to or less than 10mm in diameter and macroadenomas as greater than 10mm in diameter. They are invariably benign, with no sex predilection. Pituitary adenomas are rarely associated with parathyroid and pancreatic hyperplasia or neoplasia as part of the multiple endocrine neoplasia type I (MEN I) syndrome. Pituitary carcinomas are rare, but metastases from other solid malignancies can occur more frequently.2
| Endocrine Effects | ||
|---|---|---|
| Mass Effects | Hyperpituitarism | Hypopituitarism |
| Headaches | GH: acromegaly | GH: short stature in children, increased fat mass, decreased strength and well-being in adults |
| Chiasmal syndrome | Prolactin: hyperprolactinemia | Prolactin: failure of postpartum lactation |
| Hypothalamic syndrome | Corticotropin: Cushing’s disease Nelson’s syndrome | Corticotropin: hypocortisolism |
| Disturbances of thirst, appetite, satiety, sleep, and temperature regulation | LH/FSH: gonadal dysfunction or silent α-subunit secretion | LH or FSH: hypogonadism |
| Diabetes insipidus | Thyrotropin hyperthyroidism | Thyrotropin: hypothyroidism |
| SIADH | ||
| Obstructive hydrocephalus | ||
| Cranial nerves III, IV, V1, V2, and VI dysfunction | ||
| Temporal lobe dysfunction | ||
| Nasopharyngeal mass | ||
| CSF rhinorrhea | ||
SIADH = syndrome of inappropriate antidiuretic hormone; GH = growth hormone; LH = luteinizing hormone; FSH = follicle-stimulating hormone.
PITUITARY ADENOMAS
Approximately 50% of pituitary adenomas are prolactinomas, 15% are growth hormone (GH)-producing, 10% are corticotropin-producing, and less than 1% secrete thyrotropin. Nonfunctioning pituitary adenomas, or more appropriately named nonsecretory adenomas represent about 25% of pituitary tumors. Most of these adenomas on morphologic examination reveal granules containing hormones, typically components of glycoprotein hormones. Autopsy studies suggest that up to 20% of normal individuals harbor incidental pituitary microadenomas that are pathologically similar in distribution to those that present clinically.3
Prolactinoma
Hyperprolactinemia
Hyperprolactinemia impairs pulsatile gonadotropin (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]) release, likely through alteration in hypothalamic luteinizing hormone-releasing hormone (LHRH) secretion. Women of reproductive age usually present with oligomenorrhea, amenorrhea, galactorrhea, and infertility. Those with longstanding amenorrhea are less likely to have galactorrhea secondary to longstanding estrogen deficiency. Postmenopausal women and men usually come to medical attention because of a mass effect, such as headaches and visual field defects.4
Drug history is an important part of the initial evaluation of patients with elevated prolactin level, because some medications are associated with hyperprolactinemia and their discontinuation (if possible) will avoid any further, often expensive, workup. Other common conditions associated with elevated prolactin levels include pregnancy and hypothyroidism (Table 22-3).
| Physiologic | Pathologic | Pharmacologic |
|---|---|---|
| Pregnancy | Prolactinoma | TRH |
| Postpartum | Acromegaly (25%) | Psychotropic medications |
| Newborn | Hypothalamic disorders | Phenothiazines |
| Stress | “Chiari-Frommel” | Reserpine |
| Hypoglycemia | Craniopharyngioma | Methyldopa |
| Sleep | Metastatic disease | Estrogen therapy |
| Postprandial hypoglycemia | Pituitary stalk secretion or compression | Metoclopramide, cimetidine (especially intravenous) |
| Intercourse | Hypothyroidism | Opiates |
| Nipple stimulation | Renal failure | Verapamil |
| Liver disease | Some SSRIs, including fluoxetine and fluvoxamine | |
| Chest wall trauma (burns, shingles) |
SSRI = Selective serotonin reuptake inhibitor.
The prolactin level usually correlates well with the size of the tumor. Serum prolactin level above 200μg/L is almost always indicative of a prolactin-producing pituitary tumor. However, a serum prolactin level below 200μg/L can be seen in the presence of a large pituitary adenoma, because stalk compression from an adenoma that does not secrete prolactin can also cause hyperprolactinemia. Stimulatory tests, including the TRH stimulation test, which are performed to determine whether an elevated prolactin is a result of a prolactinoma, are nonspecific and cannot be used to diagnose or exclude a tumor. Large prolactinomas may be associated with a falsely low prolactin level. Dilution of serum will reveal the markedly elevated prolactin levels.
Observational studies in patients with microadenomas indicate that serum prolactin concentration or adenoma size increases in only a minority of patients; indeed, serum prolactin deceases in a majority of patients over time. Details of the relationship between prolactin adenomas and amenorrhea are found in Chapter 16.
Treatment
Medical therapy during pregnancy often stirs debate about the continuation of bromocriptine. Tumor-related complications are seen in about 15% of pregnancies and in only 5% of women with microadenomas. A sensible approach would be to stop bromocriptine when pregnancy begins, and then follow the clinical status with serum prolactin levels and visual field examinations. If there is significant worsening in clinical status, bromocriptine could be reinstituted. The adenoma can be followed yearly by MRI, increasing the duration between imaging studies if size is stable.5
Acromegaly
Acromegaly may occur at a rate of 3 to 4 cases per million per year, with mean age at diagnosis of 40 years in men and 45 years in women. The GH-secreting tumors tend to be more aggressive in younger patients. Classical clinical features are listed in Table 22-4. More than 95% of cases of acromegaly are caused by GH-secreting pituitary tumors. In rare cases, they are caused by ectopic GH-releasing hormone (GH-RH) secretion, mainly carcinoids and pancreatic islet cell tumors. Patients with acromegaly have a 3.5-fold increased mortality rate, with cardiovascular disease being the most common cause of death. Somatotrope adenomas appear to be monoclonal in origin. A gsp mutation in a GspIa subunit in GH cells, leading to continuous GH secretion, has been shown to cause acromegaly.6
| Coarsening of facial features |
| Prominent jaw and frontal sinus |
| Broadening of hands and feet |
| Hyperhidrosis |
| Macroglossia |
| Signs of hypopituitarism |
| Diabetes mellitus (10%–25%) |
| Skin tags (screening for colonic polyps required) |
| Hypertension (25%–30%) |
| Cardiomyopathy (50%–80%) |
| Carpal tunnel syndrome |
| Sleep apnea (5%) |
Insulin-like growth factor-I has a longer plasma half-life than GH and is an excellent initial screening test for those suspected to have acromegaly. An elevated IGF-I level in a clinical setting suggestive of acromegaly almost always confirms the diagnosis. Patients with poorly controlled diabetes and malnutrition may have falsely low serum IGF-I levels. The oral glucose tolerance test remains the gold standard test to confirm the diagnosis. Normal individuals suppress their GH level to less than 1μg/L (using chemiluminescent assays) within 2 hours after ingestion of 100 grams of oral glucose solution.
Treatment
Long-term observations of patients on somatostatin analogues have shown no evidence for tachyphylaxis. Some degree of tumor shrinkage is expected in up to 50% of patients, although in most cases there is less than 50% shrinkage in tumor size. The most common side effects are gastrointestinal, including diarrhea, abdominal pain, and nausea. The most serious side effect of somatostatin analogues is cholelithiasis, seen in up to 25% of patients. Its long-term management is similar to that for cholelithiasis in the general population, and routine ultrasonographic screening is not indicated. There have been very few reports of the use of a somatostatin analogue during pregnancy.7,8
Cushing’s Disease
Corticotropin-secreting pituitary adenoma is the most common cause of endogenous Cushing’s syndrome (60%), with the rest being adrenal (25%) or ectopic (15%) in origin. The term Cushing’s disease refers specifically to a pituitary tumor as the cause. Signs and symptoms suggestive of hypercortisolism are listed in Table 22-5. Many signs and symptoms of Cushing’s disease are nonspecific, including hypertension, abnormal glucose tolerance, menstrual irregularities, and psychiatric disturbances, including depression. Most women with Cushing’s disease have reduced fertility.9
| Clinical Feature | Approximate Prevalence (%) |
|---|---|
| Obesity | |
| General | 80–95 |
| Truncal | 45–80 |
| Hypertension | 75–90 |
| Menstrual disorders | 75–95 |
| Osteopenia | 75–85 |
| Facial plethora | 70–90 |
| Hirsutism | 70–80 |
| Impotence/decreased libido | 65–95 |
| Neuropsychiatric symptoms | 60–95 |
| Striae | 50–70 |
| Glucose intolerance | 40–90 |
| Weakness | 30–90 |
| Bruising | 30–70 |
| Kidney stones | 15–20 |
| Headache | 10–50 |
Women with Cushing’s disease typically have fine facial lanugo hair and may have acne and temporal scalp hair loss secondary to increased adrenal androgen secretion. There is usually a 3- to 6-year delay in diagnosis of patients with Cushing’s disease, and it may be possible to date the onset of the disease by determining which scars are pigmented due to excess secretion of corticotropin and other melanotropins.
Diagnosis
Twenty-four hour urinary free cortisol measurement is the single best test for diagnosis of Cushing’s syndrome (Fig. 22-1). Because of the significant overlap between normal individuals and those with Cushing’s syndrome, random serum cortisol has no role in the diagnosis of Cushing’s syndrome. A 1-mg overnight dexamethasone suppression test with a morning cortisol level below 1.8μg/dL virtually rules out the disease but is associated with an up to 40% false-positive rate.
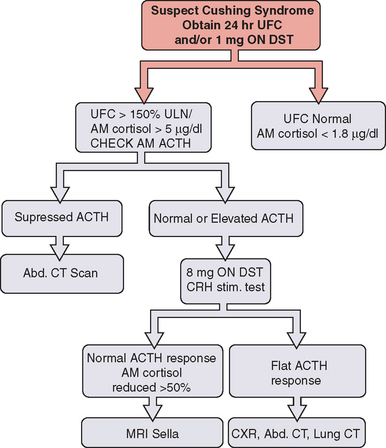
A combination of low-dose dexamethasone suppression test and corticotropin-releasing hormone (CRH) stimulation test has been shown to have 100% diagnostic accuracy in a National Institutes of Health study. This test may have a significant value in establishing the diagnosis in those with pseudo-Cushing’s and elevated 24-hour urinary free cortisol. Other tests useful in establishing the diagnosis of Cushing’s disease include midnight serum and salivary cortisol (see Fig. 22-1).
Once the diagnosis of Cushing’s syndrome has been established, the next step is to find out whether cortisol hypersecretion is corticotropin dependent (see Fig. 22-1). Although an undetectable or low corticotropin level is consistent with adrenal etiology, low-normal corticotropin may be seen in both ectopic Cushing’s syndrome and corticotropin-secreting pituitary tumor. The CRH stimulation test is used to differentiate between the two. Although corticotropin levels tend to be higher in those with ectopic Cushing’s syndrome compared to patients with pituitary disease, there is considerable overlap. The high-dose dexamethasone test or the CRH stimulation test is helpful in differentiation of the two disorders. Cortisol levels are not suppressed with the high-dose (8 mg) dexamethasone test in patients with ectopic corticotropin syndrome, and CRH stimulation may not lead to a further rise in corticotropin.10 The gold standard test to differentiate pituitary Cushing’s syndrome from ectopic corticotropin-producing tumor is inferior petrosal sinus sampling. This test should be performed by an experienced neuroradiologist; it is essential to note that it cannot be used to make the diagnosis of Cushing’s syndrome.
Nonsecretory and Glycoprotein-Secreting Pituitary Adenomas
Glycoprotein-secreting Adenomas
Rarely, pituitary adenomas can secret glycoproteins, including FSH, LH, or thyrotropin. An FSH adenoma may cause amenorrhea in a woman, or an LH adenoma may cause precocious puberty in a boy. Diagnosis is confirmed by measurement of either intact glycoprotein hormones or their α and β subunits. Levels of the α subunit tend to be inappropriately elevated, compared with those of the intact hormone itself.11
HYPOPITUITARISM
Causes of Hypopituitarism
Lymphocytic Hypophysitis
Lymphocytic hypophysitis is an autoimmune disease often presenting in women during or after pregnancy. The clinical manifestations are secondary to hypopituitarism or adrenal insufficiency or are due to a pituitary mass effect. Serum prolactin is elevated in half of patients but may be decreased. Antipituitary antibodies are present in some patients, and other autoimmune endocrine disorders, including Hashimoto’s thyroiditis and Addison’s disease, have been seen by others.12
Recent experience from Germany suggests that one should be cautiously optimistic about high-dose steroid therapy. MRI findings improved in 88% of patients treated with high-dose steroids, but clinical normalization was quite variable, with none achieving complete recovery.13
Surgery for mass effect in lymphocytic hypophysitis can lead to rapid relief of neurologic symptoms, but endocrinologic improvement was seldom reported. Indications for surgery are the presence of gross chiasma compression, ineffectiveness of corticosteroid therapy, and the impossibility of establishing the diagnosis of lymphocytic hypophysitis with sufficient certainty by conservative evaluation.14
Sheehan’s Syndrome
Sheehan’s syndrome is the result of ischemic infarction of the normal pituitary gland, leading to hypopituitarism secondary to postpartum hemorrhage and hypotension.15 Patients have a history of failure to lactate postpartum, failure to resume menses, cold intolerance, or fatigue. Some women may have an acute crisis mimicking apoplexy within 30 days postpartum. There is often subclinical central diabetes insipidus.16
ADRENAL GLAND DISORDERS
Anatomy and Physiology
The zonae reticularis and fasciculata are under the control of corticotropin released by the pituitary gland in response to hypothalamic CRH. CRH in turn is regulated by cortisol-negative feedback, stress, and a circadian rhythm. Besides increasing the synthesis of cortisol, corticotropin is trophic for the adrenal gland so that a lack of corticotropin results in atrophy of the zonae fasciculata and reticularis. Although corticotropin has some effect on aldosterone production, the zona glomerulosa is predominantly under the control of renin. Understanding the anatomy and physiology of the adrenal gland is crucial to understanding its hypofunction and hyperfunction.17
Adrenal Insufficiency
Etiology
Clinical adrenal insufficiency results from hypofunction of the adrenal cortex. This may be due to destruction of the adrenal gland itself, referred to as Addison’s disease or primary adrenal insufficiency. Alternatively, it may be due to a lack of either corticotropin (i.e., secondary adrenal insufficiency) or CRH.18
The most common cause of Addison’s disease in adults (80%) is autoimmune destruction of the adrenal gland. This is often seen in association with other autoimmune diseases, including Hashimoto’s thyroiditis, Graves’ disease, or type 1 diabetes mellitus. Adrenal insufficiency in this setting is known as type II autoimmune polyglandular syndrome. Type I autoimmune polyglandular syndrome, more commonly seen in children, consists of Addison’s disease, hypoparathyroidism, and mucocutaneous candidiasis.
Other causes associated with adrenal insufficiency are listed in Table 22-6. Currently, acquired immunodeficiency syndrome is the most common cause of infectious adrenal destruction, and the antiphospholipid syndrome (lupus anticoagulant) is increasingly being recognized as a cause of adrenal hemorrhage.
Table 22-6 Other Causes of Primary Adrenal Insufficiency in Adults
Secondary adrenal insufficiency is a result of adrenal gland atrophy from corticotropin deficiency. This most often results from pituitary corticotroph atrophy owing to previous exogenous glucocorticoid use,19 hypopituitarism, or isolated corticotropin deficiency (usually postpartum).
Clinical Presentation
The underlying etiology of adrenal insufficiency determines the clinical presentation (Table 22-7). Under the regulation of corticotropin, cortisol and adrenal androgens are lost in both primary and secondary adrenal insufficiency. Aldosterone production, predominantly regulated by renin, remains intact in secondary adrenal insufficiency. Therefore, hyperkalemia and profound dehydration with orthostatic hypotension are seen in primary adrenal insufficiency only. Likewise, hyperpigmentation of the skin or mucous membranes (secondary to increased corticotropin) is seen in primary adrenal insufficiency only. The absence of hyperkalemia or hyperpigmentation does not exclude adrenal insufficiency. In addition to hyponatremia and hyperkalemia, laboratory abnormalities in adrenal insufficiency may include hypoglycemia (usually chronic), hypercalcemia, eosinophilia, and lymphocytosis.
| Primary | Secondary | |
|---|---|---|
Diagnosis
In secondary adrenal insufficiency, however, the corticotropin stimulation test is not always abnormal. Adequate corticotropin may be present to prevent adrenal gland atrophy, thereby resulting in a response to the supraphysiologic dose of corticotropin used in the test. However, the HPA axis may not be able to respond to stress. In patients with suspected secondary adrenal insufficiency and a normal corticotropin stimulation test, CRH is now available to assess corticotropin response. In addition, the insulin tolerance test or the metyrapone test evaluate the integrity of the HPA axis by its response to hypoglycemia or inhibited cortisol synthesis, respectively. Although not widely used, some investigators find that a 1μg corticotropin stimulation test may be more sensitive at detecting mild adrenal insufficiency.20
Treatment
Cortisol 20mg in the morning and 10mg in the evening, or prednisone, 5 to 7.5mg daily, provides dramatic relief of symptoms. However, to prevent Cushing’s syndrome, the smallest dose needed to control the patient’s symptoms should be used. For a minor illness, the glucocorticoid dose should be doubled for as short a time as needed. For a major stress, parenteral hydrocortisone, 200 to 400mg daily, is given initially and then rapidly tapered. Aldosterone replacement is required in primary adrenal insufficiency only and is given as fludrocortisone acetate (Florinef Acetate), 0.05 to 0.2mg daily. The dose is adjusted according to the blood pressure and potassium level. Renin levels may be required to assess plasma volume. Adrenal androgens are not replaced.21
In undiagnosed patients with suspected adrenal crisis, dexamethasone, 2 to 4 mg intravenously or intramuscularly, should be given along with saline and glucose. Dexamethasone does not interfere with the cortisol assay. The corticotropin stimulation test should then be done as soon as possible.
Late-Onset Congenital Adrenal Hyperplasia
Women with severe forms of CAH tend to have reduced fertility rates because of oligo-ovulation. Successful conception requires careful endocrine monitoring and often ovulation induction.22
Late-Onset CAH and Pregnancy
Fertility rates may be lower in women with CAH, but with successful medical management, women, especially those with 21-hydroxylase deficiency, may conceive.23
From a fetal and neonatal standpoint, accurate prenatal diagnosis of 21-hydroxylase deficiency and 11β-hydroxylase deficiency is necessary to allow for prenatal treatment using dexamethasone in an attempt to minimize clinical problems in the neonate. Dexamethasone can cross the placenta and suppress fetal adrenal steroidogenesis and potentially prevent masculinization of affected female fetuses.24
Large quantities of estrogen are produced during normal human pregnancy, and, after the first 3 to 4 weeks of gestation, the placenta produces nearly all of the estrogen. The major precursor for estrogen production is DHEAS, which is synthesized in the fetal adrenal gland. The adrenals of the human fetus at term are as large as those of adults, weighing 8 to 10g or more. The fetal adrenals are principally composed of an inner fetal zone that accounts for 85% of the total volume. The outer zone, the neocortex, develops into the mature adrenal cortex, which is only 15% of the total volume. The capacity of the fetal adrenals for steroidogenesis is enormous, and near term, the fetal adrenals secrete 100 to 200mg of the steroid per day. The total daily steroid production by the adrenals in an unstressed adult is approximately 35mg.25
Only a small amount of the steroids in the maternal circulation reach the fetal compartment in normal pregnancy. For example, a small amount of cortisol in maternal plasma crosses the placenta, both because the reentry pathway dominates and because cortisol within the trophoblast is converted to cortisone by 11β -hydroxysteroid dehydrogenase. Circulating 19-carbon steroids in the maternal compartment, such as DHEAS, DHEA, androstenedione, and testosterone, do not reach the fetal compartment because of the presence of aromatase enzymes of the syncytiotrophoblast that are used for the conversion of 19-carbon steroids to estrogens. This mechanism protects the female fetus from possible virilization in women who may be hyperandrogenic during pregnancy.
Cushing’s Syndrome
Clinical Presentation
The clinical features of Cushing’s syndrome are listed in Table 22-5. With respect to specificity for Cushing’s syndrome, thinning of the skin, purple striae, and bruising are the best clinical signs. Hypokalemia, edema, and hyperpigmentation are more commonly seen in ectopic corticotropin secretion, in which corticotropin and cortisol levels tend to be much higher. If a pregnant woman is in a hypercortisolemic state due to exogenous glucocorticoid therapy, the fetus is at potential risk of hypoadrenalism and one needs to forewarn the neonatologist.
Diagnosis
Differential Diagnosis
Figure 22-1 will allow confirmation of hypercortisolism and assessment of potential causes. If an abnormal result is obtained by either the 24-hour urinary free cortisol or the 1-mg overnight dexamethasone suppression test, the pseudo-Cushing’s state of alcoholism or endogenous depression should first be sought by a careful history, physical examination, and laboratory evaluation. A repeat urinary free cortisol with alcohol abstention should be normal.
The corticotropin-dependent hypercortisol diseases and syndromes are:
The corticotropin-independent hypercortisol diseases and syndromes are:
Cushing’s Syndrome and Pregnancy
Although the clinical diagnosis may be difficult during pregnancy, one should seek out catabolic signs. There is also a greater likelihood of diabetes, worsening hypertension, cardiomyopathy, and psychological and muscular symptoms. The diagnostic testing, such as low- and high-dose dexamethasone testing, remains the same. One should remember that with higher cortisol binding globulin levels, the usual serum (total) cortisol levels may be falsely high.26
Poor wound healing should be anticipated as well as prolonged bleeding from any surgical procedures.
Treatment is the same as for the nonpregnant state although surgical intervention such as adrenalectomy, if required, should be performed early in the second trimester. Occasionally, ketoconazole27 has been used to medically control the hypercortisolism without adverse effects on the mother or the baby. Adrenal insufficiency can occur postoperatively and therefore preventive therapy should be initiated.28
Hyperaldosteronism
Excess aldosterone results in hyperaldosteronism with hypertension, hypokalemia, and metabolic alkalosis. This may be associated with Cushing’s syndrome, particularly in patients with adrenal carcinoma. Isolated primary hyperaldosteronism, marked by an elevated aldosterone level and suppressed plasma renin activity, accounts for 1% to 2% of patients with hypertension; the presence of spontaneous hypokalemia or a serum potassium level less than 3.0mEq/L on diuretics should prompt an evaluation.29
The next step is to differentiate adrenal adenoma from hyperplasia. An adenoma can be differentiated by CT findings, increased 18-hydroxycorticosterone levels, or bilateral adrenal vein catheterization. Spironolactone (Aldactone), an aldosterone antagonist, is the treatment of choice for patients with hyperplasia, small adenomas, or contraindications to surgery.30
Other Mineralocorticoid Excess Syndromes
Primary hyperaldosteronism is rarely observed in pregnancy.31 Plasma aldosterone levels may be normally elevated in pregnancy and urinary potassium level may be lower than that in nonpregnant patients with hyperaldosteronism due to the effects of progesterone. Plasma renin levels should be decreased in patients with primary hyperaldosteronism. In a healthy pregnancy, plasma renin activity is usually increased and decreases in the setting of primary hyperaldosteronism.32
Another dynamic test that may be used is stimulation of renin production by positioning the patient upright. In pregnant patients, prolonged standing results in a modest increase in plasma renin activity.33 If the renin activity remains suppressed, this is suggestive of primary hyperaldosteronism.
Dexamethasone-suppressible hyperaldosteronism (or glucocorticoid-remediable aldosteronism) seems to be associated with a higher likelihood of exacerbation of hypertension during pregnancy.34 There does not appear to be a higher incidence of preeclampsia.
Pheochromocytoma
Clinical Presentation
The triad of headaches, palpitations, and diaphoresis in the presence of hypertension is classic for pheochromocytoma. Other signs and symptoms are listed in Table 22-8. More frequently recognized are silent pheochromocytomas presenting as adrenal incidentalomas. Cocaine abuse may be mistaken for pheochromocytoma.
| Classic symptoms |
| Headaches, palpitations, and diaphoresis |
| Postural hypotension |
| Tachycardia |
| Weight loss |
| Pallor |
| Hyperglycemia |
| Anxiety |
| Nausea/vomiting |
| Constipation |
| Tremulousness |
Pheochromocytoma and Pregnancy
Pheochromocytoma in a pregnant woman is life-threatening, but the prognosis appears to be improving. Maternal and fetal mortality rates of 48% and 54.4%, respectively, in the late 1960s35 dropped to 17% and 26%, respectively, by the late 1980s.36 In a 1999 review of patients with pheochromocytoma in pregnancy, the maternal and fetal mortality rates were 4% and 11%, respectively. Antenatal diagnosis of pheochromocytoma reduced the maternal mortality rate to 2%.37
Surgical intervention should be performed before 24 weeks’ gestation, after achieving adequate alpha blockade. After 24 weeks’ gestation, uterine size makes abdominal exploration and access to the tumor difficult. Optimum results are obtained if surgery is delayed until fetal maturity is reached.38 At that time, with adequate alpha blockade, elective cesarean delivery may be performed, followed immediately by adrenal exploration. Vaginal delivery appears to be higher risk than cesarean delivery. Some authors have reported success with alpha and beta blockade from the beginning of the second trimester to term, with good fetal outcomes. Malignant pheochromocytoma may recur in pregnancy. Lifelong monitoring is necessary in all patients, with extra caution in those who are pregnant.
Incidentally Discovered Adrenal Mass
Incidental adrenal masses are common, detected in approximately 2% of patients having abdominal CT scan. The differential diagnosis of such masses is listed in Table 22-9.
Table 22-9 Differential Diagnosis of Incidentally Found Adrenal Masses
| Functioning or nonfunctioning adenoma |
| Functioning or nonfunctioning carcinoma |
| Pheochromocytoma |
| Metastasis from tumors at other sites (especially malignant melanoma, lung, breast, and gastrointestinal cancers) |
| Myelolipoma |
| Cyst |
| Focal enlargement in hyperplastic gland (e.g., Cushing’s disease, congenital adrenal hyperplasia) |
| Pseudoadrenal mass arising from nearby organs |
Management of an incidentaloma is controversial; clinical judgment is required. Patients should first be clinically evaluated for evidence of adrenal hormone production (cortisol, androgens, aldosterone, catecholamines). If the tumor appears to be clinically nonfunctional, most endocrinologists would still screen biochemically for pheochromocytoma.39
Despite an absence of hormone excess, nonfunctional tumors greater than 4 to 6cm should be resected owing to an increased risk of malignancy. Nonfunctional tumors measuring 4cm and smaller can be further evaluated radiographically to determine the likelihood of benign disease. The attenuation value, obtained from a noncontrast CT scan, is a measure of a tumor’s lipid content. A value less than 10 Hounsfield units (HU) suggests fat density and is specific for adenoma. Masses of indeterminate attenuation value (10 to 20 HU) can be further classified by MRI. Masses inconsistent with adenoma on CT or MRI require repeated follow-up with CT to assess growth or fine-needle aspiration biopsy.40 A biopsy, however, is rarely necessary.
THYROID GLAND DISORDERS
Thyroid Physiology
Synthesis and Secretion of Thyroid Hormone
Thyroid hormone synthesis involves six major steps:
Thyroid Hormone Transport
Congenital Thyroid Hormone Binding Protein Abnormalities
In a recent study of 15,000 consecutive patients over a period of 4 years, congenital complete TBG deficiency was reported in 1/2500 live births and partial deficiency in 1/200. TBG excess is found in 1/15,000 live births.41 Familial dysalbuminemic hyperthyroxinemia is a familial autosomal dominant syndrome caused by abnormal albumin with an increased affinity for T4 resulting in elevated serum total T4 but normal thyrotropin level. Familial dysalbuminemic hyperthyroxinemia is the most common cause of inherited euthyroid hyperthyroxinemia.
Metabolism of Thyroid Hormones
Several studies have demonstrated that there are two separate 5′- deiodinase enzymes and one 5-deiodinase. The type I 5′-deiodinase is primarily localized in the liver and kidney. The type II 5′-deiodinase is present in the anterior pituitary and has a much higher affinity for T4. The type II 5′-deiodinase is present in the central nervous system, placenta, and skin and it is the predominant deiodinase in the fetus.42
Approximately 80 to 90μg of T4 is secreted daily and T3 secretion is approximately 20 to 30μg/day. The rest comes from peripheral conversion. During illnesses, conversion of T4 can be routed preferentially to reverse T3.
Prevalence of Thyroid Disease
The National Health and Nutrition Examination Survey (NHANES III)43 measured serum thyrotropin, total serum T4, and antithyroid antibodies in a sample representative for geographic distribution of U.S. population. The prevalence of hypothyroidism was 4.3% (0.3% clinical and 4.0% subclinical) and the prevalence of hyperthyroidism was 1.3% (0.5% clinical and 0.7% subclinical). It should be noted that mean serum thyrotropin in a healthy population was 1.5mU/L. Thyrotropin levels were higher in females compared to males, higher in the white population than African Americans or Mexican Americans. Women were also more likely to have antithyroid antibodies.
In 2004, the U.S. Preventive Health Task Force44 felt that there was insufficient “good strength” evidence to support testing or routine screening of thyroid function in the general population (Table 22-10). Most experts agree that although controversial, screening is warranted in women over age 35, men over age 65, and as routine screening in women at their first prenatal visit.45,46 The main areas of disagreement between the U.S. Preventive Task Force in 2004 and the joint statement by several endocrine associations in 200547 are
| Screening Recommendations | Organization | |
|---|---|---|
| Routine screening for subclinical thyroid disease in adults, pregnant women, and women contemplating pregnancy | 2005 | Consensus statement of the American Association of Clinical Endocrinologists, The Endocrine Society, and American Thyroid Association47 |
| Uncertain whether screening for subclinical thyroid dysfunction in nonpregnant adults is beneficial | 2004 | U.S. Preventive Services Task Force44 |
| Against routine treatment of subclinical thyroid dysfunction Insufficient evidence to support population-based screening | 2004 | U.S. Preventive Services Task Force44 |
Thyroid Disease and Fertility
Thyroid disease should be considered in patients undergoing investigation for menstrual problems48 or infertility.49 A recent prospective study by Poppe and colleagues of more than 400 infertile women showed a higher prevalence of autoimmune thyroid disease (18%), determined by the presence of antimicrosomal antibodies, compared with controls (8%).50 A Finnish study reported an overall prevalence of hypothyroidism of 4% in infertile women.51 Fortunately, once treated adequately, neither hypothyroidism nor hyperthyroidism has a major impact on fertility.
The potential effect of treatment of relative thyroid hormone deficiency on infertility is unknown.52 One intervention trial in women with recurrent abortions, mild thyroid dysfunction, and positive antimicrosomal antibodies53 showed that early supplementation with thyroid hormone favorably affects the outcome of pregnancy. It is unknown whether treatment of women with antithyroid antibodies but normal thyrotropin levels can lead to better outcomes. Systematic screening54 should be considered in all women with a female cause of infertility.55
Subclinical Hypothyroidism
Subclinical hypothyroidism is defined as an elevated serum thyrotropin level associated with normal total or free levels of T4 and T3 in the absence of symptoms. The thyrotropin level is usually less than 10μU/mL. It has been also called mild hypothyroidism, preclinical hypothyroidism, mild thyroid failure, and compensated hypothyroidism56
The natural history of subclinical hypothyroidism is variable.57 In some individuals, the thyrotropin level will be normal several months later or may remain unchanged. This may be due to laboratory error or to transient silent thyroiditis. The patient may also develop overt hypothyroidism, which occurs at a rate of about 5% per year in patients with raised thyrotropin levels and detectable antithyroid antibodies.58 In some subjects with high titers of antithyroid antibodies, the risk of progression to overt disease may be closer to 20% per year.59 Consideration of these possible outcomes60 affects the decision about whether to treat or to observe without treatment.61
Potential Benefits of Treating Subclinical Hypothyroidism
Treatment may prevent progression to overt hypothyroidism, which is estimated to be 2.6% in women with increased thyrotropin levels and 4.3% if antimicrosomal antibodies are present as well. The risk of cardiovascular disease may be decreased if the lipid profile is affected,62 even though several studies failed to show a clear association between cardiovascular disease and hypothyroidism.63 There may be an improvement of symptoms if present.64
Hypothyroidism
Hypothyroidism is a common disease, especially in women, with a 1% to 2% prevalence (up to 10% in those older than age 65).43,56,65 Congenital hypothyroidism is one of the most frequent congenital diseases, with an incidence of 1/4000 newborns. Table 22-11 lists the causes of hypothyroidism.
Clinical Features
Thyroid gland examination should include the size, symmetry, consistency, tenderness, nodularity, mobility, vascularity, and any associated lymphadenopathy. The presence of goiter is not essential for diagnosis.
Other Diagnostic Methods
Lipids
An association between hypothyroidism and lipid disorder has been observed for decades, but the exact effects are still unknown. In a study on 295 patients with overt hypothyroidism, just 8.5% had a normal lipid profile.66 However, in another study of hypothyroidism in hypercholesterolemic patients (cholesterol level over 200mg/dL) the prevalence of overt hypothyroidism was 1.3% and subclinical hypothyroidism 11.2%.67 The effects of replacement with thyroxine are even more controversial. T4 replacement can reduce LDL cholesterol by 11% to 36%, depending on the degree of basal LDL elevation.63
Myxedema Coma
Myxedema coma is now extremely rare due to much earlier diagnosis of hypothyroidism. The patients have profound hypothermia, bradycardia, and typical skin and facial changes; mortality is close to 100% with or without treatment. Aggressive treatment with intravenous T4 (up to 500 μg) with possible addition of T3 can reduce mortality.68
Treatment
The thyroid hormone replacement preparations available are:
Due to its short half-life, T3 is used before some diagnostic tests. There are some data indicating that adding T3 to T4 treatment will improve mood and quality of life, but other studies don’t show any beneficial effect.69 The medication should be taken at least 1 hour from a meal. Medications such as calcium, soy, iron, aluminum, and cholestyramine can interfere with the absorption or metabolism of levothyroxine, and they should be separated by 4 hours from the thyroid hormone. In elderly patients, with or without cardiac disease, a lower dose (12.5 to 25μg/day) is started and increased slowly, by 25μg/day every 4 to 6 weeks.
The interchangeability of thyroxine products has been closely watched for several decades, but in 2004, the American Association of Clinical Endocrinologists, The Endocrine Society (TES), and the American Thyroid Association issued a joint statement addressing the introduction to the market of several generic levothyroxine products that had been approved by the U.S. Food and Drug Administration (FDA).70 The main concern was related to the FDA’s method in determining bioequivalence. Bioequivalence establishes therapeutic equivalence; this would be better reflected with an endocrine endpoint, such as serum thyrotropin, but this measurement was not used by the FDA. Thus the difference in some preparations was large (33% [uncorrected] and 12.5% to 25% [corrected for baseline values]). The panel recommended not substituting thyroxine preparations for one another, but if it is clinically indicated, then it suggested monitoring thyrotropin at 6 weeks.
Hyperthyroidism
Hyperthyroidism may be a result of either overproduction of thyroid hormone or destruction of the thyroid gland. One may also see hyperthyroidism from exogenous (oral intake) or ectopic synthesis of thyroid hormone (struma ovari) (Table 22-12).
Clinical Examination
The presence of proptosis on ocular examination or pretibial myxedema (a nonpitting edema of the shins) strongly suggests Graves’ disease72 (Table 22-13). The differential diagnosis is listed in Table 22-14.
| Symptoms (listed in order of prevalence) |
Italicized items are more specific for Graves’ disease.
Table 22-14 Differential Diagnosis of Hyperthyroid Symptoms
| Acute psychosis |
| Severe illness |
| High-altitude exposure |
| Selenium deficiency |
Treatment
Antithyroid Drugs
Thionamides, including propylthiouracil (PTU) and methimazole, are approved by the FDA for the treatment of hyperthyroidism. Both inhibit thyroid hormone synthesis by interfering with thyroid peroxidase-mediated iodination of tyrosine residues in thyroglobulin, an important step in the synthesis of T4 and T3. PTU has an additional effect in blocking the conversion of T4 to T3. The medications may also have direct immunosuppressive effects in the thyroid, accounting for some of their effects in Graves’ disease.73
Personal preference has usually determined the initial choice of antithyroid medication. Recently methimazole has become the treatment of choice due to its better side effect profile and once-a-day schedule. The usual starting dose of methimazole is 15 to 30 mg; of PTU is 300 mg daily. After starting the treatment, close follow-up testing at 4 weeks is recommended and then every 4 to 6 weeks until euthyroidism is achieved. Some authors prefer using high doses first to block the gland and supplement with exogenous thyroid hormones,74 but these benefits have not been replicated in North America.75 There do not appear to be any good predictors of clinical remission or response to antithyroid drugs. Typically antithyroid medication is used for between 12 and 18 months. Once the medication is stopped, relapse occurs, usually within the first 3 to 6 months, but remission may last as much as 40 years.
Side Effects of Antithyroid Drugs
Antithyroid drugs have a variety of minor side effects, such as skin reactions, arthralgias, gastrointestinal effects, and sialadenitis. Almost all patients taking antithyroid drugs will have transient elevation of liver transaminases within the first 2 to 6 weeks. The most fearful side effect is agranulocytosis, thought to occur in 0.3% of cases, mostly at the beginning of the treatment (first 3 months). A complete blood count is recommended after initiating therapy, but follow-up tests are not helpful because the agranulocytosis is idiosyncratic. However, all patients should be aware of this risk and should be told to stop the drug and seek urgent medical therapy if they develop sudden fever or sore throat without explanation. Rarer side effects include hepatic necrosis, vasculitis, and cholestasis.76
Radioactive Iodine
Radioactive iodine is used to definitively treat patients with hyperthyroidism.77 This will result in subsequent hypothyroidism. Radioiodine has not been shown to cause infertility or birth defects but certainly can cause fetal hypothyroidism, especially if given after the first trimester.
Thyroid Disease and Pregnancy
Glinoer reported a higher miscarriage rate in patients from Belgium with antithyroid antibodies.78 However, it is unclear whether this represents a cause-effect relationship.79
A retrospective study published in 2005 reviewing 17,000 women delivering in one center found that pregnancies in women with subclinical hypothyroidism were three times more likely to be complicated by placental abruption (relative risk [RR]: 3) and two times more predisposed to premature birth (RR, 1.8).80
Thyroid Function in Pregnancy
Pregnancy results in a number of physiologic changes81:
Total thyroid hormone (T4) is increased mostly in the first half of gestation due to a profound increase in TBG. Serum T4 levels increase between 6 and 12 weeks and slowly after that; T3 rise is more progressive. Serum T4 has a 20-fold higher affinity for TBG than T3, so the ratio between T4 and T3 is constant. The changes in TBG imply that the extrathyroidal T4 pool must increase to ensure the same free hormone level. The daily increased secretion is between 1% and 3 % until it reaches a steady state and then returns to previous levels. The free levels of T3 and T4 are maintained usually in a normal range. The indirect calculated free T4 indices are not very reliable in pregnancy. The hormonal output is regulated through the normal pituitary–thyroid feedback by thyrotropin stimulation that is minor in a patient with a normal thyroid gland.82 However, these physiologic adjustments can be “stressed” in women with any predisposition to thyroid disease.
Human chorionic gonadotropin is a weak thyroid stimulator that can bind to the thyrotropin receptor. If hCG is markedly elevated (as may happen normally in a twin pregnancy) or is a more potent molecular variant, serum free T4 concentrations may increase to hyperthyroid range with temporary thyrotropin suppression.83
Immunology of Normal Pregnancy
There are many conflicting data regarding the effect of pregnancy on T cells and B cells, with a profound interest in the Th2/Th1 ratio, which seems to be elevated in pregnancy.84 During pregnancy, there is also there is a large drop in antibody levels with no parallel reduction in B-cell number. Their reduced activity is correlated to the sex steroids. Within 6 months of delivery, total imunoglobulin G as well as autoantibodies rise above prepregnancy levels and we see an exacerbation of almost all autoimmune disorders.
Fetal Thyroid Function
The fetal thyroid begins to function after 10 to 12 weeks of pregnancy. Thyroid hormones (mostly from the fetal thyroid and a negligible amount from the mother’s thyroid) are important for development of the fetal nervous system. Iodine in the mother’s diet readily crosses the placenta and is used by the fetal thyroid gland to make thyroid hormone. Iodine deficiency can cause newborn hypothyroidism or mental retardation (cretinism) and is a major world health problem in underdeveloped countries. Because there is an overabundance of iodine in the American diet, disorders caused by a lack of dietary iodine rarely occur here. Low thyroid hormone concentrations in early gestation may be associated with lower IQ in children age 5 to 7.85 Some of the altered physiological development86 was also found in biochemically euthyroid women with elevated antimicrosomal antibodies.87
Planning Pregnancy for Women with Thyroid Disease
In patients with hypothyroidism the therapeutic aim is to maintain the thyrotropin level in the normal range, preferable close to 1 to 2μU/mL.88 If a woman is hyperthyroid before pregnancy, medical treatment with PTU should be started.89 Clearly, radioactive iodine is contraindicated in pregnancy, and nonpregnant women should be advised to avoid conception for 6 to 12 months after the treatment. If a woman conceives while taking PTU the medication can be continued but adjusted to allow the thyroid hormone levels to be at the upper end of normal.
Hypothyroidism in Pregnancy
Overt hypothyroidism is present in 0.3% to 0.7% of pregnancies and subclinical hypothyroidism in up to 2.5%. L-thyroxine is safe and is well absorbed during pregnancy. Some women require higher doses during their pregnancy. Physicians generally monitor the thyrotropin level two or three times during the pregnancy to detect even mild hypothyroidism and increase the L-thyroxine dose, if necessary. Several publications have shown that 35% to 62% of women with autoimmune hypothyroidism and up to 70% to 100% of athyroid women require an increase of levothyroxine dosage. A prospective study of 19 women found that an increase in the levothyroxine dose was necessary during 17 pregnancies.90 The mean levothyroxine requirement increased 47% during the first half of pregnancy (median onset of increase, 8 weeks’ gestation) and plateaued by week 16. This increased dose was maintained and required until delivery.
Hyperthyroidism in Pregnancy
Thyrotoxicosis (hyperthyroidism) during pregnancy, most often due to Graves’ disease, presents a challenge for diagnosis and treatment because of unique fetal and maternal considerations. The risk of miscarriage and stillbirth is increased if thyrotoxicosis goes untreated, and the overall risks to mother and baby further increase if the disease persists or is first recognized late in pregnancy.91
The main disorders that present with symptoms and signs of hyperthyroidism in pregnancy include Graves’ disease, silent thyroiditis, hyperemesis gravidarum, and molar pregnancy (Table 22-15). The diagnosis of hyperthyroidism is suggested by specific physical signs, such as prominent eyes, enlarged thyroid gland, and exaggerated reflexes, and is confirmed by markedly elevated serum thyroid hormone levels and a suppressed thyrotropin level. Radioactive investigations are not performed. However, because there is no fetal thyroid activity until after the end of the first trimester, there is very little iodide trapping by the fetus in early pregnancy. Later in pregnancy radioactive iodine can destroy the fetal thyroid, but this is probably not a sufficient reason to end the pregnancy, because recognition and treatment of hypothyroidism shortly after delivery usually ensures normal growth and development in the child.
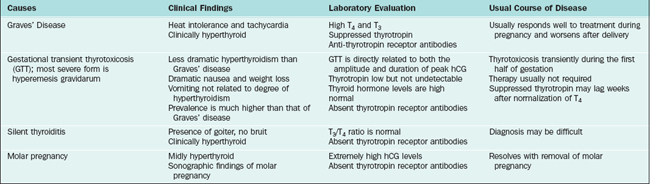
The treatment of choice for thyrotoxicosis during pregnancy is antithyroid medication, either PTU or methimazole,92 because radioactive iodine cannot be used. PTU remains the drug of choice, because methimazole has been associated with rare cases of aplasia cutis in some infants.93
The natural course of hyperthyroidism in pregnancy is for the disease to become milder or remit totally near term.94 In many patients antithyroid medications can be tapered to low levels or even discontinued. For those patients who are not so fortunate, it is important to maintain control of the hyperthyroidism throughout pregnancy to avoid severe thyrotoxicosis (thyroid storm) developing during labor and delivery.94 If this does develop, additional acute treatment with beta-adrenergic blockers such as propranolol (Inderal) and high doses of nonradioactive iodine are used. Long-term treatment with these agents is not advised in pregnancy, because beta blockers have been associated with fetal bradycardia and occasionally intrauterine growth retardation.95
In lactating mothers, both PTU and methimazole can be safely used, their concentration in breast milk being small, with no effects on thyroid functions in the breastfed infant.96
Fetal Thyroid Disease
Antithyroid medications, nonradioactive iodine, and maternal thyroid antibodies can all cross the placenta and cause hypothyroidism in the fetus. Nonradioactive iodine, which is present in some medications, including some cough medications, can cause a goiter in the fetus, making delivery difficult or causing respiratory obstruction. For this reason, iodine-containing drugs should never be used in pregnancy except in the case of thyroid storm. Unfortunately, there is no simple blood test to assess fetal thyroid function, although measurements of thyroid hormone or thyrotropin levels in the amniotic fluid sac have been used in research studies.97 Plain X-rays sometimes show delayed bone development in fetal hypothyroidism, but this test is usually not recommended. Screening for hypothyroidism at birth, now done routinely in North America on all babies, identifies the need for early short- or long- term thyroxine treatment, with excellent long-term follow-up results.
Clues to the presence of fetal hyperthyroidism are fetal heart rate consistently above the normal limit of 160 beats per minute and the presence of high levels of thyroid-stimulating antibodies in the mother’s blood. Recently, it has been suggested that ultrasonography of the fetal thyroid gland may be helpful in assessing fetal thyroid size.98
All women with a history of Graves’ disease should be tested for thyroid-stimulating antibodies late in pregnancy.99 The consequences of untreated fetal thyrotoxicosis include low birth weight and head size, fetal distress in labor, and neonatal heart failure and respiratory distress. Administration of antithyroid drugs to the mother during pregnancy can treat the baby in this situation. Close follow-up and continued treatment is required after delivery.
Postpartum Thyroid Disease in the Mother
Postpartum Thyroiditis
Postpartum thyroiditis may occur in 8% to 10% of women.100 Thyroiditis also occurs in the nonpostpartum period, as well as in men, and is probably an autoimmune thyroid disease related to Hashimoto’s thyroiditis.101
Typically, it consists of a temporary period of hyperthyroidism lasting from 6 weeks to 3 months postpartum, followed by hypothyroidism occurring between 3 and 9 months after delivery. Women at risk include those with a previous history of postpartum thyroiditis or those who can be shown to have thyroid autoantibodies.102 Usually, no treatment or only symptomatic treatment is required for the hyperthyroid phase, and a short course of thyroxine treatment for 6 to 12 months is sufficient for the hypothyroid phase. Some women who do not recover from the hypothyroid phase will require long-term thyroid replacement therapy.103
During the first 3 months after delivery, symptoms of fatigue, depression, and impairment of memory and concentration are common and often unrelated to a woman’s thyroid hormone level. Symptoms can mimic postpartum depression and thus a high index of suspicion is needed to rule out hypothyroidism.104 It is reasonable to perform a thyrotropin level in those women who do experience emotional disorders following pregnancy.
Solitary Thyroid Nodule and Thyroid Cancer in Pregnancy
A thyroid nodule may be discovered during the pregnancy, and the best way to investigate this would be an ultrasound-guided fine needle aspiration biopsy because this results in a better biopsy yield and the ultrasound can document a baseline assessment of size of the nodule. If results of the examination of the biopsy specimen is suspicious or suggestive of malignancy, necessary surgery can be performed in the second trimester; if the nodule is discovered late in pregnancy, surgery can be deferred until the postpartum period.105 History of thyroid cancer and radioactive iodide administration does not preclude conception. One normally advises a year of avoiding pregnancy after a high-dose radioactive iodine therapy for thyroid cancer. Pregnancy has not been shown to affect the course of thyroid cancer.106
MULTIPLE ENDOCRINE NEOPLASIA SYNDROMES
1 Thorner MO, Vance ML, Kaws ER, et al. The anterior pituitary. In: Wilson JD, Foster DW, editors. Williams Textbook of Endocrinology. 9th ed. Philadelphia: WB Saunders; 1998:249-340.
2 Vance ML. Hypopituitarism. NEJM. 1994;330:1651-1662.
3 Molitch ME, Thorner MO, Wilson C. Therapeutic controversy: Management of prolactinomas. J Clin Endocrinol Metab. 1997;82:996-1000.
4 Garner PR. Pituitary disorders in pregnancy. Curr Obstet Med. 1991;1:143-178.
5 Molitch M. Evaluation and management of pituitary tumours during pregnancy. Endocr Pract. 1996;2:287-295.
6 Melmed S. Acromegaly. NEJM. 1990;322:966-977.
7 Herman-Bonest, Seliverstov M, Melmed S. Pregnancy in acromegaly: Successful therapeutic outcome. J Clin Endocrinol Metab. 1998;83:727-731.
8 Landolt AM, Schmid J, Wimpfheimer C, et al. Successful pregnancy in a previously infertile woman treated with SMS 201-995 for acromegaly. NEJM. 1989;320:671-672.
9 Rees LH, Burke CW, Chard T, et al. Possible placental origin of ACTH in normal human pregnancy. Nature. 1975;254:620-622.
10 Yanovski JA, Cutler GB, Chrousos GP, et al. Corticotropin-releasing hormone stimulation following low-dose Dexamethasone administration. A new test to distinguish Cushing’s syndrome from pseudo-Cushing’s states. JAMA. 1993;269:2232-2238.
11 Robert M, Leriche-Polverelli A, Namon P, et al. Gonadotropic pituitary adenoma and pregnancy. Value of bromocriptine. Presse Med. 1991;20:503.
12 Pestell RG, Best JD, Alfard FP. Lymphocytic hypophysitis. The clinical spectrum of the disorder and evidence for an autoimmune pathogenesis. Clin Endocrinol. 1990;33:457-466.
13 Kristof RA, Van Roost D, Klingmuller D, et al. Lymphocytic hypophysitis: Noninvasive diagnosis and treatment by high dose methylprednisolone pulse therapy? J Neurol Neurosurg Psychiatry. 1999;67:398-402.
14 Reusch JE, Kleinschmidt-De Masters BU, Lillehei KO, et al. Preoperative diagnosis of lymphocytic hypophysitis unresponsive to short course dexamethasone. Neurosurgery. 1992;30:268-272.
15 Sheehan HL. The recognition of chronic hypopituitarism resulting from postpartum pituitary necrosis. Am J Obstet Gynecol. 1971;111:852-854.
16 Jialal I, Desai RK, Rajput MC. An assessment of posterior pituitary function in patients with Sheehan syndrome. Clin Endocrinol. 1987;27:91.
17 Orth DN, Kovacs WJ. The adrenal cortex. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. William’s Textbook of Endocrinology. 9th ed. Philadelphia: WB Saunders; 1998:517-664.
18 Grinspoon SK, Biller BM. Clinical review 62: Laboratory assessment of adrenal insufficiency. J Clin Endocrinol Metab. 1994;79:923-931.
19 Byny RL. Withdrawal from glucocorticoid therapy. NEJM. 1975;1:30-32.
20 Hadden DR. Adrenal disorders of pregnancy. Endocrinol Metab Clin North Am. 1995;24:139-151.
21 Perlitz Y, Varkel J, Markovitz J, et al. Acute adrenal insufficiency during pregnancy and puerperium: Case report and literature review. Obstet Gynecol Surv. 1999;54:717-722.
22 New MI. Congenital adrenal hyperplasia. In: DeGroot L, editor. Endocrinology. 3rd ed. Philadelphia: WB Saunders; 1995:1813-1835.
23 Mulaikal RM, Migeon CJ, Rock JA. Fertility rates in female patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. NEJM. 1987;316:178-182.
24 Lo JC, Grumbach MM. Pregnancy outcomes in women with congenital virilizing adrenal hyperplasia. Endocrinol Metab Clin North Am. 2001;30:207-229.
25 Mercado AB, Wilson RC, Cheng KC, et al. Prenatal treatment and diagnosis of congenital adrenal hyperplasia owing to steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1995;80:2014-2020.
26 Aron DC, Schnall AM, Sheeler LR. Cushing’s syndrome and pregnancy. Am J Obstet Gynecol. 1990;162:244-252.
27 Berwaerts J, Verhelst J, Mahler C, Abs R. Cushing’s syndrome in pregnancy treated by ketoconazole: Case report and review of the literature. Gynecol Endocrinol. 1999;13:175-182.
28 Bevan JS, Gough MH, Gillmer MD, Burke CW. Cushing’s syndrome in pregnancy: The timing of definitive treatment. Clin Endocrinol (Oxf). 1987;27:225-233.
29 Laurel MT, Kabadi UM. Primary hyperaldosteronism. Endocr Prac. 1997;3:47-53.
30 Bravo EL. Primary aldosteronism: Issues in diagnosis and management. Endocrinol Metab Clin North Am. 1994;23:271-283.
31 Baron F, Sprauve ME, Huddleston JF, Fisher AJ. Diagnosis and surgical treatment of primary aldosteronism in pregnancy: A case report. Obstet Gynecol. 1995;86:644-645.
32 Keely E. Endocrine causes of hypertension in pregnancy—when to start looking for zebras. Semin Perinatol. 1998;22:471-484.
33 Wilson M, Morganti AA, Zervoudakis I, et al. Blood pressure, the renin-aldosterone system and sex steroids throughout normal pregnancy. Am J Med. 1980;68:97-104.
34 Wyckoff JA, Seely EW, Hurwitz S, et al. Glucocorticoid-remediable aldosteronism and pregnancy. Hypertension. 2000;35:668-672.
35 Schenker JG, Chowers I. Pheochromocytoma and pregnancy. Obstet Gynecol Surv. 1971;26:734-739.
36 Harper MA, Murnaghan GA, Kennedy L, et al. Phaeochromocytoma in pregnancy. BJOG. 1989;96:594-606.
37 Ahlawat SK, Jain S, Kumari S, et al. Pheochromocytoma associated with pregnancy: Case report and review of the literature. Obstet Gynecol Surv. 1999;54:728-737.
38 Sam S, Molitch ME. Timing and special concerns regarding endocrine surgery during pregnancy. Endocrinol Metab Clin North Am. 2003;32:337-354.
39 Gross MD, Shapiro B. Clinical review 50: Clinically silent adrenal masses. J Clin Endocrinol Metab. 1993;77:885-888.
40 Hamrahian A, Ioachimescu A, Remer E, et al. Clinical utility of noncontrast CT attenuation value (HU) to differentiate adrenal adenomas/hyperplasias from nonadenomas: Cleveland Clinic experience. J Clin Endocrinol Metab. 2005;90:871-877.
41 Bhatkar S, Rajan M, Velumani A, Samuel A. Thyroid hormone binding protein abnormalities in patients referred for thyroid disorders. Ind J Med Res. 2004;120:160-165.
42 Degroot LJ, Larsen PR, Henneman G, editors. The Thyroid and Its Diseases, 6th ed., New York: Churchill Livingstone, 1996.
43 Hollowell J, Staehling N, Flanders W, et al. Serum TSH, T4, and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87:489-499.
44 Helfand M, for the U.S. Preventive Services Task Force. Screening for subclinical thyroid dysfunction in nonpregnant adults: A summary of the evidence. Ann Intern Med. 2004;140:128-141.
45 American College of Obstetricians and Gynecologists Practice Bulletin. Clinical management guidelines for obstetrician-gynecologists. Thyroid disease in pregnancy. Obstet Gynecol. 2002;100:387.
46 Poppe K, Glinoer D. Thyroid autoimmunity and hypothyroidism before and during pregnancy. Hum Reprod Update. 2003;9:149-161.
47 Gharib H, Tuttle RM, Baskin HJ, et al. Subclinical thyroid dysfunction: A joint statement on management from the American Association of Clinical Endocrinologists, the American Thyroid Association, and the Endocrine Society. J Clin Endocrinol Metab. 2005;90:581-585.
48 Stagnaro-Green A, Glinoer D. Thyroid autoimmunity and the risk of miscarriage. Best Pract Res Clin Endocrinol Metab. 2004;18:167-181.
49 Stagnaro-Green A. Postpartum thyroiditis. Best Pract Res Clin Endocrinol Metab. 2004;18:303-316.
50 Poppe K, Glinoer D, Van Steirteghem A, et al. Thyroid dysfunction and autoimmunity in infertile women. Thyroid. 2001;12:997-1001.
51 Arojoki M, Jokimaa V, Juuti A, et al. Hypothyroidism among infertile women in Finland. Gynecol Endocrinol. 2000;14:127-131.
52 Kalro BN. Impaired fertility caused by endocrine dysfunction in women. Endocrinol Metab Clin North Am. 2003;32:573-592.
53 Vaquero E, Lazzarin N, De Carolis C, et al. Mild thyroid abnormalities and recurrent spontaneous abortion: Diagnostic and therapeutical approach. Am J Reprod Immunol. 2000;43:204-208.
54 Spong CY. Subclinical hypothyroidism: Should all pregnant women be screened? Obstet Gynecol. 2005;105:235-236.
55 Hollowell J, LaFranchi S, Smallridge R, et al. 2004 where do we go from here? Summary of working group discussions on thyroid function and gestational outcomes. Thyroid. 2005;15:72-76.
56 Canaris G, Manowitz N, Mayor G, Ridgway E. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160:526-534.
57 Cooper DS. Subclinical thyroid disease: Consensus or conundrum? Clin Endocrinol. 2004;60:410-412.
58 Cooper D. Clinical practice. Subclinical hypothyroidism. NEJM. 2001;345:260-265.
59 Diez JJ, Iglesias P. Spontaneous subclinical hypothyroidism in patients older than 55 years: An analysis of natural course and risk factors for the development of overt thyroid failure. J Clin Endocrinol Metab. 2004;89:4890-4897.
60 Tuzcu A, Bahceci M, Gokalp D, et al. Subclinical hypothyroidism may be associated with elevated high-sensitive C-reactive protein (low grade inflammation) and fasting hyperinsulinemia. Endocrine J. 2005;52:89.
61 Hueston WJ, Pearson WS. Subclinical hypothyroidism and the risk of hypercholesterolemia. Ann Family Med. 2004;2:351-355.
62 Canturk Z, Cetinarslan B, Tarkun I, et al. Lipid profile and lipoprotein A as a risk factor for cardiovascular disease in women with subclinical hypothyroidism. Endocrine Res. 2003;29:307-316.
63 Biondi B, Klein I. Hypothyroidism as a risk factor for cardiovascular disease. Endocrine. 2004;24:1-13.
64 Rodondi N, Newman A, Vittinghoff E, et al. Subclinical hypothyroidism and the risk of heart failure, other cardiovascular events, and death. Arch Intern Med. 2005;165:2460-2466.
65 Tunbridge W, Evered D, Hall R, et al. The spectrum of thyroid disease in a community: The Whickham survey. Clin Endocrinol. 1977;7:481-493.
66 O’Brien T, Dinneen S, O’Brien P, Palumbo P. Hyperlipidemia in patients with primary and secondary hypothyroidism. Mayo Clinic Proc. 1993;68:860-866.
67 Bruckert E, De Gennes J, Dairou F, Turpin G. [Frequency of hypothyroidism in a population of hyperlipidemic subjects]. Presse Medicale. 1993;22:57-60.
68 Hylander B, Rosenqvist U. Treatment of myxoedema coma—factors associated with fatal outcome. Acta Endocrinologica. 1985;108:65-71.
69 Sawka A, Gerstein H, Marriott M, et al. Does a combination regimen of thyroxine (T4) and 3,5,3′-triiodothyronine improve depressive symptoms better than T4 alone in patients with hypothyroidism? Results of a double-blind, randomized, controlled trial. J Clin Endocrinol Metab. 2003;88:4551-4555.
70 Joint statement on the U.S. Food and Drug Administration’s decision regarding bioequivalence of levothyroxine sodium. Thyroid. 2004;14:486.
71 Arafah BM. Increased need for thyroxine in women with hypothyroidism during estrogen therapy. NEJM. 2001;344:1743-1749.
72 Weetman A. Graves’ disease. NEJM. 2000;343:1236-1248.
73 Cooper D. Antithyroid drugs. NEJM. 2005;352:905-917.
74 Hashizume K, Ichikawa K, Sakurai A, et al. Administration of thyroxine in treated Graves’ disease. Effects on the level of antibodies to thyroid-stimulating hormone receptors and on the risk of recurrence of hyperthyroidism. NEJM. 1991;324:947-953.
75 Rittmaster RS, Abbott EC, Douglas R. Effect of methimazole, with or without L-thyroxine, on remission rates in Graves’ disease. J Clin Endocrinol Metab. 1998;83:814-818.
76 Abraham P, Avenell A, Watson W, et al. Antithyroid drug regimen for treating Graves’ hyperthyroidism. Cochrane Database Syst Rev. 2004. CD003420.
77 Cooper DS. Hyperthyroidism. Lancet. 2003;362:459-468.
78 Glinoer D. Thyroid autoimmunity and spontaneous abortion. Fertil Steril. 1999;72:373-374.
79 Haddow J. Subclinical hypothyroidism and pregnancy outcomes. Obstet Gynecol. 2005;106:198-199.
80 Casey BM, Dashe JS, Wells CE, et al. Subclinical hypothyroidism and pregnancy outcomes. Obstet Gynecol. 2005;105:239-245.
81 Larsen PR, Schlumberger MJ, Hay ID. Thyroid. In: Wilson JD, Foster DW, editors. Williams Textbook of Endocrinology. 9th ed. Philadelphia: WB Saunders; 1998:249-340.
82 Mandel S, Spencer C, Hollowell J. Are detection and treatment of thyroid insufficiency in pregnancy feasible? Thyroid. 2005;15:44-53.
83 Hershman JM. Physiological and pathological aspects of the effect of human chorionic gonadotropin on the thyroid. Best Prac Res Clin Endocrinol Metab. 2004;18:249-265.
84 Lazarus J, Parkes A, Premawardhana L. Postpartum thyroiditis. Autoimmunity. 2002;35:169-173.
85 LaFranchi S, Haddow J, Hollowell J. Is thyroid inadequacy during gestation a risk factor for adverse pregnancy and developmental outcomes? Thyroid. 2005;15:60-71.
86 Pop VJ, Kuijpens JL, van Baar AL, et al. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol (Oxf). 1999;50:149-155.
87 Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Role of thyroid hormone during early brain development. Eur J Endocrinol. 2004;151(Suppl 3):U25-U37.
88 Mandel S. Hypothyroidism and chronic autoimmune thyroiditis in the pregnant state: Maternal aspects. Best Pract Res Clin Endocrinol Metab. 2004;18:213-224.
89 Neale D, Burrow G. Thyroid disease in pregnancy. Obstet Gynecol Clin North Am. 2004;31:893-905.
90 Alexander E, Marqusee E, Lawrence J, et al. Timing and magnitude of increases in levothyroxine requirements during pregnancy in women with hypothyroidism. NEJM. 2004;351:241-249.
91 Davis L, Lucas M, Hankins G, et al. Thyrotoxicosis complicating pregnancy. Am J Obstet Gynecol. 1989;160:63-70.
92 Wing D, Millar L, Koonings P, et al. A comparison of propylthiouracil versus methimazole in the treatment of hyperthyroidism in pregnancy. Am J Obstet Gynecol. 1994;170:90-95.
93 Milham S. Scalp defects in infants of mothers treated for hyperthyroidism with methimazole or carbimazole during pregnancy. Teratology. 1985;32:321.
94 Amino N, Tanizawa O, Mori H, et al. Aggravation of thyrotoxicosis in early pregnancy and after delivery in Graves’ disease. J Clin Endocrinol Metab. 1982;55:108-112.
95 Pruyn SC, Phelan PJ, Buchanan GC. Long-term propranolol therapy in pregnancy: Maternal and fetal outcome. Am J Obstet Gynecol. 1979;135:485-489.
96 Azizi F, Bahrainian M, Khamseh M, Khoshniat M. Intellectual development and thyroid function in children who were breast-fed by thyrotoxic mothers taking methimazole. J Pediatr Endocrinol Metab. 2003;16:1239-1243.
97 Nachum ZRY, Weiner E, Shalev E. Graves’ disease in pregnancy: Prospective evaluation of a selective invasive treatment protocol. Am J Obstet Gynecol. 2003;189:159-165.
98 Luton D, Le Gac I, Vuillard E, et al. Management of Graves’ disease during pregnancy: The key role of fetal thyroid gland monitoring. J Clin Endocrinol Metab. 2005;90:6093-6098.
99 Glinoer D. Management of hypo- and hyperthyroidism during pregnancy. Growth Hormone IGF Res. 2003;13(Suppl A):S45-S54.
100 Lazarus JH. Thyroid dysfunction: Reproduction and postpartum thyroiditis. Semin Reprod Med. 2002;20:381-388.
101 Pearce E, Farwell A, Braverman L. Thyroiditis. NEJM. 2003;348:2646-2655.
102 Hayslip C, Fein H, O’Donnell V, et al. The value of serum antimicrosomal antibody testing in screening for symptomatic postpartum thyroid dysfunction. Am J Obstet Gynecol. 1988;159:203-209.
103 Amino N, Mori H, Iwatani Y, et al. High prevalence of transient postpartum thyrotoxicosis and hypothyroidism. NEJM. 1982;306:849-852.
104 McCoy SJ, Beal JM, Watson GH. Endocrine factors and postpartum depression. A selected review. J Reprod Med. 2003;48:402-408.
105 Hamburger JL. Thyroid modules in pregnancy. Thyroid. 1992;2:165-168.
106 Moosa M, Mazzaferri E. Outcome of differentiated thyroid cancer diagnosed in pregnant women. J Clin Endocrinol Metab. 1997;82:2862-2866.
107 Ladenson P, Singer P, Ain K, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med. 2000;160:1573-1575.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 22 Management of Pituitary, Adrenal, and Thyroid Disease
PITUITARY DISORDERS
Anatomy
The optic chiasma may be in front of (15%), above (80%), or behind the sella (5%). Within this optic chiasma, nerve fibers from the nasal half of the retina cross over to the opposite optic tract while those from the temporal half remain uncrossed. The close association of the pituitary gland with the optic chiasma explains the visual symptoms associated with expanding masses in this region.1
The median eminence is an intensely vascular component at the baseline of the hypothalamus that forms the floor of the third ventricle. The pituitary stalk arises from the median eminence. The hypothalamus extends anteriorly to the optic chiasma and posteriorly to the mammillary bodies. It was not until the mid-1960s that hypothalamic releasing hormones were isolated and identified (Table 22-1).
Table 22-1 Pituitary Hormones, Hypothalamic Hormones, and Other Regulatory Factors
| Pituitary Hormones | Hypothalamic Hormones | Other Regulatory Factors |
|---|---|---|
| Thyrotropin | TRH | T4, T3, dopamine, Pit-1 |
| Corticotropin | CRH | ADH, adrenaline, cortisol |
| Luteinizing hormone | LH-RH | Estrogen, progesterone, testosterone |
| Follicle-stimulating hormone | LH-RH | Activin, estrogen, inhibin, follistatin, testosterone |
| Growth hormone | GH-RH | Somatostatin, estrogens, T4, Pit-1 |
| Prolactin | PRF | Dopamine, TRH, Pit-1, estrogen, serotonin, vasoactive intestinal peptide, GnRH-associated peptide |
TRH, thyrotropin-releasing hormone; CRH, corticotropin-releasing hormone;
LH-RH, luteinizing hormone-releasing hormone; GH-RH, growth hormone-releasing hormone; PRF, prolactin-releasing factor; Pit-1, pituitary-specific transcription factor; ADH, antidiuretic hormone.
Pituitary Tumors
Pituitary tumors may present with either hypofunction or hyperfunction, as well as symptoms directly related to the mass effect of the tumor (Table 22-2). Since the advent of computed tomography (CT), microadenomas have been arbitrarily designated as equal to or less than 10mm in diameter and macroadenomas as greater than 10mm in diameter. They are invariably benign, with no sex predilection. Pituitary adenomas are rarely associated with parathyroid and pancreatic hyperplasia or neoplasia as part of the multiple endocrine neoplasia type I (MEN I) syndrome. Pituitary carcinomas are rare, but metastases from other solid malignancies can occur more frequently.2
| Endocrine Effects | ||
|---|---|---|
| Mass Effects | Hyperpituitarism | Hypopituitarism |
| Headaches | GH: acromegaly | GH: short stature in children, increased fat mass, decreased strength and well-being in adults |
| Chiasmal syndrome | Prolactin: hyperprolactinemia | Prolactin: failure of postpartum lactation |
| Hypothalamic syndrome | Corticotropin: Cushing’s disease Nelson’s syndrome | Corticotropin: hypocortisolism |
| Disturbances of thirst, appetite, satiety, sleep, and temperature regulation | LH/FSH: gonadal dysfunction or silent α-subunit secretion | LH or FSH: hypogonadism |
| Diabetes insipidus | Thyrotropin hyperthyroidism | Thyrotropin: hypothyroidism |
| SIADH | ||
| Obstructive hydrocephalus | ||
| Cranial nerves III, IV, V1, V2, and VI dysfunction | ||
| Temporal lobe dysfunction | ||
| Nasopharyngeal mass | ||
| CSF rhinorrhea | ||
SIADH = syndrome of inappropriate antidiuretic hormone; GH = growth hormone; LH = luteinizing hormone; FSH = follicle-stimulating hormone.
PITUITARY ADENOMAS
Approximately 50% of pituitary adenomas are prolactinomas, 15% are growth hormone (GH)-producing, 10% are corticotropin-producing, and less than 1% secrete thyrotropin. Nonfunctioning pituitary adenomas, or more appropriately named nonsecretory adenomas represent about 25% of pituitary tumors. Most of these adenomas on morphologic examination reveal granules containing hormones, typically components of glycoprotein hormones. Autopsy studies suggest that up to 20% of normal individuals harbor incidental pituitary microadenomas that are pathologically similar in distribution to those that present clinically.3
Prolactinoma
Hyperprolactinemia
Hyperprolactinemia impairs pulsatile gonadotropin (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]) release, likely through alteration in hypothalamic luteinizing hormone-releasing hormone (LHRH) secretion. Women of reproductive age usually present with oligomenorrhea, amenorrhea, galactorrhea, and infertility. Those with longstanding amenorrhea are less likely to have galactorrhea secondary to longstanding estrogen deficiency. Postmenopausal women and men usually come to medical attention because of a mass effect, such as headaches and visual field defects.4
Drug history is an important part of the initial evaluation of patients with elevated prolactin level, because some medications are associated with hyperprolactinemia and their discontinuation (if possible) will avoid any further, often expensive, workup. Other common conditions associated with elevated prolactin levels include pregnancy and hypothyroidism (Table 22-3).
| Physiologic | Pathologic | Pharmacologic |
|---|---|---|
| Pregnancy | Prolactinoma | TRH |
| Postpartum | Acromegaly (25%) | Psychotropic medications |
| Newborn | Hypothalamic disorders | Phenothiazines |
| Stress | “Chiari-Frommel” | Reserpine |
| Hypoglycemia | Craniopharyngioma | Methyldopa |
| Sleep | Metastatic disease | Estrogen therapy |
| Postprandial hypoglycemia | Pituitary stalk secretion or compression | Metoclopramide, cimetidine (especially intravenous) |
| Intercourse | Hypothyroidism | Opiates |
| Nipple stimulation | Renal failure | Verapamil |
| Liver disease | Some SSRIs, including fluoxetine and fluvoxamine | |
| Chest wall trauma (burns, shingles) |
SSRI = Selective serotonin reuptake inhibitor.
The prolactin level usually correlates well with the size of the tumor. Serum prolactin level above 200μg/L is almost always indicative of a prolactin-producing pituitary tumor. However, a serum prolactin level below 200μg/L can be seen in the presence of a large pituitary adenoma, because stalk compression from an adenoma that does not secrete prolactin can also cause hyperprolactinemia. Stimulatory tests, including the TRH stimulation test, which are performed to determine whether an elevated prolactin is a result of a prolactinoma, are nonspecific and cannot be used to diagnose or exclude a tumor. Large prolactinomas may be associated with a falsely low prolactin level. Dilution of serum will reveal the markedly elevated prolactin levels.
Observational studies in patients with microadenomas indicate that serum prolactin concentration or adenoma size increases in only a minority of patients; indeed, serum prolactin deceases in a majority of patients over time. Details of the relationship between prolactin adenomas and amenorrhea are found in Chapter 16.
Treatment
Medical therapy during pregnancy often stirs debate about the continuation of bromocriptine. Tumor-related complications are seen in about 15% of pregnancies and in only 5% of women with microadenomas. A sensible approach would be to stop bromocriptine when pregnancy begins, and then follow the clinical status with serum prolactin levels and visual field examinations. If there is significant worsening in clinical status, bromocriptine could be reinstituted. The adenoma can be followed yearly by MRI, increasing the duration between imaging studies if size is stable.5
Acromegaly
Acromegaly may occur at a rate of 3 to 4 cases per million per year, with mean age at diagnosis of 40 years in men and 45 years in women. The GH-secreting tumors tend to be more aggressive in younger patients. Classical clinical features are listed in Table 22-4. More than 95% of cases of acromegaly are caused by GH-secreting pituitary tumors. In rare cases, they are caused by ectopic GH-releasing hormone (GH-RH) secretion, mainly carcinoids and pancreatic islet cell tumors. Patients with acromegaly have a 3.5-fold increased mortality rate, with cardiovascular disease being the most common cause of death. Somatotrope adenomas appear to be monoclonal in origin. A gsp mutation in a GspIa subunit in GH cells, leading to continuous GH secretion, has been shown to cause acromegaly.6
| Coarsening of facial features |
| Prominent jaw and frontal sinus |
| Broadening of hands and feet |
| Hyperhidrosis |
| Macroglossia |
| Signs of hypopituitarism |
| Diabetes mellitus (10%–25%) |
| Skin tags (screening for colonic polyps required) |
| Hypertension (25%–30%) |
| Cardiomyopathy (50%–80%) |
| Carpal tunnel syndrome |
| Sleep apnea (5%) |
Insulin-like growth factor-I has a longer plasma half-life than GH and is an excellent initial screening test for those suspected to have acromegaly. An elevated IGF-I level in a clinical setting suggestive of acromegaly almost always confirms the diagnosis. Patients with poorly controlled diabetes and malnutrition may have falsely low serum IGF-I levels. The oral glucose tolerance test remains the gold standard test to confirm the diagnosis. Normal individuals suppress their GH level to less than 1μg/L (using chemiluminescent assays) within 2 hours after ingestion of 100 grams of oral glucose solution.
Treatment
Long-term observations of patients on somatostatin analogues have shown no evidence for tachyphylaxis. Some degree of tumor shrinkage is expected in up to 50% of patients, although in most cases there is less than 50% shrinkage in tumor size. The most common side effects are gastrointestinal, including diarrhea, abdominal pain, and nausea. The most serious side effect of somatostatin analogues is cholelithiasis, seen in up to 25% of patients. Its long-term management is similar to that for cholelithiasis in the general population, and routine ultrasonographic screening is not indicated. There have been very few reports of the use of a somatostatin analogue during pregnancy.7,8
Cushing’s Disease
Corticotropin-secreting pituitary adenoma is the most common cause of endogenous Cushing’s syndrome (60%), with the rest being adrenal (25%) or ectopic (15%) in origin. The term Cushing’s disease refers specifically to a pituitary tumor as the cause. Signs and symptoms suggestive of hypercortisolism are listed in Table 22-5. Many signs and symptoms of Cushing’s disease are nonspecific, including hypertension, abnormal glucose tolerance, menstrual irregularities, and psychiatric disturbances, including depression. Most women with Cushing’s disease have reduced fertility.9
| Clinical Feature | Approximate Prevalence (%) |
|---|---|
| Obesity | |
| General | 80–95 |
| Truncal | 45–80 |
| Hypertension | 75–90 |
| Menstrual disorders | 75–95 |
| Osteopenia | 75–85 |
| Facial plethora | 70–90 |
| Hirsutism | 70–80 |
| Impotence/decreased libido | 65–95 |
| Neuropsychiatric symptoms | 60–95 |
| Striae | 50–70 |
| Glucose intolerance | 40–90 |
| Weakness | 30–90 |
| Bruising | 30–70 |
| Kidney stones | 15–20 |
| Headache | 10–50 |
Women with Cushing’s disease typically have fine facial lanugo hair and may have acne and temporal scalp hair loss secondary to increased adrenal androgen secretion. There is usually a 3- to 6-year delay in diagnosis of patients with Cushing’s disease, and it may be possible to date the onset of the disease by determining which scars are pigmented due to excess secretion of corticotropin and other melanotropins.
Diagnosis
Twenty-four hour urinary free cortisol measurement is the single best test for diagnosis of Cushing’s syndrome (Fig. 22-1). Because of the significant overlap between normal individuals and those with Cushing’s syndrome, random serum cortisol has no role in the diagnosis of Cushing’s syndrome. A 1-mg overnight dexamethasone suppression test with a morning cortisol level below 1.8μg/dL virtually rules out the disease but is associated with an up to 40% false-positive rate.
A combination of low-dose dexamethasone suppression test and corticotropin-releasing hormone (CRH) stimulation test has been shown to have 100% diagnostic accuracy in a National Institutes of Health study. This test may have a significant value in establishing the diagnosis in those with pseudo-Cushing’s and elevated 24-hour urinary free cortisol. Other tests useful in establishing the diagnosis of Cushing’s disease include midnight serum and salivary cortisol (see Fig. 22-1).
Once the diagnosis of Cushing’s syndrome has been established, the next step is to find out whether cortisol hypersecretion is corticotropin dependent (see Fig. 22-1). Although an undetectable or low corticotropin level is consistent with adrenal etiology, low-normal corticotropin may be seen in both ectopic Cushing’s syndrome and corticotropin-secreting pituitary tumor. The CRH stimulation test is used to differentiate between the two. Although corticotropin levels tend to be higher in those with ectopic Cushing’s syndrome compared to patients with pituitary disease, there is considerable overlap. The high-dose dexamethasone test or the CRH stimulation test is helpful in differentiation of the two disorders. Cortisol levels are not suppressed with the high-dose (8 mg) dexamethasone test in patients with ectopic corticotropin syndrome, and CRH stimulation may not lead to a further rise in corticotropin.10 The gold standard test to differentiate pituitary Cushing’s syndrome from ectopic corticotropin-producing tumor is inferior petrosal sinus sampling. This test should be performed by an experienced neuroradiologist; it is essential to note that it cannot be used to make the diagnosis of Cushing’s syndrome.
Nonsecretory and Glycoprotein-Secreting Pituitary Adenomas
Glycoprotein-secreting Adenomas
Rarely, pituitary adenomas can secret glycoproteins, including FSH, LH, or thyrotropin. An FSH adenoma may cause amenorrhea in a woman, or an LH adenoma may cause precocious puberty in a boy. Diagnosis is confirmed by measurement of either intact glycoprotein hormones or their α and β subunits. Levels of the α subunit tend to be inappropriately elevated, compared with those of the intact hormone itself.11
HYPOPITUITARISM
Causes of Hypopituitarism
Lymphocytic Hypophysitis
Lymphocytic hypophysitis is an autoimmune disease often presenting in women during or after pregnancy. The clinical manifestations are secondary to hypopituitarism or adrenal insufficiency or are due to a pituitary mass effect. Serum prolactin is elevated in half of patients but may be decreased. Antipituitary antibodies are present in some patients, and other autoimmune endocrine disorders, including Hashimoto’s thyroiditis and Addison’s disease, have been seen by others.12
Recent experience from Germany suggests that one should be cautiously optimistic about high-dose steroid therapy. MRI findings improved in 88% of patients treated with high-dose steroids, but clinical normalization was quite variable, with none achieving complete recovery.13
Surgery for mass effect in lymphocytic hypophysitis can lead to rapid relief of neurologic symptoms, but endocrinologic improvement was seldom reported. Indications for surgery are the presence of gross chiasma compression, ineffectiveness of corticosteroid therapy, and the impossibility of establishing the diagnosis of lymphocytic hypophysitis with sufficient certainty by conservative evaluation.14
Sheehan’s Syndrome
Sheehan’s syndrome is the result of ischemic infarction of the normal pituitary gland, leading to hypopituitarism secondary to postpartum hemorrhage and hypotension.15 Patients have a history of failure to lactate postpartum, failure to resume menses, cold intolerance, or fatigue. Some women may have an acute crisis mimicking apoplexy within 30 days postpartum. There is often subclinical central diabetes insipidus.16
ADRENAL GLAND DISORDERS
Anatomy and Physiology
The zonae reticularis and fasciculata are under the control of corticotropin released by the pituitary gland in response to hypothalamic CRH. CRH in turn is regulated by cortisol-negative feedback, stress, and a circadian rhythm. Besides increasing the synthesis of cortisol, corticotropin is trophic for the adrenal gland so that a lack of corticotropin results in atrophy of the zonae fasciculata and reticularis. Although corticotropin has some effect on aldosterone production, the zona glomerulosa is predominantly under the control of renin. Understanding the anatomy and physiology of the adrenal gland is crucial to understanding its hypofunction and hyperfunction.17
Adrenal Insufficiency
Etiology
Clinical adrenal insufficiency results from hypofunction of the adrenal cortex. This may be due to destruction of the adrenal gland itself, referred to as Addison’s disease or primary adrenal insufficiency. Alternatively, it may be due to a lack of either corticotropin (i.e., secondary adrenal insufficiency) or CRH.18
The most common cause of Addison’s disease in adults (80%) is autoimmune destruction of the adrenal gland. This is often seen in association with other autoimmune diseases, including Hashimoto’s thyroiditis, Graves’ disease, or type 1 diabetes mellitus. Adrenal insufficiency in this setting is known as type II autoimmune polyglandular syndrome. Type I autoimmune polyglandular syndrome, more commonly seen in children, consists of Addison’s disease, hypoparathyroidism, and mucocutaneous candidiasis.
Other causes associated with adrenal insufficiency are listed in Table 22-6. Currently, acquired immunodeficiency syndrome is the most common cause of infectious adrenal destruction, and the antiphospholipid syndrome (lupus anticoagulant) is increasingly being recognized as a cause of adrenal hemorrhage.
Table 22-6 Other Causes of Primary Adrenal Insufficiency in Adults
Secondary adrenal insufficiency is a result of adrenal gland atrophy from corticotropin deficiency. This most often results from pituitary corticotroph atrophy owing to previous exogenous glucocorticoid use,19 hypopituitarism, or isolated corticotropin deficiency (usually postpartum).
Clinical Presentation
The underlying etiology of adrenal insufficiency determines the clinical presentation (Table 22-7). Under the regulation of corticotropin, cortisol and adrenal androgens are lost in both primary and secondary adrenal insufficiency. Aldosterone production, predominantly regulated by renin, remains intact in secondary adrenal insufficiency. Therefore, hyperkalemia and profound dehydration with orthostatic hypotension are seen in primary adrenal insufficiency only. Likewise, hyperpigmentation of the skin or mucous membranes (secondary to increased corticotropin) is seen in primary adrenal insufficiency only. The absence of hyperkalemia or hyperpigmentation does not exclude adrenal insufficiency. In addition to hyponatremia and hyperkalemia, laboratory abnormalities in adrenal insufficiency may include hypoglycemia (usually chronic), hypercalcemia, eosinophilia, and lymphocytosis.
| Primary | Secondary | |
|---|---|---|
Diagnosis
In secondary adrenal insufficiency, however, the corticotropin stimulation test is not always abnormal. Adequate corticotropin may be present to prevent adrenal gland atrophy, thereby resulting in a response to the supraphysiologic dose of corticotropin used in the test. However, the HPA axis may not be able to respond to stress. In patients with suspected secondary adrenal insufficiency and a normal corticotropin stimulation test, CRH is now available to assess corticotropin response. In addition, the insulin tolerance test or the metyrapone test evaluate the integrity of the HPA axis by its response to hypoglycemia or inhibited cortisol synthesis, respectively. Although not widely used, some investigators find that a 1μg corticotropin stimulation test may be more sensitive at detecting mild adrenal insufficiency.20
Treatment
Cortisol 20mg in the morning and 10mg in the evening, or prednisone, 5 to 7.5mg daily, provides dramatic relief of symptoms. However, to prevent Cushing’s syndrome, the smallest dose needed to control the patient’s symptoms should be used. For a minor illness, the glucocorticoid dose should be doubled for as short a time as needed. For a major stress, parenteral hydrocortisone, 200 to 400mg daily, is given initially and then rapidly tapered. Aldosterone replacement is required in primary adrenal insufficiency only and is given as fludrocortisone acetate (Florinef Acetate), 0.05 to 0.2mg daily. The dose is adjusted according to the blood pressure and potassium level. Renin levels may be required to assess plasma volume. Adrenal androgens are not replaced.21
In undiagnosed patients with suspected adrenal crisis, dexamethasone, 2 to 4 mg intravenously or intramuscularly, should be given along with saline and glucose. Dexamethasone does not interfere with the cortisol assay. The corticotropin stimulation test should then be done as soon as possible.
