[level-membership-for-critical-care-medicine-category]
CHAPTER 89 MANAGEMENT OF COAGULATION DISORDERS IN THE SURGICAL INTENSIVE CARE UNIT
Surgeons commonly encounter coagulation disorders in the course of caring for patients, especially those with serious injury and those undergoing or recovering from surgery. Whereas bleeding is a condition well known to man since the beginnings of time, understanding the pathophysiology of bleeding and coagulation and developing effective therapies for them have come relatively recently and continue to undergo change as more is learned about the complex mechanism of blood coagulation and fibrinolysis. The ability to treat hemorrhage effectively had to await the discovery of blood types A, B, and O by Karl Landsteiner in 1900 and the AB blood type by Alfred Decastello and Adriano Sturli in 1902.
INCIDENCE
Congenital Bleeding Disorders
Von Willebrand Disease
1-deamino-8-D-arginine vasopressin (DDAVP) may be used to stimulate production of vWf and increase factor VIII levels in type 1 and type 2a disease. It is ineffective in type 3, however, and contraindicated in type 2b due to risk of thrombocytopenia and increased bleeding. Concentrates of factor VIII vWf are virus inactivated and are used commonly in types 2 and 3, but also in type 1 that is unresponsive to DDAVP. Cryoprecipitate contains vWf and factor VIII, and may be used in all types of vWD. However, it is pooled and not virus inactivated. It is only recommended as a third-line therapy. Antifibrinolytic amino acids, such as aminocaproic acid and tranexamic acid, are used as adjuvant therapy in all types of vWD along with the previously cited treatments.
Acquired Bleeding Disorders
Acidosis
Metabolic acidosis has long been recognized as a consequence of, and contributor to, coagulopathy. However, the specific pathways whereby acidosis impairs coagulation have yet to be clearly defined. Animal data suggest that hypothermia induces a delayed onset of thrombin formation, whereas acidosis decreases the overall thrombin generation rate. The association of severe acidosis (pH < 7.1) with hypotension and hypothermia in severely injured patients virtually guarantees life-threatening coagulopathy. Therapy is again directed at the cause of acidosis and not merely the correction of the pH. While simultaneously addressing the inciting events, lactic acidosis is treated with fluid resuscitation to optimize tissue perfusion. It can be guided by following the trend in base deficit or lactate level. Sodium bicarbonate administration is ineffective and potentially harmful in lactic acidosis, and is not recommended.
Traumatic Brain Injury
The importance of the hemostatic changes may lie in promotion of secondary brain injury through cerebral microvascular thrombosis, which may exacerbate cerebral ischemia. Currently, it is not known whether therapy should target hemostasis to prevent further bleeding in the injured brain or block part of the coagulation pathway to prevent microthrombosis and ischemia. Until the pathophysiology is better understood, treatment should be directed only toward clinical endpoints and maintenance of platelets and clotting factors as necessary for hemostasis (as outlined elsewhere in this chapter).
DIAGNOSIS
Clinical Evaluation
Bleeding in a critically ill patient should be evaluated in a systematic fashion to detect the cause and direct the treatment of the bleeding (Figure 1). In the surgical ICU, the first critical decision in a bleeding patient is to differentiate surgical bleeding from nonsurgical coagulopathic microvascular bleeding. This may be one of the most difficult decisions a surgeon can face. A nontherapeutic operation on a coagulopathic patient may exacerbate the vicious cycle, but leaving a surgically correctable source of bleeding untouched can prove fatal.
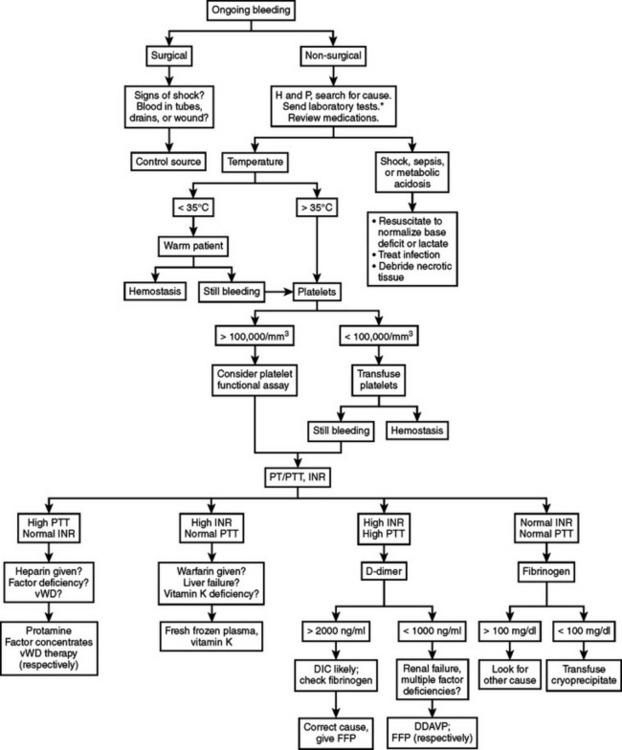
Coagulation disorders may be grouped into those affecting primary hemostasis (formation of initial platelet plug) or secondary hemostasis (clotting factors and the coagulation cascade). These groups may be subdivided into qualitative defects (e.g., dysfunctional platelets, factor inhibition by heparin) or quantitative defects (e.g., thrombocytopenia, factor deficiencies). Furthermore, these conditions may be congenital or acquired. The algorithm presented in Figure 1 represents one example of a systematic approach that can help guide therapy in most surgical patients, even if the exact cause of the coagulopathy is not evident. It is intended to aid rapid assessment and initiation of treatment in the ICU, rather than as a definitive guide to diagnosis of specific bleeding disorders.
Laboratory Tests of Coagulation
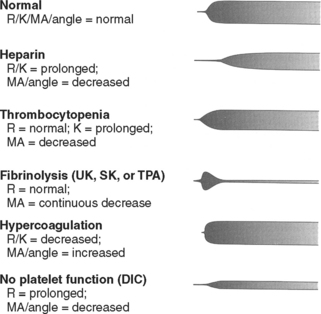
TEG: TEG is reported as a graph of clot formation in a sample of whole blood (Figure 2). The TEG tracing is drawn based on several factors, including rate of clot formation, fibrin cross-linking, and platelet-fibrin interaction. By measuring various parameters of the tracing, TEG provides an assessment of platelet function, coagulation enzyme activity, and the overall degree of coagulability. It can identify conditions such as primary fibrinolysis, consumptive coagulopathy, anticoagulant therapy, and the effect of hypothermia. TEG is used frequently during cardiopulmonary bypass, liver transplantation, and in intensive care settings due to its rapid availability and ability to assess the components of coagulation in an integrated fashion.
MANAGEMENT
Blood Product Transfusion
Reversal of Warfarin
Reversal of warfarin is guided by the INR and is best managed by standardized evidence-based guidelines. Vitamin K takes 8–12 hours to take effect, and is the first-line choice for nonemergent treatment of a high INR. However, it has a long half-life and high or repeated doses should be avoided if re-anticoagulation with warfarin is anticipated in the next several days. Oral vitamin K is preferred for nonemergent reversal of warfarin, whereas the subcutaneous route is not recommended because of inefficient absorption. Patients receiving intravenous vitamin K should have continuous cardiac monitoring due to the risk of anaphylaxis. FFP is the standard therapy for patients with a high INR and significant bleeding or need for invasive procedures.
Many elderly patients on warfarin have concomitant heart disease, and caution must be used to avoid precipitating congestive heart failure with overly aggressive fluid loading. In our experience, an INR greater than 2 is rarely normalized with only one or two units of FFP. In addition, patient factors vary considerably—resulting in an unpredictable and nonlinear dose-response relationship. Recombinant activated factor VII (rFVIIa) may be used when immediate reversal of anticoagulation is required in emergent situations such as severe TBI or life-threatening bleeding. However, data on its proper use in these conditions is limited. rFVIIa’s half-life is only a fraction of that of warfarin, and thus it must be used in conjunction with FFP and vitamin K to maintain normal coagulation. Table 1 is an example of a management scheme for patients on warfarin with an elevated INR and risk of bleeding.
Table 1 Management Options for Patients with Warfarin Anticoagulation and Bleeding Risk
| Clinical Context | Treatment Options |
|---|---|
| INR < 5, no significant bleeding | Hold warfarin |
| INR > 5, no significant bleeding | Vitamin K 1–5 mg orally |
| INR > 1.5, bleeding or high risk of bleeding, nonemergent | FFP in 2- to 4-unit doses, recheck INR after each dose, until bleeding stopped or INR < 1.5 |
| Consider vitamin K 5 mg orally | |
| INR > 1.5, life-threatening bleeding or emergent surgery or invasive procedure required | FFP in 4-unit doses, recheck INR after each dose, and vitamin K 10 mg slow IV infusion |
| Consider factor 7a (repeat in 2–3 hours if still bleeding) |
CONCLUSIONS
The use of blood components should be guided by objective evidence of coagulation abnormalities (including clinical findings and laboratory data) rather than resorting to formula-based replacement. Selective use of components (especially platelet transfusion) will yield safer and more effective therapy. Such an approach should lead to more effective management of coagulopathy and more judicious use of blood component therapy. Continued advances in the field present novel opportunities to affect coagulation, but the fundamental principles still apply in the patient with hemorrhage: control bleeding rapidly, expeditiously resuscitate from shock, manage temperature carefully, and monitor the patient for clinical and laboratory evidence of coagulation abnormalities.
Aird WC. Sepsis and coagulation. Crit Care Clin. 2005;21:417.
Ansell J, et al. The pharmacology and management of the vitamin K antagonists: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest. 2004;126(Suppl 3):204S.
Bernard GR, et al. Safety and dose relationship of recombinant human activated protein C for coagulopathy in severe sepsis. Crit Care Med. 2001;29:2051.
Boffard KD, et al. Recombinant factor VIIa as adjunctive therapy for bleeding control in severely injured trauma patients: two parallel randomized, placebo-controlled, double-blind clinical trials. J Trauma. 2005;59:8.
Boccardo P, et al. Platelet dysfunction in renal failure. Semin Thromb Hemost. 2004;30(5):579.
Cable R, et al. Practice guidelines for blood transfusion: a compilation from recent peer-reviewed literature. American National Red Cross, 2002. http://www.redcross.org/services/biomed/profess/pgbtscreen.pdf
Committee on Trauma of the American College of Surgeons. Advanced Trauma Life Support Course for Doctors, 7th ed., Chicago: American College of Surgeons; 2004:69.
Cosgriff N, et al. Predicting life-threatening coagulopathy in the massively transfused trauma patient: hypothermia and acidoses revisited. J Trauma. 1997;42:857.
Demetriades D, et al. Liver cirrhosis in patients undergoing laparotomy for trauma: effect on outcomes. J Am Coll Surg. 2004;199:539.
Fakhry SM, et al. Hematologic principles in surgery. In: Townsend CM, Beauchamp RD, Evers BM, Mattox KL, editors. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 16th ed. Philadelphia: WB Saunders; 2001:68.
Forsythe SM, Schmidt GA. Sodium bicarbonate for the treatment of lactic acidosis. Chest. 2000;117:260.
Gando S. Disseminated intravascular coagulation in trauma patients. Semin Thromb Hemost. 2001;27(6):585.
Gando S, et al. Coagulofibrinolytic changes after isolated head injury are not different from those in trauma patients without head injury. J Trauma. 1999;46:1070.
Gentilello LM, et al. Is hypothermia in the victim of major trauma protective or harmful? A randomized, prospective study. Ann Surg. 1997;226(4):439.
Holcomb JB. Use of recombinant activated factor VII to treat the acquired coagulopathy of trauma. J Trauma. 2005;58:1298.
Jurkovich GJ, et al. Hypothermia in trauma victims: an ominous predictor of survival. J Trauma. 1987;27:1019.
Kashuk JL, et al. Major abdominal vascular trauma—a unified approach. J Trauma. 1982;22:672.
Kujovich JL. Hemostatic defects in end stage liver disease. Crit Care Clin. 2005;21:563.
Levi M. Disseminated intravascular coagulation: what’s new? Crit Care Clin. 2005;21:449.
Mannucci PM. Treatment of von Willebrand’s disease. N Engl J Med. 2004;351:683.
Mina AA, et al. Complications of preinjury warfarin use in the trauma patient. J Trauma. 2003;54:842.
Oung CM, et al. In vivo study of bleeding time and arterial hemorrhage in hypothermic versus normothermic animals. J Trauma. 1993;35:251.
Owings JT, Gosselin RC. Bleeding and transfusion. In: Fink MP, Jurkovich GJ, Kaiser LR, Pearce WH, Pemberton J, Soper NJ, editors. ACS Surgery: Principles & Practice. New York: Web MD Corp, 2003. www.acssurgery.com
Rodgers RPC, Levin J. A critical reappraisal of the bleeding time. Semin Thromb Hemost. 1990;16:1.
Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479.
The American Society of Anesthesiologists Task Force on Blood Component Therapy. practice guidelines for blood component therapy. Anesthesiology. 1996;84(3):732.
Valeri CR, et al. Hypothermia induced reversible platelet dysfunction. Ann Surg. 1987;205:175.
Watts DD, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma. 1998;44:846.
Wolberg AS, et al. A systematic evaluation of the effect of temperature on coagulation enzyme activity and platelet function. J Trauma. 2004;56:1221.
Wojcik R, et al. Preinjury warfarin does not impact outcome in trauma patients. J Trauma. 2001;51:1147.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
CHAPTER 89 MANAGEMENT OF COAGULATION DISORDERS IN THE SURGICAL INTENSIVE CARE UNIT
Surgeons commonly encounter coagulation disorders in the course of caring for patients, especially those with serious injury and those undergoing or recovering from surgery. Whereas bleeding is a condition well known to man since the beginnings of time, understanding the pathophysiology of bleeding and coagulation and developing effective therapies for them have come relatively recently and continue to undergo change as more is learned about the complex mechanism of blood coagulation and fibrinolysis. The ability to treat hemorrhage effectively had to await the discovery of blood types A, B, and O by Karl Landsteiner in 1900 and the AB blood type by Alfred Decastello and Adriano Sturli in 1902.
INCIDENCE
Congenital Bleeding Disorders
Von Willebrand Disease
1-deamino-8-D-arginine vasopressin (DDAVP) may be used to stimulate production of vWf and increase factor VIII levels in type 1 and type 2a disease. It is ineffective in type 3, however, and contraindicated in type 2b due to risk of thrombocytopenia and increased bleeding. Concentrates of factor VIII vWf are virus inactivated and are used commonly in types 2 and 3, but also in type 1 that is unresponsive to DDAVP. Cryoprecipitate contains vWf and factor VIII, and may be used in all types of vWD. However, it is pooled and not virus inactivated. It is only recommended as a third-line therapy. Antifibrinolytic amino acids, such as aminocaproic acid and tranexamic acid, are used as adjuvant therapy in all types of vWD along with the previously cited treatments.
Acquired Bleeding Disorders
Acidosis
Metabolic acidosis has long been recognized as a consequence of, and contributor to, coagulopathy. However, the specific pathways whereby acidosis impairs coagulation have yet to be clearly defined. Animal data suggest that hypothermia induces a delayed onset of thrombin formation, whereas acidosis decreases the overall thrombin generation rate. The association of severe acidosis (pH < 7.1) with hypotension and hypothermia in severely injured patients virtually guarantees life-threatening coagulopathy. Therapy is again directed at the cause of acidosis and not merely the correction of the pH. While simultaneously addressing the inciting events, lactic acidosis is treated with fluid resuscitation to optimize tissue perfusion. It can be guided by following the trend in base deficit or lactate level. Sodium bicarbonate administration is ineffective and potentially harmful in lactic acidosis, and is not recommended.
