[level-membership-for-critical-care-medicine-category]
CHAPTER 90 MANAGEMENT OF ENDOCRINE DISORDERS IN THE SURGICAL INTENSIVE CARE UNIT
The endocrine system as a part of the neuroendocrine axis (hypothalamic-pituitary axis) influences the response to stress and critical illness. Endocrine abnormalities within this axis change and modify the physiologic response to trauma and stress. Critically ill patients with a known diagnosis of an endocrine problem are treated with replacement therapy. However, an unrecognized endocrine abnormality often creates management difficulties and increases morbidity. Endocrine problems occur at all levels of the neuroendocrine axis from primary or secondary disease, medications, or end-organ failure.
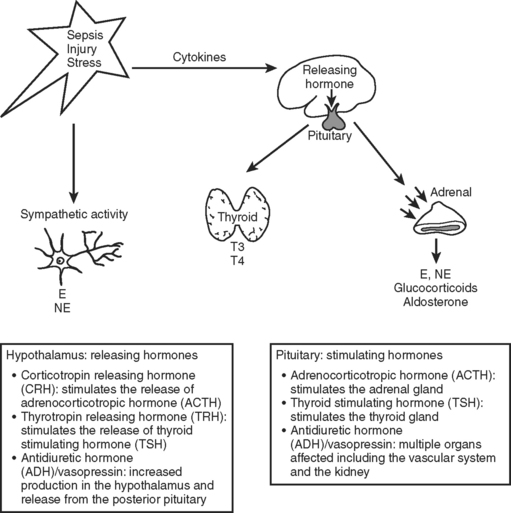
The neuroendocrine axis is responsible for the stress response and is controlled by the hypothalamus, pituitary, and the autonomic nervous system (Figure 1). This axis is activated by baroreceptor response to intravascular volume, sympathetic response from tissue injury, and inflammatory mediators released from tissue trauma. The hormones released in response to injury act through binding to cell surface receptors or intracellular receptors and produce a complex series of responses and feedback loops that maintain cellular processes.1 This chapter addresses abnormalities in the endocrine system that affect the course of critically ill patients.
BRAIN PROBLEMS: ABNORMALITIES IN HYPOTHALAMIC/PITUITARY RESPONSE
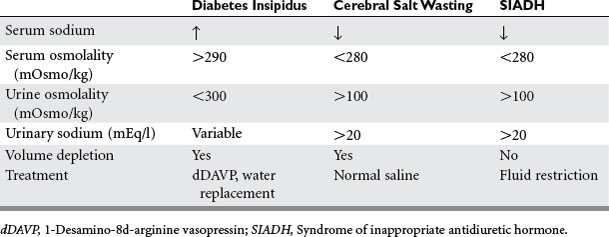
Injuries that affect the brain can interrupt the hypothalamus or pituitary production of hormone. Head injury, brain surgery, mass lesions or infiltrative diseases, vascular or hypoxic injuries, and cerebral infections cause failure of the releasing and pituitary hormones—resulting in single or combined abnormalities. Cerebral edema or increased intracranial pressure is thought to restrict the blood flow to the hypothalamic pituitary area. Frequently encountered abnormalities are diabetes insipidus (DI), syndrome of inappropriate antidiuretic hormone (SIADH), and cerebral salt wasting. These syndromes cause abnormalities of sodium and water balance. Evaluation of volume status, urine and serum sodium, and osmolality are required to determine which syndrome is present. This evaluation is important because the treatment depends on which abnormality is present (Table 1).
Diabetes Insipidus
DI is caused by lack of vasopressin (antidiuretic hormone [ADH]), which causes water diuresis of more than 3 l/day, dehydration, and hypernatremia. The urine is dilute with urine osmolality of less than 300 mOsm/kg and urine specific gravity of less than 1.005. Urine osmolality greater than 800 mOsm/kg excludes DI. The diagnosis is usually made when dilute urine output exceeds 200 ml for 2 consecutive hours.2 A dramatic rise in serum sodium occurs in the ICU patient population unless fluids are aggressively replaced. In the neurosurgery patient population, the incidence of DI is 3.7% with a mortality of 70%.2 DI commonly occurs in association with severe brain injury and herniation. The treatment is fluid rehydration and vasopressin replacement via IV dDAVP 2 to 4 mcg/day or intranasally 10 to 60 mcg/day. Frequent monitoring of electrolytes and central venous pressure monitoring are necessary. The water deficit is calculated and slowly replaced. Caution should be used when severe hypernatremia is present, with half the water deficit replaced in 24 hours to avoid demyelination (Table 2).
SIADH and Cerebral Salt Wasting
The syndrome of inappropriate antidiuretic hormone and cerebral salt wasting are linked through a common cause—traumatic brain injury—and common result—hypotonic hyponatremia. Cerebral salt wasting is most often associated with subarachnoid hemorrhage, whereas SIADH is also associated with brain injury, tumors, and medications (Table 3).3 The cause of SIADH is excessive release of ADH that leads to water retention and an increase in extracellular fluid volume. Volume expansion increases renal sodium excretion, producing hyponatremia. Cerebral salt wasting is thought to involve disruption of the neural input to the kidney or central secretion of a natriuretic factor. Sodium is wasted by the kidney, causing extracellular volume depletion and stimulation of ADH secretion. Hyponatremia develops with extracellular volume depletion.
Table 3 Medications That Interfere with Thyroid Hormone and ADH
| Drugs Causing Syndrome of Inappropriate Antidiuretic Hormone (SIADH): |
|
Stimulating ADH release: opiates, opioids, barbiturates, nicotine, thiazides, isoproterenol, cyclic antidepressants, MAO inhibitors, haloperidol, risperidone, acetylcholine
|
NSAID, Nonsteroidal anti-inflammation drug.
In both syndromes, the diagnosis begins with serum sodium less than 135 mmol/l. Measurement of serum osmolality is less than 280 mOsm/kg, and the urine osmolality is greater than 100 mOsm/kg in both diseases. The differentiating factor is the effective blood volume, which is normal in SIADH and low in cerebral salt wasting. Low effective blood volume causes orthostatic blood pressure, tachycardia, low central venous pressure (CVP), low urine sodium, chloride, and fractional excretion of sodium with high BUN. Both disease states have low uric acid level. With correction of the salt deficit, uric acid levels normalize in SIADH and not in cerebral salt wasting.3 The treatment of SIADH is fluid restriction (800–1000 ml/day) and occasional hypertonic saline. Demelocycline, phenytoin, and lithium are used for chronic SIADH. The treatment for cerebral salt wasting is normal saline fluid replacement to expand the extracellular fluid compartment (see Table 1).
ABNORMALITIES IN THYROID RESPONSE
Untreated or unrecognized thyroid problems (excess or deficit) create life-threatening illness in critically ill patients. Thyroid hormones are responsible for the metabolic rate in all tissues. Critical illness may alter the production of thyroid hormone through thyroid-stimulating hormone (TSH) regulation, peripheral metabolism, or alteration in binding proteins. Cytokines as well as commonly used intensive care unit (ICU) medications (see Table 3) affect thyroid hormone function.
Thyroid Excess
Hyperthyroidism with a 3% incidence in out-patients is caused by Grave’s disease, goiter, and adenoma. Treatment with amiodarone increases the incidence to 9%.4 Thyroid storm (severe hyperthyroidism) was first recognized after thyroidectomy in unprepared patients and now encompasses 1%–2% of all admissions for thyrotoxicosis (with a mortality of 20%–30%).5 It is precipitated by physiologic stress related to specific events such as surgery, trauma, childbirth, severe illness, overdose of thyroid medication, iodine in medications, and contrast. The classic symptomatology includes fever, cardiovascular abnormalities, and mental status changes. The tachycardia is out of proportion to the fever. Fever is the hallmark of this disease, with temperatures to 106° F. A state of high-output cardiac failure can develop with bounding pulses, rales and hepatomegaly, and thyroidal bruit. Atrial fibrillation and congestive heart failure are common in elderly patients with hyperthyroidism and can occur without fever (thyrocardiac crisis). The mental status changes include a broad spectrum, from anxiety to coma. There can be nonspecific gastrointestinal (GI) complaints and clinical findings of Grave’s disease.
This syndrome is recognized by clinical signs and symptoms. Laboratory test turnaround time is long, and treatment should be started based on clinical suspicion. The TSH is not detectable, and both T3 and T4 are elevated (with T3 > T4).6 There is an associated elevation of white blood counts (WBCs), calcium, blood glucose, and liver function tests. The treatment is directed toward decreasing the production of thyroid hormone and preventing its release, blocking the peripheral action, supportive care, and treating the cause (Table 4).7
|
Inhibit hormone synthesis: propylthiouracil 1000 mg loading dose, and then 300 mg orally every 6 hours
|
Thyroid Deficit
Hypothyroidism occurs in patients over age 50 years, 8% females and 2% males.8 Previous surgery, radiation, and autoimmune disease are the most common causes. Myxedema coma is severe hypothyroidism. Myxedema coma is rare, but has a mortality rate of more than 60%.5 It is precipitated by physiologic stress of trauma, surgery, burns, infections, cardiovascular events or cold temperatures, or failing to take thyroid medication. Medications can decrease hormone production and function (see Table 3). The cardinal findings relate to reduced metabolic rate and oxygen consumption and include hypothermia, bradycardia, hypotension, hypoventilation, and mental status changes.8 The mental status changes range from lethargy to coma and are associated with decreased deep tendon reflexes. Low cardiac output with both right- and left-sided failure and decreased myocardial contractility occur. There is slowing of the GI system, with constipation or ileus. Skin, nail, and hair changes associated with hypothyroidism are present. The name myxedema comes from the infiltration of mucopolysaccharide infiltration of the skin. This is a clinical diagnosis with elevated TSH, very low T4 and hyponatremia, low blood glucose, elevated CPK, low PaO2, and elevated PCO2. There are characteristic EKG changes (Table 5).
The treatment of myxedema coma includes supportive care in an ICU setting and replacement of thyroid hormone. ICU care includes EKG monitoring and possibly cardiac pacing, arterial blood gas monitoring with possible intubation and ventilation, warming with blankets and external warmers and IV fluid with glucose, temperature monitoring, and neurologic checks. Thyroxine is given 300 to 500 mcg IV on day 1 and then 50 to 100 mcg IV every day until oral replacement is started.7,8 Adrenal insufficiency should be tested for with the rapid cosyntropin test and treated with 100 mg of hydrocortisone IV every 8 hours.5
Sick Euthyroid Syndrome
Sick euthyroid or nonthyroidal illness is a common finding in the ICU patient population. It is not completely clear if this syndrome represents a pathologic process or a means of adapting to critical illness. There are three patterns of abnormal thyroid function, which represent a progression of disease severity: decreased T3, decreased T3 and T4, and decreased T3, T4, and TSH (Table 6).7 Patients with decreased T3 alone usually have mild to moderate illness. This pattern is the most common pattern of the sick euthyroid syndrome. The peripheral conversion of T4 to T3 is decreased. Mainly this pattern is the effect of medications (see Table 3). Serum T4 levels are increased early in acute illness related to decreased conversion or increased binding levels. This pattern is seen with elderly patients and patients with psychiatric problems. The most severe pattern has decreased T3, T4, and TSH. The free T4 may be low, normal, or elevated. In addition to conversion problems, the binding proteins are low (as is TSH). The decline in T4 correlates with prognosis. Mortality increases as the T4 level drops below 4 mcg/dl and is 80% at T4 levels of 2 mcg/dl.1,7

The diagnosis requires trending thyroid function tests. These tests may serve as markers of the severity of disease rather than treatable thyroid disease. The treatment of sick euthyroid syndrome is unclear. Both thyroid replacement and no treatment are advocated.9 Animal studies show improvement with thyroid replacement, but human studies have not shown similar results. Therefore, currently treatment is not advised.
ABNORMALITIES OF ADRENAL FUNCTION
Pheochromocytoma
Pheochromocytomas produce excess catecholamines and follow the rule of 10s: 10% are malignant, 10% are extraadrenal, 10% are incidental findings on radiographic studies, and 10% are multiple. Less than 0.2% of patients with hypertension have pheochromocytoma as their diagnosis.10 The diagnosis is usually not made in the ICU, but patients require ICU management for hypertensive crisis or perioperative care. The patients usually have the classic triad of headache, sweating, and tachycardia or palpitations. Weekly paroxysms of hypertension occur in at least 75% of patients.11 This is due to rapid release of catecholamines from an inciting event. Other symptoms include blurred vision, orthostatic hypotension, weight loss, polyuria, and polydipsia.
Clinical suspicion leads to urinary and plasma evaluation for catecholamines and metabolites. These tests confirm the diagnosis 95% of the time.12 If these tests are inconclusive, a clonidine suppression test is performed with 0.3 mg of clonidine given orally 12 hours after antihypertensives have been stopped. No beta blockers, diuretics, or tricyclic antidepressants can be used. Alpha blockers will not affect the test. The patients without a pheochromocytoma have a fall of the plasma catecholamines to less than 500 pg/ml.13 The tumor is localized with a computed tomography (CT) scan or magnetic resonance imaging (MRI). The CT has a risk of exacerbating the hypertension with its contrast. Failure to detect the tumor with these studies leads to an MIBG, octreoscan, total body MRI, or selected venous sampling.
Acute hypertensive crisis requires treatment with sodium nitroprusside or phentolamine. Phentolamine is given as an IV bolus of 2.5–5 mg, repeated every 5 minutes until the blood pressure is controlled. Nitroprusside is dosed at 0.5–10 mcg/kg/minute.11
Adrenal Insufficiency
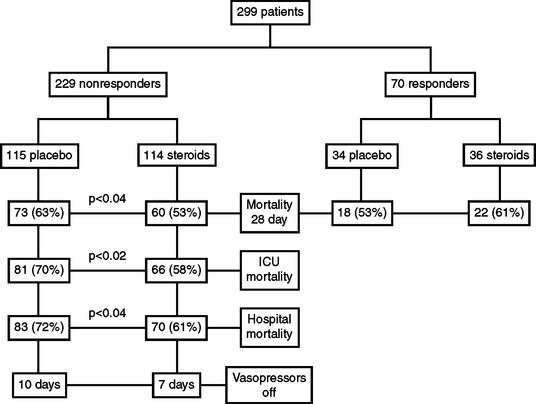
Controversy exists over the incidence, diagnosis, and treatment of adrenal insufficiency in ICU patients.14 The incidence is less than 0.01% in the general population but up to 28% in seriously ill patients.15 Reported mortality is as high as 25%, but this mortality rate may be reduced with early recognition to 6%–11%.16 Cortisol is needed in the critically ill patient for appropriate response to acute inflammation and vasomotor stability. Cortisol has metabolic, catabolic, anti-inflammatory, and vasoactive properties. The most common cause of adrenal insufficiency remains adrenal suppression from the administration of steroids. This effect lasts up to a year after the discontinuation of steroids. In the ICU, adrenal gland destruction can occur from infection, bleeding, or system inflammation. However, decreased cortisol concentration during critical illness without anatomic disruption is the more common cause. This secondary adrenal insufficiency is usually related to sepsis.7,17
In critically ill patients, the usual signs and symptoms of adrenal insufficiency are not usually apparent. These critically ill patients often have hemodynamic instability despite fluid resuscitation and vasopressor use. This is a tip-off to adrenal insufficiency and should lead to evaluation of cortisol level and treatment. The most common abnormalities in ICU patients with adrenal insufficiency are listed in Table 7.18 Several signs and symptoms exist if the patient had preexisting adrenal insufficiency. These include fatigue, weight loss, nausea, abdominal pain, arthralgias, syncope, hyperpigmentation of the skin, vitiligo, anorexia, and decreased libido.
In ICU patients, clinical suspicion leads to a random cortisol test. If the cortisol level is low with a random level less than 15 mcg/dl, adrenal insufficiency is present and treatment begins. With a random level greater than 35 mcg/dl, adrenal insufficiency is unlikely. If the level is between these two values, a stimulation test is performed with 250-mcg IV synthetic ACTH. Cortisol levels are drawn at 0, 30, and 60 minutes after the ACTH. If the cortisol level changes 9 mcg/dl or more, adrenal insufficiency is unlikely. If the cortisol changes less than 9 mcg/dl, adrenal insufficiency is likely and treatment occurs.17
The patient is treated immediately after a stimulation test is performed. Dexamethasone is used. It will not interfere with the stimulation test as hydrocortisone does. Do not wait for the results. If the stimulation test comes back as normal, the steroids can be discontinued. Hydrocortisone 50 mg IV every 6 hours with 50 mcg fluorocortisone daily orally is used for those patients with adrenal insufficiency in shock on vasopressors. This therapy is continued for 7 days (Figure 2).19 For patients with proven adrenal insufficiency but not in shock, hydrocortisone 50 mg IV every 6 hours is given. This dose can then be tapered over several days as the illness subsides. Retesting after the illness can determine if this was a primary adrenal problem and transient or if it is a secondary adrenal insufficiency and requires further workup and treatment.
PROBLEMS WITH HYPERGLYCEMIA
Hyperglycemia occurs in most critically ill patients. Diabetes and undiagnosed diabetes may contribute to hyperglycemia. However, in critically ill patients increased sympathetic activity and the activation of the cytokine cascade are the major causes of elevated blood glucose. Medications (total parenteral nutrition, beta blockers, cyclosporine, catecholamines, and glucocorticoids) promote hyperglycemia. Electrolyte imbalance (hypokalemia) decreases insulin release and contributes. The stress-induced increased sympathetic activity leads to increased glycogenolysis, increased hepatic gluconeogenesis, and peripheral insulin resistance—which cause an increase in the serum glucose. Cytokines produced by the inflammatory process promote insulin resistance and/or activate the hypothalamic-pituitary-adrenal axis.14 All or many of these factors come into play in the critically ill patient and produce an imbalance between glucose production and uptake, resulting in hyperglycemia.
Regardless of the mechanism of hyperglycemia, organ system dysfunction occurs in the cardiovascular, cerebrovascular, neuromuscular, and immunologic systems. Hyperglycemia in the cardiovascular population has been widely studied.20,21 An increased risk of in-hospital mortality has been found in patients with myocardial infarction and hyperglycemia. Insulin deficiency/resistance is associated with increased free fatty acids, which the heart uses as a fuel source. These free fatty acids are toxic to the ischemic myocardial cells and lead to arrhythmias. Hyperglycemia also causes an osmotic diuresis, leading to volume depletion and increased oxygen consumption from increased contractility.7
Hyperglycemic patients with ischemic stroke and head injury have worse outcomes than patients who are euglycemic. With elevated blood glucose levels, there is an increased risk of in-hospital mortality after ischemic stroke in nondiabetic patients. Persistent hyperglycemia is an independent predictor of infarct expansion and is associated with worse functional outcome in patients with ischemic stroke.22 There may be an association with tissue plasminogen activator and hemorrhagic transformation in hyperglycemic patients with ischemic stroke. In patients with head injury, both the Glasgow coma scale and mortality appear related to blood glucose levels—with an inverse relationship to the Glasgow coma scale and survival.23
Increased brain glucose levels contribute to the acidosis from the glycolysis induced by anaerobic metabolism of the ischemic brain tissue. The lactic acidosis promotes the formation and accumulation of free radicals, which impede mitochondrial activity. This effect is most important at the edge of the infarct, where neurons that may survive are recruited into the infarct. Many animal models have shown infarct size increasing with hyperglycemia. Both disruption of the blood/brain barrier and hemorrhagic infarct conversion are suggested mechanisms for deterioration of brain function.24
Polyneuropathy of critical illness appears to be related to blood glucose levels. Van den Berghe et al. screened for polyneuropathy in critically ill patients and found that patients with control of hyperglycemia were less likely to have critical-illness neuropathy.25 They also found that in those patients who developed neuropathy there was more rapid resolution. There was a positive linear correlation between blood glucose levels and risk of polyneuropathy. The mechanism of polyneuropathy and its association with hyperglycemia has yet to be defined.
Postoperative wound infections, pneumonias, urinary tract infections, and bacteremias are increased in patients with hyperglycemia. The immunologic system is impaired by hyperglycemia through action on the white blood cells. Polymorphonuclear cells have impaired chemotaxis, phagocytosis, and oxidative burst pathways. Macrophages have impaired phagocytosis and deceased complement fixation. Collagen deposition is impaired, granulocyte adherence is irregular, and there is decreased bactericidal activity.7
Evidence is accumulating that control of blood glucose in the normal range improves outcome from critical illness (Figure 3). Van den Berghe et al. showed that tight glycemic control of blood glucose 80 to 110 mg/dl reduced morbidity and mortality in the surgical ICU, with reduction of episodes of septicemia and length of stay in the ICU.25 Two large patient population studies showed that control of blood glucose to less than 200 mg/dl in cardiothoracic surgery patients reduced the incidence of deep wound infections.26,27 Improved survival after myocardial infarction occurs with glycemic control. Evidence is lacking for improved neurologic outcomes with glycemic control.
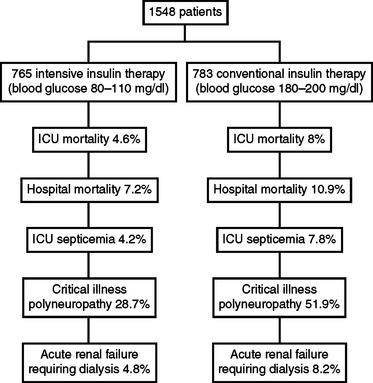
Figure 3 Outcome of tight glycemic control.
(Data from Van Den Berghe, et al: Intensive insulin therapy in critically ill patients. N Engl J Med 345(19):1359–1367, 2001.)
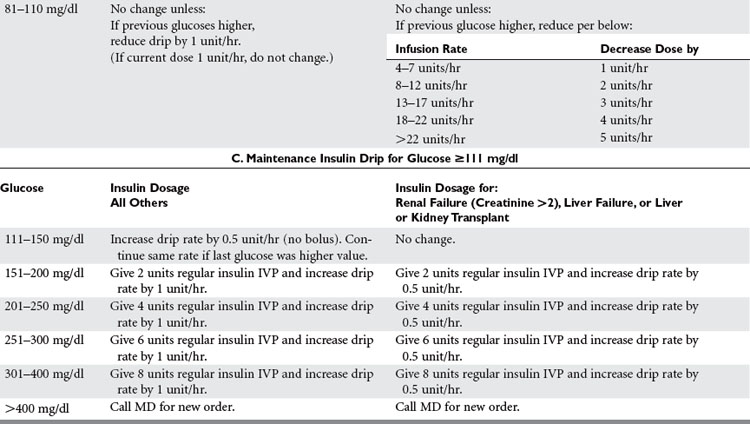
The hyperglycemia should be rapidly controlled with IV insulin to a normal glucose range (Table 8). Rapid control can easily be achieved with continuous insulin infusion.28 Patients with renal failure, liver failure/resection, and transplantation are susceptible to hypoglycemia.

1 Nylen ES, Muller B. Endocrine changes in critical illness. J Intensive Care Med. 2004;19(2):67-82.
2 Boughey JC, Yost MJ, Bynoe RP. Diabetes insipidus in the head-injured patient. Am Surg. 2004;70(6):500-503.
3 Palmer BF. Hyponatremia in a neurosurgical patient: syndrome of inappropriate antidiuretic hormone secretion versus cerebral salt wasting. Nephrol Dial Transplant. 2000;15:262-268.
4 Braithwaite SS. Thyroid Disorders. In: Parrillo JE, Dellinger RP, editors. Critical Care Medicine: Priniciples of Diagnosis and Management in the Adult. 2nd ed. St. Louis: Mosby; 2002:1235-1256.
5 Sarlis NJ. Loukas gourgiotis: thyroid emergencies. Rev Endocr Metabol Disord. 2003;4:129-136.
6 Franklyn J. Thyrotoxicosis. Clin Med. 2003;3(1):11-14.
7 Goldberg PA, Inzucchi SE. Clinical issues in endocrinology. Clin Chest Med. 2003;24:583-606.
8 Ringel MD. Management of hypothyroidism and hyperthyroidism in the intensive care unit. Crit Care Clin. 2001;17(1):59-75.
9 Stathotos N, Levetan C, Burman KD, et al. The controversy of the treatment of critically ill patients with thyroid hormone. Best Pract Res Clin Endocrinol Metab. 2001;15(4):465-478.
10 Rusnak RA. Adrenal and Pituitary Emergencies. The Emeregency Medicine Clinic of North America. 1989;7(4):903-925.
11 Brouwers FM, Lenders JW, Eisenhofer G, et al. Pheochromocytoma as an endocrine emergency. Rev Endocr Metabol Disord. 2003;4:121-128.
12 Vaughn ED. Diseases of the adrenal gland. Med Clin North Am. 2004;88:443-466.
13 Grossman E, Goldstein DS, Hoffman A, Keiser A. Glucagon and clonidine testing in the diagnosis of pheochromocytoma. Hypertension. 1991;17:733.
14 Gropper MA. Evidence-based management of critically ill patients: analysis and implementation. Anesth Analg. 2004;99(2):566-572.
15 Rivers EP, Gaspari M, Saad GA, et al. Adrenal insufficiency in high-risk surgical ICU patients. Chest. 2001;119:889-896.
16 Barquist E, Kirton O. Adrenal insufficiency in the surgical intensive care unit. J Trauma. 1997;42(1):27-31.
17 Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348(8):727-734.
18 Angelis M, Yu M, Takanishi D, et al. Eosinophilia as a marker of adrenal insufficiency in the surgical intensive care unit. J Am Coll Surg. 1996;183:589-596.
19 Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288(7):862-871.
20 Capes SE, Hunt D, Malmberg, et al. Stress hyperglycemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355:773-778.
21 Malmberg K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. BMJ. 1997;314(7093):1512-1515.
22 Baird TA, Parsons MW, Phanh T, et al. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke. 2003:2208-2214.
23 Laird AM, Miller PR, Kilgo PD, et al. Relationship of early hyperglycemia to mortality in trauma patients. J Trauma. 2004;56(5):1058-1062.
24 De Courten-Myers GM, Kleinholz M, Holm P, et al. Hemorrhagic infarct conversion in experimental stroke. Annals of Emergency Medicine. 1992;21(2):120-126.
25 Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345(19):1359-1367.
26 Furnary AP, Zerr KJ, Grunkemeier GL, et al. Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures. Ann Thorac Surg. 1999;67:352-362.
27 Zerr KJ, Furnary AP, Grunkemeier GL, et al. Glucose control lowers the risk of wound infection in diabetics after open heart operations. Ann Thorac Surg. 1997;63:356-361.
28 Zimmerman CR, Mlynarek ME, Jordan JA, et al. An insulin infusion protocol in critically ill cardiothoracic surgery patients. Ann Pharmacother. 2004;38:1123-1129.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
CHAPTER 90 MANAGEMENT OF ENDOCRINE DISORDERS IN THE SURGICAL INTENSIVE CARE UNIT
The endocrine system as a part of the neuroendocrine axis (hypothalamic-pituitary axis) influences the response to stress and critical illness. Endocrine abnormalities within this axis change and modify the physiologic response to trauma and stress. Critically ill patients with a known diagnosis of an endocrine problem are treated with replacement therapy. However, an unrecognized endocrine abnormality often creates management difficulties and increases morbidity. Endocrine problems occur at all levels of the neuroendocrine axis from primary or secondary disease, medications, or end-organ failure.
The neuroendocrine axis is responsible for the stress response and is controlled by the hypothalamus, pituitary, and the autonomic nervous system (Figure 1). This axis is activated by baroreceptor response to intravascular volume, sympathetic response from tissue injury, and inflammatory mediators released from tissue trauma. The hormones released in response to injury act through binding to cell surface receptors or intracellular receptors and produce a complex series of responses and feedback loops that maintain cellular processes.1 This chapter addresses abnormalities in the endocrine system that affect the course of critically ill patients.
BRAIN PROBLEMS: ABNORMALITIES IN HYPOTHALAMIC/PITUITARY RESPONSE
Injuries that affect the brain can interrupt the hypothalamus or pituitary production of hormone. Head injury, brain surgery, mass lesions or infiltrative diseases, vascular or hypoxic injuries, and cerebral infections cause failure of the releasing and pituitary hormones—resulting in single or combined abnormalities. Cerebral edema or increased intracranial pressure is thought to restrict the blood flow to the hypothalamic pituitary area. Frequently encountered abnormalities are diabetes insipidus (DI), syndrome of inappropriate antidiuretic hormone (SIADH), and cerebral salt wasting. These syndromes cause abnormalities of sodium and water balance. Evaluation of volume status, urine and serum sodium, and osmolality are required to determine which syndrome is present. This evaluation is important because the treatment depends on which abnormality is present (Table 1).
Diabetes Insipidus
DI is caused by lack of vasopressin (antidiuretic hormone [ADH]), which causes water diuresis of more than 3 l/day, dehydration, and hypernatremia. The urine is dilute with urine osmolality of less than 300 mOsm/kg and urine specific gravity of less than 1.005. Urine osmolality greater than 800 mOsm/kg excludes DI. The diagnosis is usually made when dilute urine output exceeds 200 ml for 2 consecutive hours.2 A dramatic rise in serum sodium occurs in the ICU patient population unless fluids are aggressively replaced. In the neurosurgery patient population, the incidence of DI is 3.7% with a mortality of 70%.2 DI commonly occurs in association with severe brain injury and herniation. The treatment is fluid rehydration and vasopressin replacement via IV dDAVP 2 to 4 mcg/day or intranasally 10 to 60 mcg/day. Frequent monitoring of electrolytes and central venous pressure monitoring are necessary. The water deficit is calculated and slowly replaced. Caution should be used when severe hypernatremia is present, with half the water deficit replaced in 24 hours to avoid demyelination (Table 2).
SIADH and Cerebral Salt Wasting
The syndrome of inappropriate antidiuretic hormone and cerebral salt wasting are linked through a common cause—traumatic brain injury—and common result—hypotonic hyponatremia. Cerebral salt wasting is most often associated with subarachnoid hemorrhage, whereas SIADH is also associated with brain injury, tumors, and medications (Table 3).3 The cause of SIADH is excessive release of ADH that leads to water retention and an increase in extracellular fluid volume. Volume expansion increases renal sodium excretion, producing hyponatremia. Cerebral salt wasting is thought to involve disruption of the neural input to the kidney or central secretion of a natriuretic factor. Sodium is wasted by the kidney, causing extracellular volume depletion and stimulation of ADH secretion. Hyponatremia develops with extracellular volume depletion.
Table 3 Medications That Interfere with Thyroid Hormone and ADH
| Drugs Causing Syndrome of Inappropriate Antidiuretic Hormone (SIADH): |
|
Stimulating ADH release: opiates, opioids, barbiturates, nicotine, thiazides, isoproterenol, cyclic antidepressants, MAO inhibitors, haloperidol, risperidone, acetylcholine
|
NSAID, Nonsteroidal anti-inflammation drug.
In both syndromes, the diagnosis begins with serum sodium less than 135 mmol/l. Measurement of serum osmolality is less than 280 mOsm/kg, and the urine osmolality is greater than 100 mOsm/kg in both diseases. The differentiating factor is the effective blood volume, which is normal in SIADH and low in cerebral salt wasting. Low effective blood volume causes orthostatic blood pressure, tachycardia, low central venous pressure (CVP), low urine sodium, chloride, and fractional excretion of sodium with high BUN. Both disease states have low uric acid level. With correction of the salt deficit, uric acid levels normalize in SIADH and not in cerebral salt wasting.3 The treatment of SIADH is fluid restriction (800–1000 ml/day) and occasional hypertonic saline. Demelocycline, phenytoin, and lithium are used for chronic SIADH. The treatment for cerebral salt wasting is normal saline fluid replacement to expand the extracellular fluid compartment (see Table 1).
ABNORMALITIES IN THYROID RESPONSE
Untreated or unrecognized thyroid problems (excess or deficit) create life-threatening illness in critically ill patients. Thyroid hormones are responsible for the metabolic rate in all tissues. Critical illness may alter the production of thyroid hormone through thyroid-stimulating hormone (TSH) regulation, peripheral metabolism, or alteration in binding proteins. Cytokines as well as commonly used intensive care unit (ICU) medications (see Table 3) affect thyroid hormone function.
Thyroid Excess
Hyperthyroidism with a 3% incidence in out-patients is caused by Grave’s disease, goiter, and adenoma. Treatment with amiodarone increases the incidence to 9%.4 Thyroid storm (severe hyperthyroidism) was first recognized after thyroidectomy in unprepared patients and now encompasses 1%–2% of all admissions for thyrotoxicosis (with a mortality of 20%–30%).5 It is precipitated by physiologic stress related to specific events such as surgery, trauma, childbirth, severe illness, overdose of thyroid medication, iodine in medications, and contrast. The classic symptomatology includes fever, cardiovascular abnormalities, and mental status changes. The tachycardia is out of proportion to the fever. Fever is the hallmark of this disease, with temperatures to 106° F. A state of high-output cardiac failure can develop with bounding pulses, rales and hepatomegaly, and thyroidal bruit. Atrial fibrillation and congestive heart failure are common in elderly patients with hyperthyroidism and can occur without fever (thyrocardiac crisis). The mental status changes include a broad spectrum, from anxiety to coma. There can be nonspecific gastrointestinal (GI) complaints and clinical findings of Grave’s disease.
This syndrome is recognized by clinical signs and symptoms. Laboratory test turnaround time is long, and treatment should be started based on clinical suspicion. The TSH is not detectable, and both T3 and T4 are elevated (with T3 > T4).6 There is an associated elevation of white blood counts (WBCs), calcium, blood glucose, and liver function tests. The treatment is directed toward decreasing the production of thyroid hormone and preventing its release, blocking the peripheral action, supportive care, and treating the cause (Table 4).7
|
Inhibit hormone synthesis: propylthiouracil 1000 mg loading dose, and then 300 mg orally every 6 hours
|
