[level-membership-for-anesthesiology-category]
Chapter 18 Management of cardiac arrhythmias
CARDIAC ELECTROPHYSIOLOGY
The electrophysiological properties of cardiac cells are important in understanding cardiac arrhythmias and their management. Cardiac cells undergo cyclical depolarisation and repolarisation to form an action potential. The shape and duration of each action potential are determined by the activity of ion channel protein complexes on the myocyte surface. These highly selective ion channels determine the rate of ion flux which in turn determines the magnitude and rate of change of myocyte membrane potential. Many of these ion channels are the molecular targets for antiarrhythmic drugs.
Ion channel function can be affected by:
The spectrum of cardiac action potentials varies from fast-response cells – conducting and contractile myocytes (Figure 18.1a) – to slow-response cells of pacemaker myocytes – sinoatrial (SA) and atrioventricular (AV) nodes (Figure 18.1b). Fast myocytes lose their characteristic action potential and behave more like slow myocytes when ischaemic. The action potential is divided into five phases, as follows.
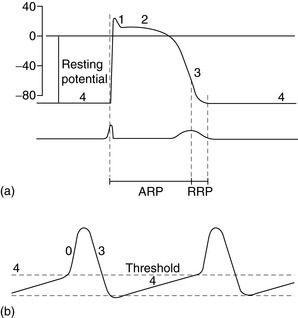
PHASE 0
In fast myocytes (Figure 18.1a) rapid depolarisation occurs due to activation of voltage-dependent Na+ channels. Activation is initiated in an all-or-none response once the threshold is reached. The Na+ channels are inactivated as membrane potential rises to +30 mV and remain inactivated until repolarisation occurs. Rapidity of depolarisation determines speed of conduction. In slow-response myocytes depolarisation does not involve Na+ channels and the slower rate of depolarisation is due to a slow inward Ca2+ current via L- and T-type voltage-dependent Ca2+ channels.
PHASE 1
Early rapid incomplete repolarisation to approximately 0 mV occurs due to activation of transient outward current from ITO1 and ITO2 K+ channels. Slow myocytes do not exhibit phase 1 or 2 characteristics (Figure 18.1b).
PHASE 4
This is a stable electrical state in fast non-pacemaker myocytes. In slow pacemaker myocytes the resting membrane potential (RMP) slowly depolarises until the action potential threshold is reached (Figure 18.1b). This inward or pacemaker current is due to If K+ channels.
Fast-response and slow-response myocytes also have important differences in properties of refractoriness. In fast myocytes, Na+ channels are progressively reactivated during phase 3 repolarisation as the membrane potential becomes more negative. When an extra stimulus occurs during phase 3, the magnitude of the resulting inward Na+ current and likelihood of impulse propagation depend on the number of reactivated Na+ channels. Refractoriness is therefore determined by the voltage-dependent recovery of Na+ channels. The absolute refractory period (Figure 18.1) is that minimum time needed for recovery of sufficient Na+ channels for a stimulus to result in impulse propagation. However, once propagation in fast myocytes occurs, conduction velocity is normal. In contrast, slow-response or Ca2+ channel-dependent myocytes exhibit time-dependent refractoriness. Even after full repolarisation further time is needed before all Ca2+ channels are reactivated. Stimuli during this period produce reduced Ca2+ current and the propagation velocity of any resulting impulse is reduced. The conduction velocity independence of premature action potentials with fast-response myocytes is lost in the setting of Na+ channel-blocking drugs or ischaemia because they behave increasingly like slow-response myocytes with resulting slowed impulse conduction.
GENETIC BASIS TO ARRHYTHMIA1
MOLECULAR BASIS TO ARRHYTHMIA1
Tachycardic remodelling of the atrium is associated with:
Heart failure is associated with:
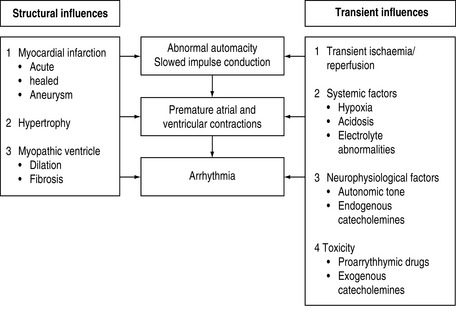
ARRHYTHMOGENIC MECHANISMS2–4
Many factors in isolation or combination give rise to the substrate of arrhythmogenesis (Figure 18.2). Arrhythmia may arise from abnormalities of impulse generation or conduction. Table 18.1 demonstrates the relationship between mechanism and type of arrhythmia, and desired antiarrhythmic effect.
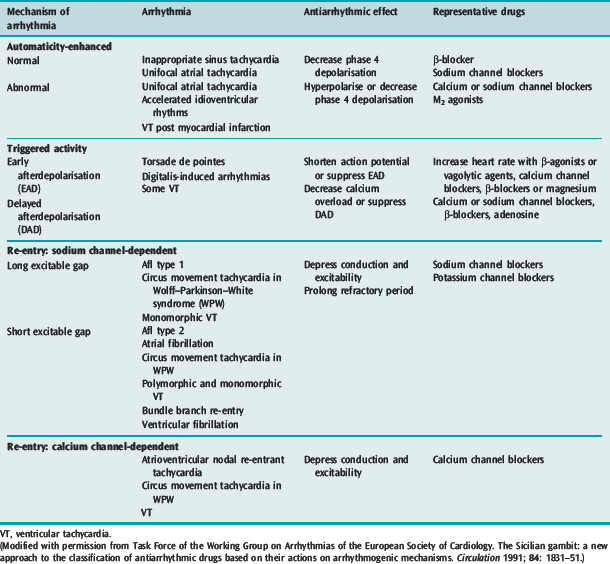
ABNORMAL IMPULSE GENERATION (Table 18.2)
ENHANCED NORMAL AUTOMATICITY
| Enhanced normal automaticity | Adrenergic stimulation |
| Abnormal automaticity | Ischaemia |
| Early afterdepolarisations | Hypoxia |
| Hypercapnia | |
| Catecholamines | |
| Class IA antiarrhythmic drugs | |
| Class III antiarrhythmic drugs | |
| Other drugs that prolong repolarisation | |
| Delayed afterdepolarisations | Digoxin toxicity |
| Increased intracellular Na+ | |
| Decreased extracellular K+ | |
| Increased intracellular Ca2+ | |
| Intracellular Ca2+ overload | |
| Myocardial infarction | |
| Reperfusion after ischaemia |
TRIGGERED ACTIVITY
ABNORMAL IMPULSE CONDUCTION5
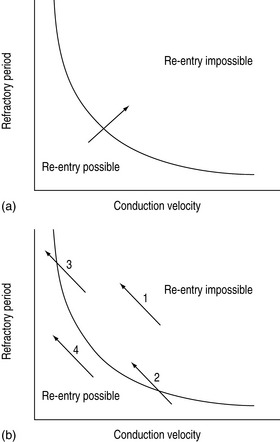
Abnormal impulse conduction may cause an arrhythmia by the phenomena of re-entry. Re-entry describes the re-excitation of an area or entire heart by a circulating impulse. Although the classic ‘bifurcating Purkinje fibre’ model of Schmitt and Erlanger has given way to a much more complex picture, the essential electrophysiological requirements for re-entrant excitation remain. Requirements for re-entry are (Figure 18.3):
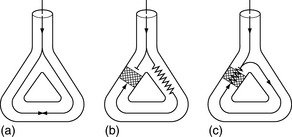
When these properties are present, the chance of a circulating impulse producing re-entrant excitation depends on pathway geometry, the electrical properties and length of the depressed area and conduction velocity within each component. The segment of the re-entry pathway that is initially refractory and therefore blocks conduction down one limb and recovers in time to conduct the return impulse is termed the ‘excitable gap’. Therefore, the generation and subsequent maintenance of a circuit depend on this excitable gap of non-refractory tissue circulating between the advancing depolarising wave front and the repolarising tail. The resulting re-entrant impulse can be self-terminating, causing ectopic beats, or lead to atrial or ventricular tachyarrhythmias.
Re-entry may be terminated by:
The cellular properties that lead to impaired conduction include:
ELECTROLYTE ABNORMALITIES AND ARRHYTHMIA6
POTASSIUM
Hyper- and hypokalaemia both cause arrhythmia mediated by the resultant changes in RMP (Table 18.3). In ischaemia, hyperkalaemia at the local tissue level caused by a pathological extracellular shift of K+ is the major factor contributing to ventricular arrhythmia in this setting. In hypokalaemia, the dispersion of pacemaker activity and the effect on repolarisation are similar to the electrophysiological effects of cardiac glycosides and β-adrenergic agonists, and it is not surprising that a combination of these factors is associated with an increased incidence of arrhythmia. The increased risk of death in hypertensive patients treated with thiazide diuretics has been attributed to hypokalaemic (and possibly hypomagnesaemic)-induced arrhythmia (Multiple Risk Factor Intervention Trial). Thiazide-induced hypokalaemic ventricular ectopy is worsened by exercise.7 Hypokalaemia is associated with VF and VT following acute myocardial infarction (AMI). The increased incidence of VF/VT with a serum K+ less than 3.5 mmol/l is clearly established and the probability of VT increases as the serum K+ decreases. During AMI the incidence of VF/VT was 15% at 4.5 mmol/l, 38% at 3.5 mmol/l, 55% at 3.0 mmol/l and 67% at 2.5 mmol/l.8
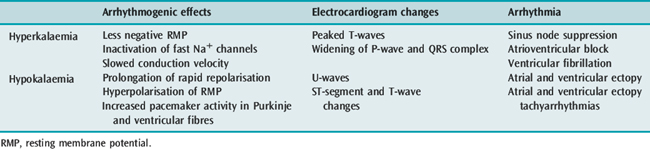
MAGNESIUM
The antiarrhythmic properties of Mg2+ are clearly established but a causal relationship between hypomagnesaemia and arrhythmia is largely circumstantial. Decreased extracellular Mg2+ by itself has little effect on the electrophysiological properties of myocytes or the ECG. Hypomagnesaemia has been implicated in the genesis of VT/VF in patients with hypertension and heart failure receiving thiazide or loop diuretics, acute alcohol intoxication or withdrawal and possibly with AMI. The product of K+ and Mg2+ is the best predictor of arrhythmia in hypertensive patients taking thiazide diuretics.9
AUTONOMIC NERVOUS SYSTEM AND VENTRICULAR ARRHYTHMIA10
PROARRHYTHMIC EFFECTS OF ANTIARRHYTHMIC DRUGS11,12
Concomitant proarrhythmia with the use of antiarrhythmic drugs is increasingly recognised. The ‘quinidine syncope’ due to VF and polymorphic VT at therapeutic concentrations was also seen with disopyramide. The Cardiac Arrhythmia Suppression Trial (CAST) clearly defined the magnitude of this deleterious side-effect in drugs that were previously perceived to be of benefit.13 This study, which involved flecainide, encainide and morizicine (a class IA drug), was terminated early because of adverse outcome in the flecainide and encainide groups (relative risk of arrhythmic death or non-fatal cardiac arrest of 3.6, 95% confidence interval (CI) 1.7–8.5). Proarrhythmia is reported between 5.9% and 15.8% depending on agent, clinical setting and definition of proarrhythmia, and now considered ubiquitous with all antiarrhythmic drugs.
Proarrhythmia has been defined as an increase in frequency of ventricular ectopic beat (VEB) or aggravation of the target arrhythmia on Holter monitor or exercise test. Manifestations of proarrhythmia not only include VEB, monomorphic and polymorphic VT and VF, but also bradyarrhythmias and Afl with 1:1 AV conduction. Most proarrhythmic events occur soon after starting the drug, but late arrhythmias are also a significant problem.
The mechanism of drug proarrhythmia is probably via both slowing of conduction and abnormal automaticity. Paradoxically slowing conduction, which may block a re-entry circuit, may also create the very substrate needed for re-entry, unidirectional block and an excitable gap. The existence of a re-entrant circuit requires the circulating wave front of the impulse not to catch up with the refractory tissue behind the tail. Re-entry is more likely to occur with a shorter refractory period and reduced conduction velocity (Figure 18.4).11
Antiarrhythmic drugs are effective at suppressing abnormal automaticity, with the exception of triggered automaticity due to EAD. Class IA, class III and many non-antiarrhythmic drugs can produce proarrhythmia via EAD. These drugs increase not only the frequency of EAD, but also the likelihood of them leading to triggered tachyarrhythmias. Slowing repolarisation, which leads to QT prolongation and slower heart rate, is central to this increased frequency and sensitivity to EAD. EAD manifests as prominent and bizarre T-U waves on the ECG and, if triggered activity results, VEB and ventricular tachyarrhythmias may occur. Torsade de pointes is the classical resulting arrhythmia, although less classical polymorphic VT and VF result. Risk of proarrhythmia via this mechanism correlates with the degree of QT prolongation.
Antiarrhythmic drug proarrhythmia is facilitated by several factors, which are frequently found in patients on antiarrhythmic drugs or with heart disease (Table 18.4).
Table 18.4 Factors facilitating antiarrhythmic drug proarrhythmia
| Toxic blood levels due to excessive dose or reduced clearance from old age, heart failure, renal disease or hepatic disease |
| Severe left ventricular dysfunction. Ejection fraction less than 35% |
| Pre-existing arrhythmia or arrhythmia substrate |
| Digoxin therapy |
| Hypokalaemia or hypomagnesaemia |
| Bradycardia |
| Combinations of antiarrhythmic drugs and concomitant drugs with similar toxicity |
(Adapted from Campbell TJ. Proarrhythmic actions of antiarrhythmic drugs: a review. Aust NZ J Med 1990; 20: 275–82, with permission.)
MANAGEMENT OF THE PATIENT WITH A CARDIAC ARRHYTHMIA
INVESTIGATIONS
A 12-lead ECG should be recorded with a longer rhythm strip (usually lead II or V1). If P-waves are not visible, atrial activity may be recorded using an oesophageal electrode or pacing lead, or via a central venous catheter or the right atrial injectate port of a pulmonary artery catheter, using 20% saline and a bedside monitor.14
EPS, which involves invasive electrophysiological testing with programmed electrical stimulation, attempts to reproduce the spontaneously occurring arrhythmia.15,16 EPS is not clearly superior to Holter monitoring in evaluating drug treatment for ventricular arrhythmias.
Other investigative techniques being studied include signal-averaged ECG, heart rate variability and electrical alternans measurement.10,17
MANAGEMENT OF SPECIFIC ARRHYTHMIAS
Treatment has two aspects: acute termination of the arrhythmia and long-term prophylaxis. The decision whether to treat depends on the rhythm diagnosis, haemodynamic consequences, aetiology of the arrhythmia and the prognosis (e.g. risks of sudden death or long-term complications).
PREMATURE VENTRICULAR ECTOPIC BEATS
ECG
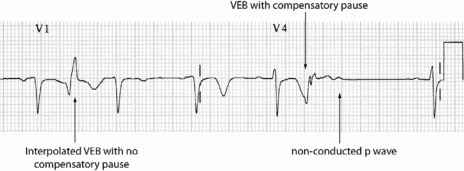
Occasionally VEB may not produce any pause, and are said to be interpolated (see Figure 18.6). Interpolated VEB occur when the background sinus rhythm is slow. The retrograde conduction into the AV node renders it partially refractory to the next impulse and its conduction through the AV node is slowed and the PR interval is prolonged. A VEB following each sinus beat is ventricular bigeminy (Figure 18.7). Ventricular trigeminy refers to recurring sequences of a VEB followed by two sinus beats. Two VEB in succession are a couplet (Figure 18.8), and three, a triplet.
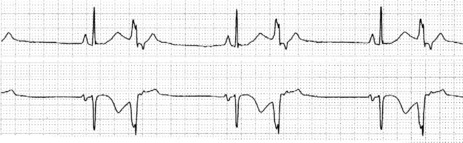
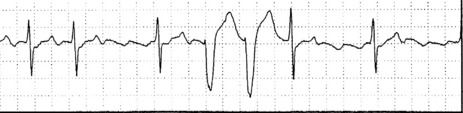
CLINICAL
Even when frequent, complex, or in short runs of non-sustained VT, VEB are not associated with risk of sudden death in asymptomatic healthy adults.18 However, there is increased risk of cardiovascular death with:
SUPRAVENTRICULAR TACHYCARDIAS22,23 (Table 18.5)
| Atrioventricular (AV) node-dependent |
| AV nodal re-entry tachycardia: re-entry within the AV node |
| AV re-entry tachycardia: re-entry includes accessory pathway between atria and ventricles |
| Accelerated idionodal rhythm: increased automaticity of AV nodea |
| AV node-independent |
| Atrial flutter: re-entry confined to atria |
| Atrial fibrillation: multiple re-entry circuits confined to atria |
| Unifocal atrial tachycardia: usually due to increased automaticity |
| Multifocal atrial tachycardia: increased automaticity or triggered activity |
| Others: sinus node re-entry tachycardia |
A clinically useful classification divides SVT into AV node-dependent and AV node-independent.
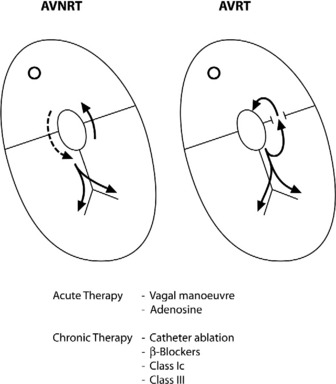
AV NODAL RE-ENTRY TACHYCARDIA (AVNRT)(Figure 18.9)
ECG
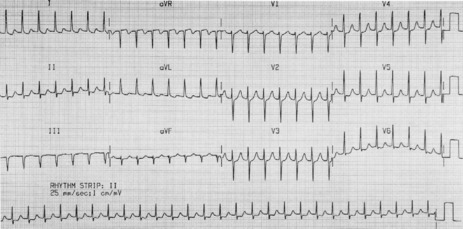
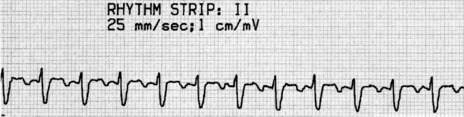
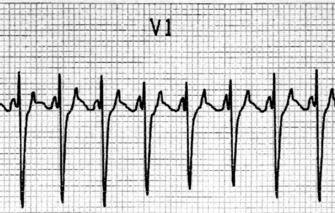
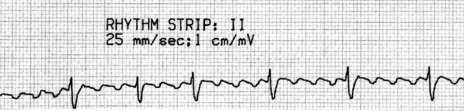
There is regular narrow-complex tachycardia (140–220 beats/min) with abrupt onset and termination. P-waves are not usually observed as they are buried in the QRS complexes (Figure 18.10).
TREATMENT
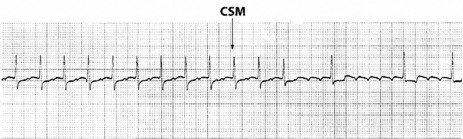
Vagal manoeuvres slow conduction through the AV node and may ‘break’ the tachycardia. If carotid sinus massage fails, adenosine is the drug of choice and nearly all AVNRT will revert with adenosine.24,25 Verapamil has been used in the past, but causes hypotension, which may be prolonged if cardiac function is depressed or patients are receiving β-adrenergic blockers. Sotalol, amiodarone and flecainide may also be effective but are rarely used. Rapid atrial pacing will usually terminate AVNRT but is rarely needed.
AV RE-ENTRY TACHYCARDIA (SEE Figure 18.9)
ECG
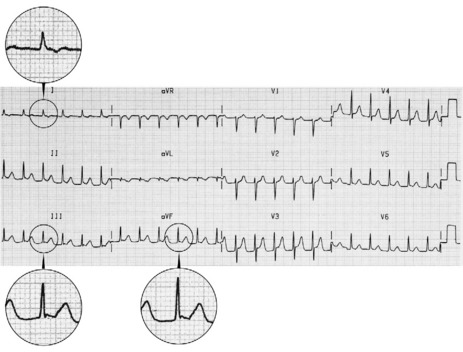
The ECG is similar to AVNRT. The length of the re-entry circuit is however greater, and the accessory AV pathway is some distance from the AV node. It therefore takes longer for the impulse to be conducted backwards to the atria, and so the retrograde P-wave usually occurs after the QRS, sometimes at some distance, and is inverted in leads II, III and aVF (Figure 18.11 and 18.12).
ACCELERATED IDIONODAL RHYTHM
ECG
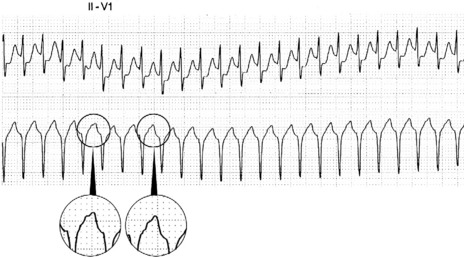
There are narrow complexes on the ECG at a regular rate (60–130 beats/min) (Figure 18.13), often with independent atrial activity. With isorhythmic dissociation, P-wave is either fixed relative to the QRS complex (usually just after) or oscillates to and fro across the QRS in a rhythmical manner.
UNIFOCAL ATRIAL TACHYCARDIA
ECG
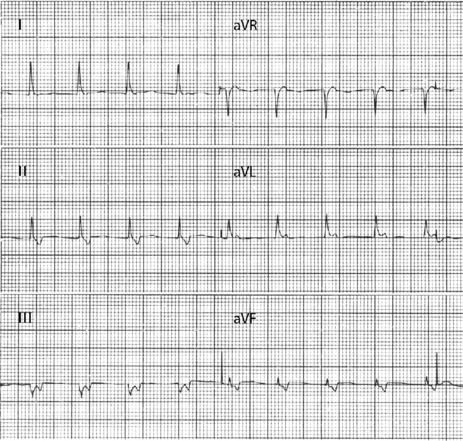
P-wave morphology is abnormal but monomorphic. Atrial rate is often 130–160 beats/min, and may occasionally exceed 200 beats/min. Atrial rate distinguishes unifocal atrial tachycardia from atrial flutter (Afl), with Afl greater than 250 beats/min. The QRS complexes will usually be narrow (Figure 18.14). AV block is common (Figure 18.15).
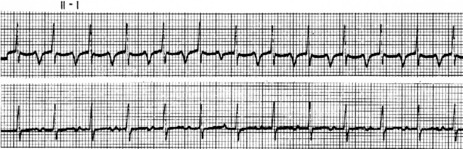
CLINICAL
Digitalis intoxication is the most common cause, especially when AV block is present. Other causes include myocardial infarction, chronic lung disease and metabolic disturbances.
MULTIFOCAL ATRIAL TACHYCARDIA28
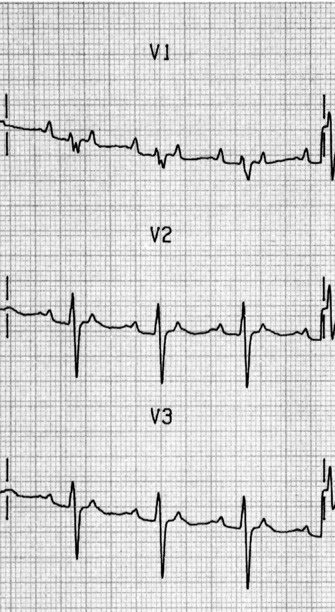
ECG
There are irregular atrial rates, usually 100–130 beats/min, with varying P-wave morphology (at least three different P-wave morphologies and varying PR interval) and some degree of AV block (Figure 18.16). Most P-waves are conducted to the ventricles, usually with narrow QRS complexes.
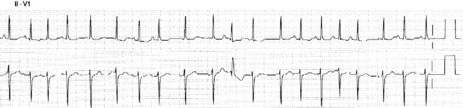
TREATMENT
Treatment should correct the underlying cause (e.g. treatment of cardiorespiratory failure, electrolyte and acid–base abnormalities and theophylline toxicity). Spontaneous reversion is common, and few patients require antiarrhythmic therapy. Magnesium is the drug of choice for acute control.29 β-Blockers are probably more effective than diltiazem, but because of the common association of MAT with obstructive lung disease have limited utility.30 Digoxin and cardioversion are ineffective, which highlights the need to differentiate MAT from AF. Longer-term control is best achieved with diltiazem in patients with good left ventricular (LV) function and amiodarone in those without.
ATRIAL FLUTTER31
ECG
Afl waves (characteristic sawtooth appearance with no isoelectric baseline) are best seen in V1 (Figure 18.17) or aVF, but leads II and III may also be useful. The flutter waves are usually negative in aVF. Rapid QRS waves may obscure typical flutter waves, and vagal manoeuvres may unmask them (see Figure 18.5). AV conduction block (usually 2:1) is usually present, so that alternate flutter waves are conducted to the ventricles, with a ventricular rate close to 150 beats/min. Frequently flutter waves are not obvious and a ventricular rate of 150 beats/min leads to the presumption of Afl (Figure 18.18). Type II Afl results in greater atrial and ventricular rates (Figure 18.19). Treatment with drugs that affect AV node conduction may lead to higher degrees of AV block (Figure 18.20) and/or variable AV block with irregular QRS duration. Rarely, Afl with 1:1 conduction occurs. This is usually associated with sympathetic overactivity or class I antiarrhythmic drugs (which slow atrial discharge rate to 200 beats/min, thereby allowing each atrial impulse to be conducted) (Figure 18.21). QRS complexes are usually narrow, as conduction through the bundle branches is normal.
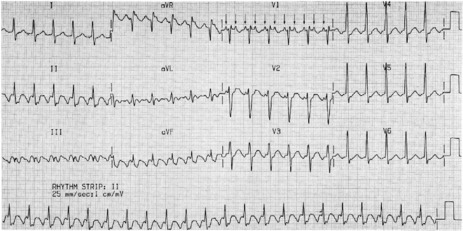
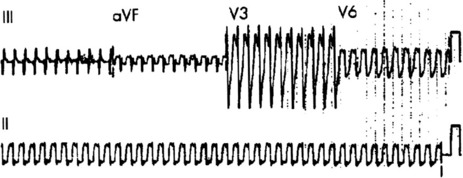
TREATMENT25
No drug will reliably terminate Afl, although ibutillide and dofetilide have been shown to be most likely to result in pharmacological reversion. Attempts at slowing ventricular rate by drugs that will increase the degree of AV block are worthwhile in the first instance. Drugs such as digoxin, diltiazem, β-adrenergic blockers, sotalol and amiodarone may be tried; the choice depends on LV function. Flecainide and procainamide may occasionally be effective at terminating Afl. However, class IA and IC drugs may lead to 1:1 AV conduction. Class I drugs should probably be avoided unless ventricular response has been slowed with calcium channel or β-adrenergic blocking drugs.
Anticoagulation guidelines are the same as that for AF, although there are less supporting data.
PREVENTION
Prevention is difficult. Drugs used include sotalol and amiodarone at low doses. Class IC agents (e.g. flecainide) may be used in patients without significant structural heart disease. Increasingly recurrent or refractory Afl may be cured by radiofrequency ablation to create a linear lesion between the inferior tricuspid annulus and the eustachian ridge at the anterior margin of the inferior vena cava to interrupt the re-entry circuit.26
ATRIAL FIBRILLATION32
ECG
Atrial activity is chaotic with rapid (350–600 beats/min) and irregular depolarisations varying in amplitude and morphology (fibrillation waves). Ventricular response is irregularly irregular (Figure 18.22). Most atrial impulses are not conducted to the ventricles, resulting in an untreated ventricular rate of 100–180 beats/min. QRS complexes will usually be narrow. When the ventricular rate is very rapid or very slow, ventricular irregularity may be missed (Figure 18.23).
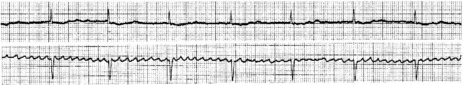
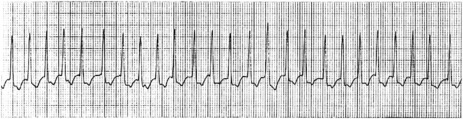
CLINICAL
TREATMENT25,34,35
The goals of treatment include ventricular rate control, anticoagulation where appropriate and conversion to sinus rhythm. There is increasing evidence available on the ‘rate versus rhythm’ control debate. Results from several recent major studies have challenged the previous belief that achievement of sinus rhythm is important in the long term (Table 18.6). When comparing control of ventricular rate versus reversion to sinus rhythm no clear survival benefit is apparent. However composite end-points of death, stroke and recurrent hospilisation favour rate control only.36–39
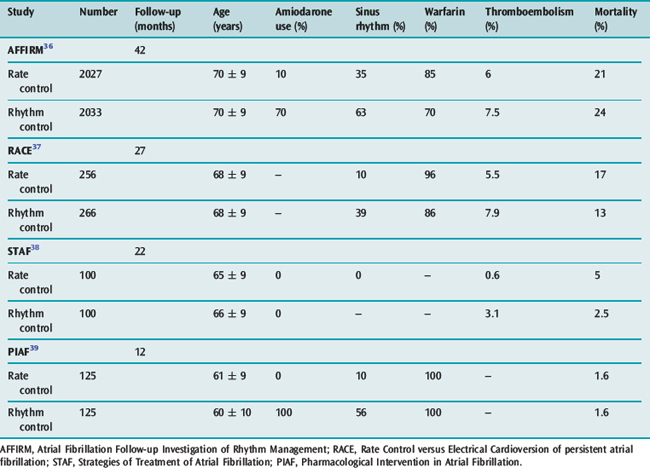
The possible reasons why rhythm control has not been shown to be superior include:
However rhythm control (if possible) appears superior in patients with LV dysfunction, with both amiodarone and dofetilide reducing mortality when sinus rhythm is achieved.40,41 The paucity of data in younger patients (less than 60 years) favours initial attempts at rhythm control, particularly in those with structurally normal hearts, in the hope that progressive atrial electrical and anatomical remodelling is prevented.
RECENT ONSET OR PAROXYSMAL AF
VENTRICULAR RATE CONTROL
CONVERSION TO SINUS RHYTHM
Antiarrhythmic drugs or DC shock cardioversion can be used. The likelihood of short- and long-term success depends on the clinical situation. Conversion to sinus rhythm is more important in young patients and those with heart failure. Maintenance of sinus rhythm is problematic: sinus rhythm at 1 year is 60% with amiodarone and 40% with sotalol, and associated with significant drug cardiac and extracardiac toxicities. The risk of stroke and need for antithrombotic therapy due to frequent AF recurrences, which may be asymptomatic, remain. Achieving sinus rhythm (especially greater than 60 years) is less important than previously thought.36
DC SHOCK CARDIOVERSION
Critically ill patients who are septic, postoperative or on drugs such as catecholamines are likely to relapse.
ANTIARRHYTHMIC DRUGS
Quinidine is more effective than placebo, but increases mortality through proarrhythmia (generally class IA and IC antiarrhythmic drugs are contraindicated). Ibutilide and dofetilide are newer antiarrhythmic drugs with particular success at pharmacological cardioversion. Pretreatment with ibutilide increased DC shock cardioversion from 72% for placebo to 100%. In placebo failures, cross-over to ibutilide resulted in a 100% success rate with subsequent cardioversion. Ibutilide also resulted in reduction in DC shock energy required from 228 ± 93 J to 166 ± 80 J. However, ibutilide was associated with a 3% incidence of sustained polymorphous VT.45 Dofetilide appears to cardiovert AF and Afl pharmacologically in about a third of patients (intravenous (IV) better than oral, recent onset better than prolonged and Afl may be more responsive than AF). Dofetilide is far superior to placebo and sotalol, with similar recurrence rates to amiodarone.46
Other drugs currently used to promote onset of sinus rhythm and prevent AF relapse include amiodarone, sotalol, procainamide, flecainide and propafenone. Amiodarone was found to be superior in preventing AF recurrence with a recurrence rate of 35% compared to a recurrence rate of 63% for sotalol and propafenone.47
The factors dictating choice are:
When using amiodarone for prevention of AF recurrence there was an 18% incidence of adverse effects versus 11% for sotalol and propafenone.47
ATRIAL FIBRILLATION ABLATION THERAPY
Ablation techniques for AF have been continuously refined since the original Maze III surgical procedure which involved numerous atrial incisions to form a maze-like pattern of scarring, blocking propagation of arrhythmia. The ultility of this procedure was limited because it was surgical, with longer bypass times, postoperative bleeding and impaired atrial contractility. The magnitude of this original procedure was based on the belief that the entire atrium was involved in the initiation and maintenance of the fibrillatory conduction. This may be true for long-standing AF but paroxysmal AF appears to originate primarily at the junction of the left atrium and pulmonary veins. AF in 94% of patients is initiated by rapid discharges from one or more foci at or near the pulmonary vein orifices.48 Atrial tissue in this area has heterogeneous electrophysiological properties and there is also clustering of vagal inputs, which creates substrate for rapid discharges that initiate microre-entrant circuits or ‘rotors’. These high-frquency periodic rotors send spiral wave fronts of activation into surrounding atria. Localised ablation of a single dominant foci and rotor is inadequate as there are usually multiple foci.
Left atrial catheter (transatrial septum) AF ablation isolating all four pulmonary veins using radiofrquency is being heralded as the possible AF cure. Results are improving as all pulmonary veins are now isolated and the encircling lesion is clear of the pulmonary vein antrum (reducing pulmonary vein stenosis). Success rates of 81% (75–88%) free of AF and off drugs are reported. Success appears long-term as recurrence occurs early. A further 10–20% may become responsive to antiarrhythmic drugs which were previously ineffective. Repeating the procedure can increase success to > 90% with failure only in patients found to have extensive atrial scarring (predicting and excluding patients with this extensive atrial scarring is a major future challenge). Although not yet the universal cure the results are two- to threefold better than antiarrhythmic drugs alone.
Complication rates are also falling associated with:
Transient ischaemic attacks, strokes, tamponade/perforation and symptomatic pulmonary vein stenosis are all well below 1% respectively. Proarrhythmia resulting from re-entrant tachycardias from incomplete ablative lesions is more common. Some are advocating ablation as first-line treatment whereas most are selecting younger patients (less than 70 years) with paroxysmal AF for whom antiarrhythmic therapy has failed, left atrial diameter is less than 5 cm and ejection fraction is greater than 40%.26 Head-to-head studies comparing ablation and antiarrhythmic drugs are appearing with suggested survival benefit, improved quality of life, reduced adverse effects and cost-effectiveness after approximately 3 years with catheter AF ablation therapy.49,50
ANTICOAGULATION FOR CHRONIC ATRIAL FIBRILLATION
Consider for all patients, especially those with risk factors (Table 18.7).
Table 18.7 Prognostic factors for ischaemic stroke and systemic embolism in patients with atrial fibrillation
| High | Previous stroke, transient ischaemic attack, systemic embolism |
| Mitral stenosis | |
| Prosthetic heart valve | |
| Moderate | Age > 75 years |
| Left atrial size > 45 mm | |
| Hypertension | |
| Congestive cardiac failure | |
| Diabetes mellitus | |
| Left ventricular ejection fraction < 35% | |
| Low | Female |
| Age 65–74 years | |
| Coronary artery disease | |
| Thyrotoxicosis |
NON-VALVULAR ATRIAL FIBRILLATION
The risk of stroke is determined by CHADS2 score (assign 1 point for congestive heart failure, hypertension, age = 75 years and diabetes mellitus, and 2 points for stroke/TIA)51 (Tables 18.8 and 18.9).
Table 18.8 CHADS2 score stroke risk stratification in non-valvular atrial fibrillation
| Prognostic factor | Relative risk* | CHADS2 score |
|---|---|---|
| Congestive heart failure (ejection fraction < 35%) | 1.4 | 1 |
| History of hypertension | 1.6 | 1 |
| Age ≥ 75 years | 1.4 | 1 |
| Diabetes mellitus | 1.7 | 1 |
| Stroke or transient ischaemic attack in past | 2.5 | 2 |
CHADS2, congestive heart failure, hypertension, age = 75 years and diabetes mellitus, and 2 points for stroke/transient ischaemic attack.
* Relative risk without any antithrombotic treatment compared to atrial fibrillation patients without these prognostic factors.
Table 18.9 The adjusted annual stroke rate in non-valvular atrial fibrillation without any antithrombotic treatment
| CHADS2 score | Adjusted stroke rate | |
|---|---|---|
| %/year | 95% CI | |
| 0 | 1.9 | 1.2–3.0 |
| 1 | 2.8 | 2.0–3.8 |
| 2 | 4.0 | 3.1–5.1 |
| 3 | 5.9 | 4.6–7.3 |
| 4 | 8.5 | 6.3–11.1 |
| 5 | 12.5 | 8.2–17.5 |
| 6 | 18.2 | 10.5–27.4 |
CHADS2, congestive heart failure, hypertension, age = 75 years and diabetes mellitus, and 2 points for stroke/transient ischaemic attack; CI, confidence interval.
WARFARIN
Adjusted-dose warfarin reduces relative risk of stroke by 62%. The absolute risk reduction is 2.8% per year for primary and 8.4% per year for secondary prevention, although intracranial haemorrhage occurs (0.3% per year). Low-dose warfarin (INR 1.5–2.0) is less effective than an INR of 2.0–3.0 but has fewer haemorrhagic complications. Embolic stroke rate doubles as INR falls from 2.0 to 1.7, and is markedly higher at an INR of 1.3 compared to 2.0.43,52
ASPIRIN
Aspirin has reduced efficacy when compared with warfarin, with a 22% relative risk reduction and an absolute risk reduction of 1.5% and 2.5% per year for primary and secondary prevention. Warfarin compared with aspirin for AF will prevent 23 strokes and result in nine additional major bleeds per 1000 patients per year.
PRE-EXCITATION SYNDROME
ECG
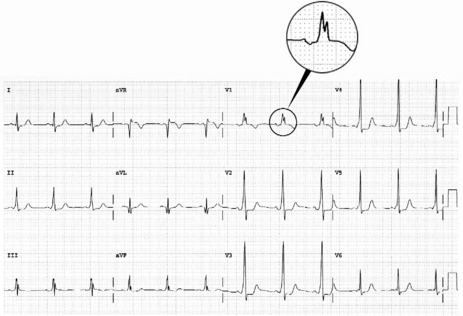
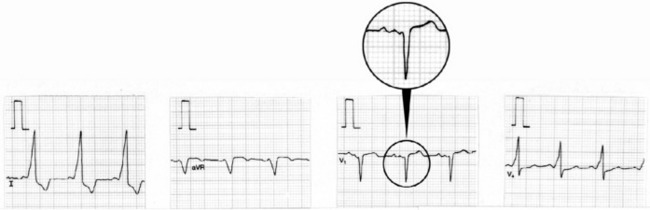
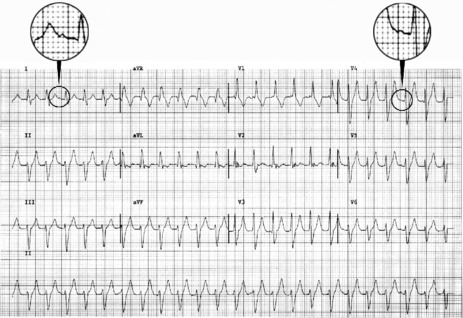
During sinus rhythm, an atrial impulse will reach the ventricles via both the AV node and the accessory AV pathway. The latter conducts the atrial impulse to the ventricles before the AV node, resulting in ventricular pre-excitation and a short PR interval. On reaching the ventricles, the pre-excitation impulse is not conducted via the specialised conducting system. Hence, early ventricular activation will be slowed, resulting in a slurred upstroke of the QRS complex, the so-called delta (δ) wave (Figure 18.24). The abnormal ventricular activation also gives rise to secondary S-T segment and T-wave abnormalities. δ-wave polarity in a 12-lead ECG may help localise the anatomical position of the accessory pathway. Type A WPW is characterised by upright QRS deflections in the right precordial leads (tall R-waves in V1 and V2) (Figure 18.25). In type A WPW the accessory pathway is usually situated on the left with pre-excitation of the left ventricle. Type B WPW has a dominantly negative QRS complex in V1 and the accessory pathway tends to be on the right with pre-excitation of the right ventricle (Figure 18.26).
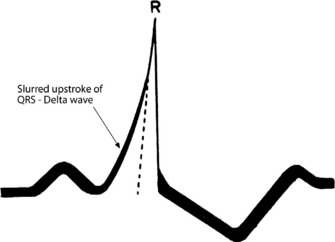
Figure 18.24 Ventricular pre-excitation via accessory pathway giving rise to a slurred upstroke and widened QRS complex.
CLINICAL
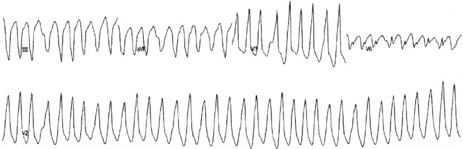
AVRT or AF can occur with WPW. During AVRT, the re-entry impulse usually travels down the AV node and back up the accessory pathway. Ventricular activation is via the normal conducting pathways and the QRS will be narrow. Occasionally, the re-entry impulse may pass in the opposite direction (down the accessory pathway and up the AV node), resulting in a wide QRS-complex tachycardia due to abnormal slow ventricular activation. Treatment is the same as for AVRT (i.e. IV adenosine). AF is uncommon in WPW, but may be life-threatening. Most impulses are conducted via the accessory pathway, leading to wide QRS complexes. The ECG of WPW with AF usually shows rapid, irregular QRS complexes with variable QRS width (Figure 18.27). Ventricular response is very rapid, leading to hypotension or cardiogenic shock. This arrhythmia may degenerate to VF.
TREATMENT25
Long-term management by radiofrequency ablation of the accessory pathway is effective in selected patients.25
VENTRICULAR TACHYCARDIA
VT is defined as three or more VEB at a rate greater than 130 beats/min, and may exceed 300 beats/min. VT lasting over 30 s is considered to be sustained. Non-sustained VT may not cause symptoms, but is associated with increased mortality in certain patients (e.g. after myocardial infarction). VT may be monomorphic (i.e. same QRS morphology) (Figure 18.28) or polymorphic (varying QRS morphology).
MONOMORPHIC VENTRICULAR TACHYCARDIA
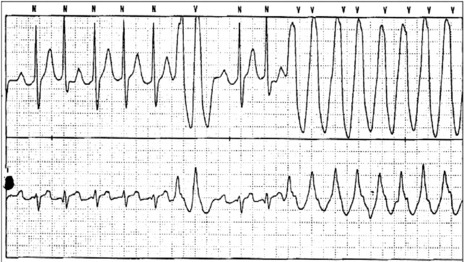
This is the most common form of VT. It is commonly associated with previous myocardial infarction, and often causes symptoms (e.g. palpitations, shortness of breath, chest pain or syncope). It may result in cardiac arrest, due to the tachycardia itself or degeneration into VF. The most common mechanism is re-entry secondary to inhomogeneous activation of the myocardium and slow conduction through scar tissue from a previous myocardial infarction. AV dissociation (i.e. independent atrial and ventricular activity) (Figure 18.29) is present in about 75% of instances, whereas retrograde ventricle to atrial conduction occurs in about 25%. AV dissociation is virtually diagnostic for VT during a wide-complex tachycardia, but ECG recognition of independent (and slower) atrial activity can be difficult (Figure 18.30). VT is the most common cause of a wide-complex tachycardia (QRS > 120 ms) and any such tachycardia should be considered VT until proven otherwise. Mistakes in diagnosis are common: SVT with aberrant conduction is often mistaken for VT. Inappropriate treatment based on incorrect diagnosis can have disastrous consequences.
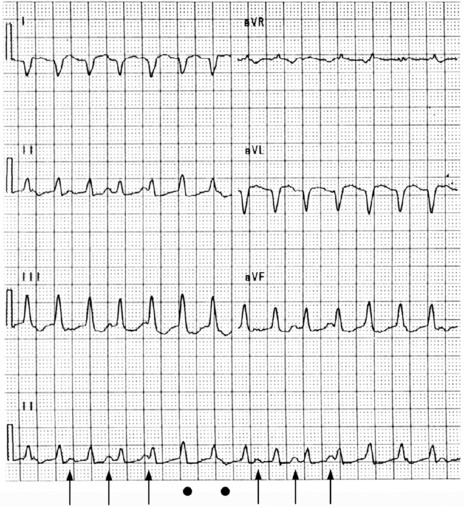
Figure 18.29 Ventricular tachycardia with obvious arteriovenous dissociation and independent P-waves are highlighted with arrows in lead II.
ECG
Older criteria (e.g. QRS > 140 ms and extreme electrical axis changes) are unhelpful in rhythm diagnosis.53 ECG criteria initially proposed by Wellens and revised by Brugada et al. permit accurate diagnosis in four sequential steps (Figure 18.31).54–56

Figure 18.31 Algorithm proposed by Brugada et al.54 to diagnose ventricular tachycardia in the setting of a broad QRS-complex tachycardia.
The sensitivity of these four consecutive steps was 0.987, with a specificity of 0.965.
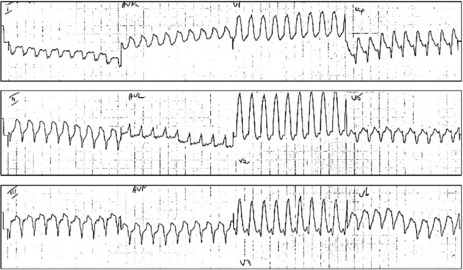
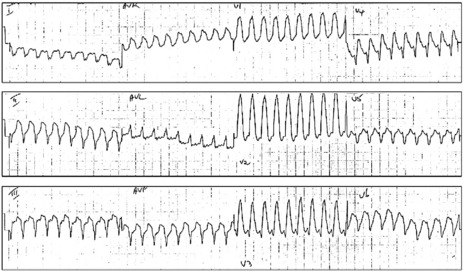
Figure 18.32 (a, b) Ventricular tachycardia. Broad QRS-complex tachycardia with absence of RS complex in all precordial leads.
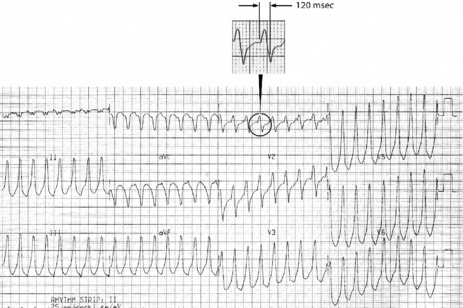
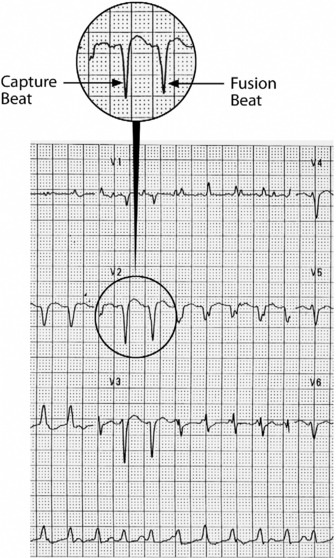
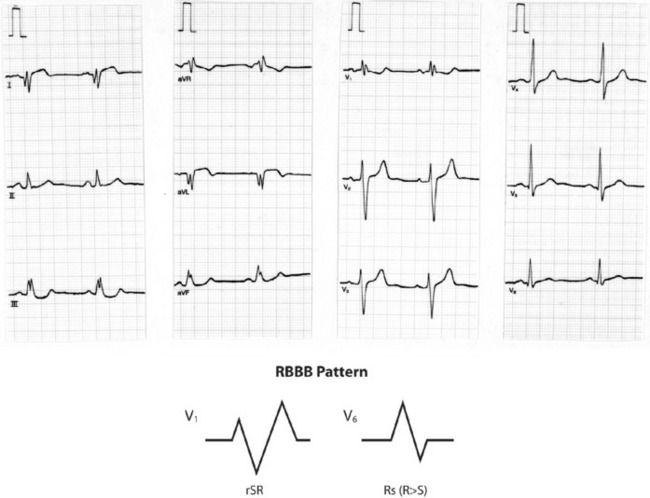
Figure 18.38 Right bundle branch block (RBBB) with characteristic M-shaped rSR complex in lead V1 and Rs complex in V6.
Termination of a wide-complex tachycardia by IV adenosine strongly suggests the arrhythmia as SVT. However, adenosine in this setting has the risk of destabilising VT when blood pressure is barely compensated by vasodilatation or acceleration of accessory pathway conduction, and is not recommended by International Liaison Committee on Resuscitation (ILCOR) as a diagnostic strategy in wide-complex tachycardia.25 Demonstration of AV dissociation by intracardiac ECG from a central venous catheter or a transvenous pacing lead signifies VT.
POLYMORPHIC VENTRICULAR TACHYCARDIA AND TORSADE DE POINTES
This arrhythmia has QRS complexes at 200 beats/min or greater, which change in amplitude and axis so that they appear to twist around the baseline (Figure 18.35). Torsade de pointes usually has prolonged QT during sinus rhythm, and U-waves are often present (see section on long-QT syndrome, below). However, polymorphic VT may be associated with a normal QT interval in settings such as myocardial ischaemia, infarction or post cardiac surgery.
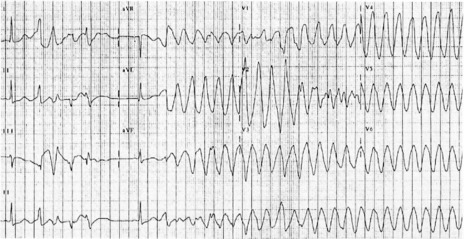
ACCELERATED IDIOVENTRICULAR RHYTHM (AIVR)
VENTRICULAR FIBRILLATION

TREATMENT57
If DC shock sequence fails, basic and advanced life support aiming to maximise coronary blood flow with chest compressions and vasopressors is crucial to cardiac success. Until recently, any role of antiarrhythmic drugs in DC shock-resistant VF has been traditional rather than proven. Recommendations have varied from lidocaine and bretyllium to amiodarone. The ILCOR currently recommends consideration of a range of antiarrhythmic drugs, including amiodarone, lidocaine, magnesium and procainamide. Recent studies indicate amiodarone as the drug of choice for DC shock-resistant VF. Amiodarone (300 mg) was superior to lidocaine and, in another study, 5 mg/kg followed by 2.5 mg/kg if required was superior to lidocaine. There is an incidence of bradycardia and hypotension but no difference in adverse effect profile between lidocaine and this sizeable amiodarone dose.58,59 After return of circulation, appropriate antiarrhythmic therapy is less clear but the role of lidocaine continues to disappear. Precipitating factors should be sought and treated (for long-term management issues, see the section on sudden cardiac death, below).
HEMIBLOCKS
The left branch of the bundle of His divides into the left anterosuperior division supplying the anterior superior lateral wall of the left ventricle and the posteroinferior division supplying the posterior inferior diaphragmatic surface of the left ventricle. Although block can occur in either division it is more common in the anterosuperior division, as it is more vulnerable to disease processes due to its longer course and thinner dimension. The anterosuperior division runs close to the aortic valve and tends to be involved in degenerative processes affecting this valve. The posteroinferior division is shorter and thicker and, unlike the anterosuperior division, has a double blood supply.
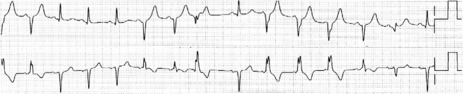
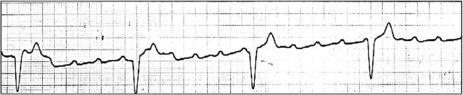
HYPERKALAEMIA
A high serum K+ can produce ECG changes (Figure 18.41). Early changes consist of tall peaked T-waves with reduced P-wave amplitude. Progressive widening of the QRS may be confused with BBB. Cardiac arrest may eventually occur.
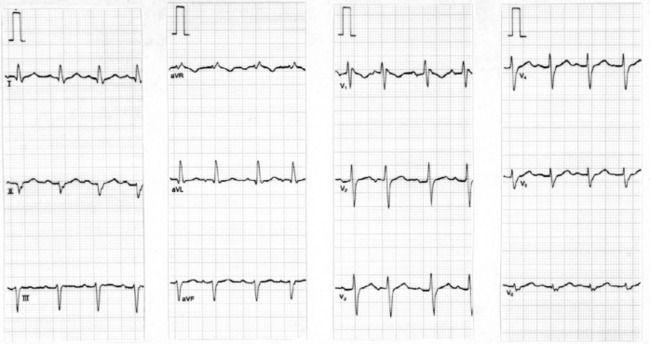
FIRST-DEGREE AV BLOCK
ECG
PR interval (measured from the onset of the P-wave to the onset of the QRS) exceeds 200 ms (Figure 18.42). Each P-wave is followed by a QRS. PR intervals may be prolonged to such a degree that the P-wave is buried in the previous T-wave or even QRS.
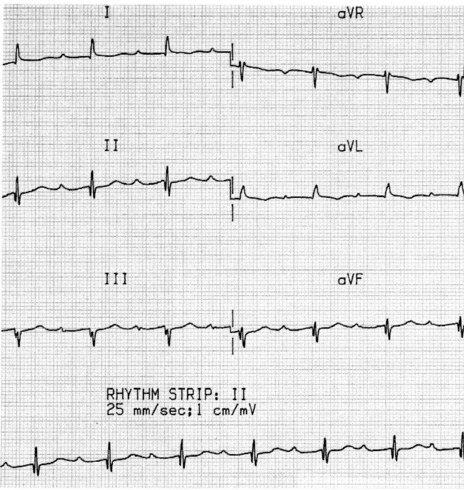
MOBITZ TYPE I (WENCKEBACH)
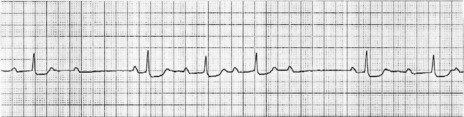
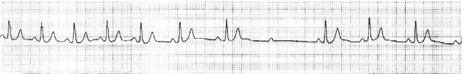
MOBITZ TYPE II
There is intermittent failure of conduction of atrial impulses to the ventricles without preceding increases in the PR interval. The ratio of conducted to non-conducted atrial impulses varies, e.g. every second or fourth atrial impulse may be conducted (i.e. 2:1 or 4:1 second-degree AV block). The lesion causing type ?? is usually situated in the bundle of His and is always pathological.
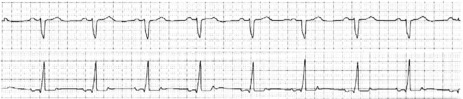
THIRD-DEGREE (COMPLETE) AV BLOCK
This rhythm occurs when no atrial impulses are conducted to the ventricles; atrial and ventricular contraction are dissociated. The SA node usually continues to depolarise the atria, whereas ventricular activation depends on a standby escape pacemaker located below the block. The escape pacemaker may be close to the His bundle (narrow QRS, stable pacemaker usually 40–60 beats/min) (Figure 18.46), or more distal in ventricular tissue (wide QRS, relatively unstable pacemaker with a rate of 20–40 beats/min) (Figure 18.47). If no ectopic escape pacemaker emerges, ventricular asystole will occur, resulting in a Stokes–Adams attack, or death if the episode is prolonged. Torsade de pointes may also occur associated with the bradycardia.
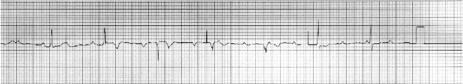
ECG
The ECG shows normal regular P-waves completely dissociated from QRS complexes. The QRS rate is always significantly slower than the P-wave rate and may be very slow at times.
SICK-SINUS SYNDROME
This consists of a number of sinus node abnormalities, including inappropriate sinus bradycardia, SA blocks or sinus arrest. When sinus bradycardia or SA block occurs, junctional escape rhythms are common. There may also be abnormalities of AV conduction. Paroxysms of AF or Afl may alternate with episodes of bradycardia (bradycardia–tachycardia syndrome) (Figure 18.48).
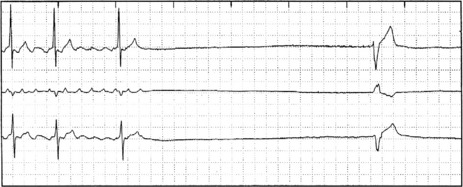
CRITICALLY ILL PATIENTS AND ARRHYTHMIA60
In the general population or critically ill patients, excluding acute coronary syndromes and cardiac surgical patients, arrhythmia is common. The documented incidence is as high as 78%; however, the incidence of arrhythmia that requires treatment is much lower, at 15–30%. SVT are by far the most common arrhythmia that requires treatment. AF, Afl and unifocal atrial tachycardias are the most frequent, in descending order. These SVTs are rarely the cause of admission but develop early in the admission, the majority by day 2. In critically ill patients, SVTs often result in:
TREATMENT OF SUPRAVENTRICULAR TACHYCARDIA IN CRITICALLY ILL PATIENTS
Continuing arrhythmogenic and chronotropic factors make rate control difficult.
MYOCARDIAL INFARCTION AND ARRHYTHMIA21
MANAGEMENT OF ACUTE MYOCARDIAL INFARCTION AND ARRHYTHMIA CONTROL
The early work demonstrating survival benefit of magnesium was initially thought to be due to the prevention of arrhythmia.64 However, the Leicester Intravenous Magnesium Intervention Trial (LIMIT-2) found the improved survival not to be related to a reduction in arrhythmia.65 Subsequent studies in the thrombolytic era have failed to show any benefit at all with magnesium, although debate regarding optimal time of administration persists. Magnesium may have a role in patients in whom β-adrenergic blockers or thrombolytic therapy are contraindicated.
ELECTROLYTE CONCENTRATIONS AND ARRHYTHMIA FOLLOWING ACUTE MYOCARDIAL INFARCTION
Serum potassium following AMI is negatively correlated with the incidence of VEB and VT, with the probability of VT falling until serum potassium exceeds 4.5 mmol/l.8 There is no evidence that magnesium levels in this setting have any effect on ventricular arrhythmia. Nonetheless, ILCOR recommendations not only include the maintenance of serum potassium greater than 4.0 mmol/l, but also serum magnesium levels greater than 1.0 mmol/l.
BRADYARRHYTHMIAS POST ACUTE MYOCARDIAL INFARCTION
Treatment is only indicated for sinus bradycardia associated with symptoms, hypotension or signs of poor cardiac output. Most often, first- and second-degree block also do not need treatment. Mobitz type I second-degree block may require treatment and atropine is indicated. However, in Mobitz type II, atropine usually has no effect on infranodal block and may precipitate third-degree block by increasing sinus rate and enhancing block. Atropine may improve heart rate with AV block occurring at the AV node, as demonstrated by a narrow QRS complex, by improving AV conduction or accelerating escape rhythm. Atropine is not indicated for infranodal third-degree block, which is diagnosed by the presence of a new wide QRS complex. When required, atropine is administered, 0.5–1.0 mg every 3 min until signs or symptoms are resolved, up to a maximum of 0.03–0.04 mg/kg. If atropine is not indicated or effective, cardiac pacing is required (Table 18.10). Transcutaneous pacing is indicated for initial management as a bridge until a transvenous temporary pacing wire can be inserted safely and with appropriate sterile technique. With the ready availability of transcutaneous pacing, IV catecholamines for bradyarrhythmias are to be avoided in the setting of AMI.
Table 18.10 Indications for pacing following myocardial infarction
| Haemodynamically unstable bradycardia (< 50 beats/min) |
| Mobitz type II second-degree atrioventricular block |
| Third-degree heart block |
| Bilateral bundle branch block |
| Left anterior fascicular block |
| New left bundle branch block |
| Bundle branch block and first-degree atrioventricular block |
VENTRICULAR ARRHYTHMIA POST ACUTE MYOCARDIAL INFARCTION20,66,67
Thrombolytic therapy has reduced VF incidence. The Gruppo Italiano per lo Studio della Streptochinasi nell’infarto miocardico (GISSI) study66 found an incidence of primary VF of 3.6% and secondary VF of 0.6%. The overall incidence of ventricular arrhythmia in the Global Utilization of Streptokinase and Tissue plasminogen activator to treat Occluded arteries (GUSTO-1) report was VF, 4.1%, VT, 3.5% and both, 2.7%.
Lidocaine reduces primary VF by 33% but mortality is increased by a similar amount such that there is no net benefit and the third International Study of Infarct Survival (ISIS-3) reported an overall trend to increased mortality.68 Being more selective as to which patients receive lidocaine has not been possible, as only 50% of patients who develop VF have ‘warning’ ventricular arrhythmia. In the ‘thrombolytic and β-blocker’ era of treatment of AMI, the use of prophylactic lidocaine, or any other antiarrhythmic drug, to prevent VF will have even less benefit. There are no conclusive data to support the use of lidocaine to prevent recurrent VF in those patients who have already suffered an episode of VF. Despite this, a short period of 6–24 h of lidocaine has been advocated.
Class I agents showed no overall benefit on all-cause mortality and class IC agents have excess mortality despite arrhythmia suppression.13
Class II antiarrhythmics, β-blockers, have an established and broadening role, with recent evidence showing significant benefit.69
Class III agents lack a consistent class effect. Sotalol (Survival with oral D-sotalol: SWORD) was found to increase all-cause mortality and arrhythmia deaths.70 Amiodarone (Canadian Amiodarone Myocardial Infarction Arrhythmia Trial: CAMIAT) reduces all-cause mortality in patients with frequent VEB post myocardial infarction71 but another study (European Myocardial Infarction Amiodarone Trial: EMIAT) evaluated amiodarone in patients with ejection fraction less than 40% and found no effect on all-cause mortality but a 35% reduction in arrhythmia deaths.72 Subsequent analysis of combined CAMIAT and EMIAT data has emphasised the importance of β-blockers. The combination of amiodarone and β-blockers in these postinfarct patients was better than either drug alone.73
CARDIOTHORACIC SURGERY AND ARRHYTHMIA
PREVENTION OF SUPRAVENTRICULAR TACHYCARDIA75,76
Preoperative β-blocker treatment should be continued postoperatively. β-Blockers consistently reduce SVT across many studies with differing agents.
Amiodarone prophylaxis after elective cardiac surgery reduced postoperative AF from 53% to 25%. Diltiazem has also been shown to be effective at SVT prevention and there was associated improvement in haemodynamic variables and rates of myocardial ischaemia. Verapamil is not effective. Esmolol was found to be more effective than diltiazem. Sotalol is also effective but problematic bradycardia and hypotension are greater.77 Atorvastatin is also effective.78
TREATMENT OF SUPRAVENTRICULAR TACHYCARDIA
Treatment is aimed at control of ventricular rate, prevention of thromboembolism and cardioversion.
Digoxin, atenolol, diltiazem and magnesium are appropriate choices to control rate.
VENTRICULAR ARRHYTHMIA FOLLOWING CARDIAC SURGERY
Ventricular arrhythmia requiring treatment, DC shock or drug therapy is common following cardiac surgery and occurs in 23% of patients.79 Arrhythmias requiring DC shock occur in the first 36 h and are associated with:
LONG-QT SYNDROME80
The traditional criteria of prolonged QT is corrected heart rate (QTc, Bazett’s formula, QT divided by the square root of the RR interval) and a QTc greater than 0.44 s. This should also be adjusted for age and gender. The causes of LQTS can be divided into acquired and idiopathic (Table 18.11). Common to all causes is a prolongation of repolarisation which creates the substrate for random re-entry, giving rise to polymorphous VT (classically of the torsade de pointes type), particularly under conditions of acute adrenergic arousal.
| Acquired |
| Drugs |
| Class IA antiarrhythmic drugs |
| Quinidine, procainamide |
| Class III antiarrhythmic drugs |
| Amiodarone, sotalol |
| Tricyclic antidepressants |
| Macrolide antibiotics |
| Phenothiazines |
| Antihistamines |
| Cisapride |
| Myocardial ischaemia/infarction |
| Hypokalaemia |
| Cardiomyopathy |
| Acute myocarditis |
| Mitral valve prolapse |
| Acute cerebral injury |
| Hypothermia |
| Idiopathic |
| Familial: 90% |
| Linked to a DNA marker on the short arm of chromosome 11 |
| Autosomal-dominant in most cases |
| Some cases linked to congenital deafness and autosomal-recessive |
| Sporadic: 10% |
| Non-familial – related to new gene mutation |
Prolonged action potential in LQTS prediposes to arrhythmia in two ways:
IDIOPATHIC LQTS81
The other described genetic mutations leading to LQTS are rare.
MANAGEMENT
The first line of management of polymorphous VT with shock is DC shock, with magnesium being the antiarrhythmic of choice.83
Strategies for prevention of recurrence in idiopathic LQTS depend on the presentation. Patients who present with or have a history of syncope or aborted sudden death have a high risk of recurrence (5% per year). β-Blockers are the first line of treatment with the goal of reducing the exercise heart rate to less than 130 beats/min. Symptomatic bradycardia following adequate β-blockade requires a permanent pacemaker. Patients with recurrence despite these measures and those with an early malignant course need stellate sympathetic ganglionectomy. In 5% of these high-risk patients triple therapy fails and an implantable defibrillator is required. Asymptomatic patients with incidental LQTS (< 0.5% per year) and asymptomatic family members (0.5% per year) have a very low risk of syncope or sudden death. It is also very rare for the first episode to be fatal in these two groups so prophylactic measures are generally not required and close follow-up is sufficient.
SUDDEN CARDIAC DEATH84
Arrhythmic causes of SCD can be divided into three categories:
The causes of these arrhythmia can be divided into three general categories:
INVESTIGATIONS15–17
PREVENTION OF RECURRENT VENTRICULAR ARRHYTHMIA IN SURVIVORS OF SUDDEN CARDIAC DEATH
MYOCARDIAL REVASCULARISATION AND SUDDEN CARDIAC DEATH
The Coronary Artery Surgery Study (CASS) registry looked at 13 476 patients with significant operable coronary artery disease and showed an incidence of SCD of 5.2% in the medical arm compared to 1.8% in those assigned to surgery.85 The precise mechanism of the benefit of surgery in primary prevention is unclear but is probably associated with prevention of ischaemia rather than arrhythmia control. In summary, the near-universal practice of surgical coronary revascularisation in SCD survivors with critical stenoses is based upon this primary prevention data (and the central role that ischaemia and infarction are known to play in arrhythmia substrate) rather than data demonstrating arrhythmia control.
IMPLANTABLE CARDIOVERTER DEFIBRILLATORS AND SUDDEN CARDIAC DEATH
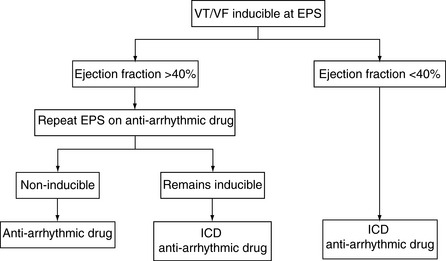
Representative data from several studies are shown in Table 18.12. The benefit of ICD on survival persists for at least 8 years. Current indications are expanding, but the benefit is clear in the following groups of survivors of SCD and patients with documented VT/VF outside the early postinfarct phase:
Table 18.12 Mortality data for the various treatment regimens for sudden cardiac death
| Sudden death (%) | Total mortality (%) | |
|---|---|---|
| Implantable cardioverter defibrillator | 3.5 | 13.6 |
| Empirical amiodarone | 12.0 | 34.0 |
| Electrophysiological study-directed drug treatment | 14.0 | 24.0 |
| Surgery | 3.7 | 37.0 |
Further developments currently taking place include:
Although one of the proposed advantages of ICD is the avoidance of antiarrhythmic drug side-effects, particularly the myocardial depressant effects, current practice usually combines ICD with low-dose amiodarone. This enables improved arrhythmia control by reducing atrial tachyarrhythmia and slowing VT rate, and prolonging battery life by reducing the frequency of arrhythmia. The role of ICD in the management algorithms of SCD is shown in Figure 18.49 and 18.50. Given the current lack of available ICD, some advocate restriction to patients less than 75 years. With wider availability of ICD, fewer patients will be managed on drug-only strategies in the future.
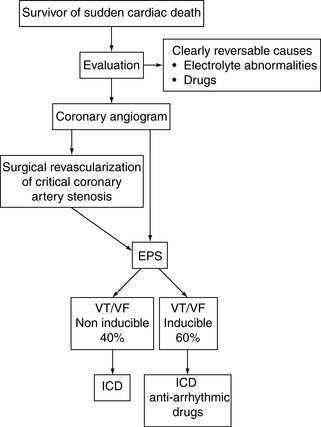
CLASSIFICATION OF ANTIARRHYTHMIC DRUGS
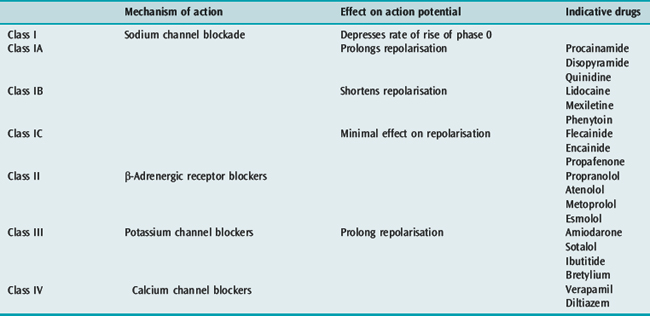
The time-honoured physiologically based classification of antiarrhythmic drugs is the Vaughan-Williams classification (Table 18.13), which has been modified over the years. This classification is a hybrid, with classes I and IV representing ion channel blockers, class II representing a receptor blocker and class III representing a change in an electrophysiological variable. The prolongation of repolarisation, the defining effect of class III agents, can be produced by a block of any one of several K+ ionic channels (the delayed rectifier potassium current IKr responsible for phase 3 repolarisation is the ionic channel target of most class III agents) or from modification of Na+ or Ca+ channel function. This classification is also incomplete and does not include cholinergic agonists, digitalis, magnesium and adenosine. For this reason, an alternative classification has been advocated based upon molecular targets for drug action that include ion channels, receptors and pumps/carriers (Table 18.14).2

ANTIARRHYTHMIC DRUGS68
DIGOXIN
Digoxin is a muscurinic subtype 2 receptor (M2) agonist and a highly potent Na+,K+-ATPase pump-blocking agent. Digoxin exerts its antiarrhythmic activity predominantly at the AV node where at lower doses conduction is slowed by the M2 vagotonic effect. This effect is easily reversed by enhanced sympathetic tone in the setting of exercise, critical illness and postoperative state. At higher concentrations, digoxin has a direct effect on AV node conduction by the Na+,K+-ATPase pump blockade and is more resistant to sympathomimetic effects. The decrease in [K+]i and increase in [Na+]i results in hyperpolarisation, shortening of atrial action potential and an increase in AV nodal refractoriness. There is also increased availability of intracellular Na+ for the Na+-Ca2+ exchanger, increasing [Ca2+]i which results in the positive inotropic effects of digoxin, making it an ideal agent in the setting of LV dysfunction. However, the positive inotropic effects of Na+,K+-ATPase blockade are deleterious in the setting of myocardial ischaemia and other causes of diastolic dysfunction. Digoxin also has weak vasopressor properties when administered as a slow bolus.61 The major ECG effects of digoxin are PR prolongation and a non-specific alteration in ventricular repolarisation with characteristic reverse-tick S-T segments.
CONTRAINDICATIONS
Relative contraindications include myocardial ischaemia/infarction, diastolic heart failure due to hypertrophy and ischaemia, renal failure and hyperkalaemia, planned DC shock cardioversion and tachycardia–bradycardia syndromes. Coadministration with other drugs that effect AV nodal conduction, typically β-adrenergic and calcium channel blockers, requires caution.
β-ADRENERGIC BLOCKERS
β-Adrenergic blocking or class II antiarrhythmic drugs have differing properties such as relative cardioselectivity (atenolol, metoprolol), non-cardioselectivity (propranolol), intrinsic sympathomimetic activity (pindolol), lipid solubility and central activity (metoprolol, propranolol) and membrane-depressant effects (propranolol). The antiarrhythmic properties appear to be a class effect and no agent has been shown to be superior. There are data to suggest that the survival benefit of β-adrenergic blockers post myocardial infarction may relate to some extent to the central modulation of autonomic tone of the more lipid-soluble agents. The direct membrane-stabilising or ‘quinidine-like’ effect of propranolol requires doses far greater than those used clinically and is of negligible clinical significance. β-Adrenergic blockers competitively inhibit catecholamine binding at the β-adrenergic receptor sites, which reduces the phase 4 slope of the action potential of pacemaker cells, prolongs their refractoriness and slows conduction in the AV node. Refractoriness and conduction in the His–Purkinje system are unchanged. β-Adrenergic blockers are most effective in arrhythmia associated with increased cardiac adrenergic stimulation (postoperative states, sepsis, thyrotoxicosis, phaeochromocytoma, exercise or emotion).
INDICATIONS
Supraventricular Tachycardias
β-Adrenergic blockers may terminate SVT when the AV node is an intrinsic part of the re-entry circuit (AVNRT and AVRT); adenosine is more effective. AF and Afl do not revert with β-adrenergic blockers but the ventricular rate will be slowed. β-adrenergic blockers are effective at preventing SVT following cardiac surgery. Studies involving propranolol and atenolol have produced the best results. β-Adrenergic blockers are effective in MAT, but, as this arrhythmia is most often seen in patients with severe chronic airflow limitation and cor pulmonale, their utility is limited.30
ATENOLOL
ESMOLOL
CALCIUM CHANNEL BLOCKERS
Calcium channel blockers or class IV antiarrhythmic drugs block the slow calcium channels in cardiac tissue. Verapamil and diltiazem have similar electrophysiological properties. The dihydropyridine group of calcium channel blockers, which include nifedipine, does not have any significant electrophysiological properties. Calcium channel blockers depress the slope of diastolic depolarisation in the SA node cells, the rate of rise of the phase 0 and action potential amplitude in the SA and AV nodal cells. They also slow conduction and prolong the refractory period of the AV node, which results in their main antiarrhythmic actions. Refractoriness of atrial, ventricular and accessory pathway tissue is unchanged. The sinus rate does not usually change significantly because calcium channel blockers induce peripheral vasodilatation, which causes reflex sympathetic stimulation of SA node. Verapamil particularity has marked negative inotropic actions and hypotension is often seen; however, cardiac index is generally maintained because of afterload reduction. Diltiazem has less negative inotropic effect than verapamil.
INDICATIONS
MAGNESIUM
Magnesium is an emerging antiarrhythmic agent with a range of indications; however, as an antiarrhythmic agent, magnesium has largely defied classification. Magnesium has many reported electrophysiological effects, including blocking voltage-dependent L-type Ca2+ channels. Magnesium is a necessary cofactor for the membrane enzyme Na+/K+-ATPase that provides energy for the membrane Na/K channels.89 The consequences of magnesium deficiency are as follows:
Magnesium in pharmacological doses decreases RMP, resulting in a reduction in automaticity. However, once depolarisation occurs, the maximum rate of depolarisation and action potential amplitude is increased, thereby improving conduction. Action potential duration is increased, thereby increasing absolute refractory period and reducing relative refractory period. The net result is a reduction in the vulnerable period and more synchronous conduction. All of these electrophysiological effects are augmented in the setting of increased extracellular potassium. It is not surprising that the utility of magnesium appears greatest in the setting of ischaemia where loss of potassium from the cell is a major consequence. A secondary effect is the reduction in the availability of intracellular sodium to contribute to inward calcium flux, producing triggered activity. Magnesium has also been shown to elevate VF and ectopy threshold.90
INDICATIONS
DOSE
ADVERSE EFFECTS
PROCAINAMIDE
CLINICAL USE
Procainamide is used to treat both atrial and ventricular arrhythmias.
LIDOCAINE
Lidocaine, long considered an important antiarrhythmic drug for ventricular tachyarrhythmia, is now relegated to lower-choice options or missing from most treatment algorithms. Na+ channel-blocking effect is increased in myocardial ischaemia and is unproven outside acute ischaemia settings. It has no effect on SA node automaticity, but it depresses automaticity in other pacemaker tissues. Normally, lidocaine has little or no effect on conduction. The ECG shows no changes in sinus rate, PR interval, QRS width or QT interval with lidocaine.
CLINICAL USE
FLECAINIDE
CLINICAL USE
Flecainide can be administered IV or orally. If flecainide is deemed necessary for prophylaxis of ventricular arrhythmias, therapy should start with ECG monitoring. In CAST, suppression of VEBs after myocardial infarction with flecainide was associated with increased mortality.13
ADVERSE EFFECTS
AMIODARONE
Amiodarone is a potent antiarrhythmic agent with a complex electrophysiological and pharmacological profile. The appealing broad spectrum and haemodynamic stability of amiodarone have resulted in it emerging as the most frequently used antiarrhythmic in critically ill patients. In this setting short-term use predominates and the formidable side-effect profile is much less significant. Amiodarone:
SUPRAVENTRICULAR TACHYCARDIA
Amiodarone is effective in terminating and suppressing recurrences of AVNRT and AVRT tachycardias, although adenosine (for acute reversion) or verapamil (acute termination and long-term prophylaxis) is superior. Amiodarone IV is less effective in reverting Afl or AF, but the ventricular rate will slow. Administration over a longer time span (days to weeks) may be more effective in reverting recent-onset AF. In preventing recurrence of AF after reversion, amiodarone is comparable in efficacy to quinidine and flecainide but superior to sotalol and propafenone.47
DOSE
VENTRICULAR TACHYARRHYTHMIAS
Amiodarone IV may be effective in treating life-threatening ventricular tachyarrhythmias refractory to other drugs, especially in myocardial infarction and poor LV function. Amiodarone’s efficacy in DC shock-resistant VF further confirms a prominent role in this setting.58,59 Long-term oral amiodarone is useful is controlling symptomatic VT and VF, especially when other conventional antiarrhythmics have failed. The absence of negative inotropic effect is useful in those with severely depressed LV function, but its many adverse effects limit widespread use. The Cardiac Arrest in Seattle, Conventional versus Amiodarone Drug Evaluation (CASCADE) study demonstrated that empirical amiodarone treatment was superior to guided (non-invasive Holter or EPS) class I drugs in survivors of VF unassociated with AMI. Amiodarone prevented arrhythmia recurrence and decreased the incidence of sudden death. Amiodarone, presumably because it is better tolerated in patients with poor LV function and having less proarrhythmia, can be considered as first-line drug to prevent life-threatening ventricular tachyarrhythmias.71–73 The rate of arrhythmia control is often slower with ventricular arrhythmia and may take several days to achieve. This delay appears to be independent of dose. The pharmacodynamic basis for this relates to the fact that much of the class III or K+ channel-blocking activity is due to the amiodarone metabolite, desethylamiodarone, whereas the predominant actions of acute administration of amiodarone are due to its class I and class II activity. The full potency of the class III activity requires several days, at least, for the effects of desethylamiodarone to appear.
PHARMACOKINETICS
Oral bioavailabiliy of amiodarone is variable at 40–70% with a delayed onset of action (days to weeks); however loading doses reduce this interval. Initial IV dose recommendations were 5–7 mg/kg over 30 min, followed by 50 mg/h. The kinetics of amiodarone have been modelled to four compartments when given acutely. Following a slow IV bolus over 15 min, amiodarone is rapidly distributed to the active compartment (t1/2a = 4.2 min), with demonstrable prolongation of QTc and antiarrhythmic effect within 2–5 min. Subsequently, amiodarone is progressively redistributed to ‘deeper’ compartments (t1/2b = 36.6 min, t1/2g = 4.5 h and t1/2d = 33.6 h).91 This is in contrast to the very prolonged terminal half-life – greater than 50 days – after long-term treatment. This redistribution of amiodarone explains why repeated boluses are often required in the first 24–48 h of treatment. Transient loading-dose hypotension due to myocardial depression or vasodilatation is dose-, rate- and patient-dependent. A loading dose of 5 mg/kg over 20 min resulted in significant further myocardial depression and a fall in cardiac index, whereas in patients without evidence of heart failure, 5 mg/kg over 1 min resulted in hypotension due to systemic vasodilatation and an increase in cardiac index.92,93
ADVERSE EFFECTS
SOTALOL
SUPRAVENTRICULAR TACHYCARDIA
VENTRICULAR ARRHYTHMIAS
Sotalol is superior to lidocaine to terminate sustained VT, and should be considered a first-line drug in patients without heart failure. Oral sotalol is more effective than class I drugs for the long-term prevention of VT or VF. Guided (Holter or EPS) sotalol and empirical amiodarone are first-line drugs to prevent recurrences of VT and VF over the long term; however, the worse outcome with sotalol in SWORD has significantly reduced the role of sotalol in this setting.70
IBUTILIDE
Ibutilide is an IKr K+ channel blocker, which prolongs action potential and increases the refractory period. This agent, with class III effect, is recommended for acute pharmacological conversion of AF and Afl, or as an adjunct to improve success of DC shock cardioversion. The success rate for ibutilide alone is higher for Afl (50–70%) than that with AF (30–50%) and, as expected, efficacy is greatest in short-term AF/Afl without structural heart disease. The major role of ibutilide appears to be cardioversion pretreatment to facilitate reversion to sinus rhythm from AF.45 Ibutilide has minimal effect on blood pressure and heart rate and the major adverse effect is proarrhythmia, with torsade de pointes occurring in 3–6% of patients. For this reason, patients should be monitored for at least 6 h after administration. Ibutilide has a short duration of action and other antiarrhythmic drugs are required to maintain sinus rhythm.
DOFETILIDE
Dofetilide is one of the latest class III IKr K+ channel blockers being investigated in the hope of finding an antiarrhythmic drug for long-term use with amiodarone’s efficacy but without its side-effect profile. Dofetilide’s emerging role is again in pharmacological cardioversion of AF and Afl (superior to sotalol, similar to amiodarone but inferior to ibutilide) in about 30% of patients. Prevention of recurrence is similar to amiodarone and long-term oral therapy is not associated with increased mortality in post-MI and heart failure patients. It also appears safe in patients with LV dysfunction. Early (first 3 days) proarrhythmia (3.3% of patients developed torsade de pointes) also necessitates monitoring with commencement of therapy.40,46,94,95
ADENOSINE
CLINICAL USE
DIRECT CURRENT CARDIOVERSION96
MECHANISM OF ACTION
The exact mechanism of action is unknown. DC shocks need to produce a current density that depolarises a critical mass of myocardium, thereby leaving insufficient myocardium to maintain the re-entrant tachycardia and prevent reinitiation. For VF and AF, the critical mass involves the entire ventricles or atria, whereas for the more organised tachyarrhythmias, VT and Afl, which involve specific re-entrant circuits, regional depolarisation in the path of their circulating wave fronts is all that is required. DC shocks also prolong the refractoriness of myocardium and this effect will contribute to the arrhythmia termination and prevention of immediate recurrence.
DIRECT CURRENT SHOCK DOSAGE
Recommendations for energy doses are changing, as biphasic waveform generators become more widely available. Where studied, BTE shocks have been consistently as effective as higher-energy MDS shocks. Recommended doses are always a balance between that energy likely to generate a critical current flow and an energy not likely to cause functional and morphological damage. Electrical energies greater than 400 J have been reported to cause myocardial necrosis.
ANTICOAGULATION FOR CARDIOVERSION96
1 Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517-2529.
2 Task Force of the Working Group on Arrhythmias of the European Society of Cardiology. The Sicilian gambit: a new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Circulation. 1991;84:1831-1851.
3 Hoffman BF, Rosen MR. Cellular mechanisms for cardiac arrhythmias. Circ Res. 1981;49:1-15.
4 Binah O, Rosen MR. Mechanisms of ventricular arrhythmias. Circulation. 1992;85(suppl. 1):25-31.
5 Wit AL, Cranefield PF. Reentrant excitation as a cause of cardiac arrhythmias. Am J Physiol. 1978;235:H1-17.
6 Gettes LS. Electrolyte abnormalities underlying lethal and ventricular arrhythmias. Circulation. 1992;85(suppl. 1):70-76.
7 Hollifield JW. Thiazide treatment of hypertension: effects of thiazide diuretics on serum potassium, magnesium and ventricular ectopy. Am J Med. 1986;80:8-12.
8 Nordrehaug JE, Johannessen K-A, von der Lippe G. Serum potassium concentration as a risk factor of ventricular arrhythmias early in acute myocardial infarction. Circulation. 1985;71:645-649.
9 Hollifield JW. Thiazide treatment of systemic hypertension: effects on serum magnesium and ventricular ectopic activity. Am J Cardiol. 1989;63:G22-25.
10 Schwartz PJ, La Rovere MT, Vanoli E. Autonomic nervous system and sudden cardiac death. Circulation. 1992;85(suppl. 1):77-91.
11 Campbell TJ. Proarrhythmic actions of antiarrhythmic drugs: a review. Aust NZ J Med. 1990;20:275-282.
12 Dhein S, Muller A, Gerwin R, et al. Comparative study on the proarrhythmic effects of some antiarrhythmic agents. Circulation. 1993;87:617-630.
13 The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:227-233.
14 Donovan KD, Power BM, Hockings BE, et al. Usefulness of atrial electrograms recorded via central venous catheters in the diagnosis of complex cardiac arrhythmias. Crit Care Med. 1993;21:532-537.
15 Mason JW. A comparison of electrophysiologic testing with Holter monitoring to predict antiarrhythmic drug efficacy for ventricular tachyarrhythmias. N Engl J Med. 1993;329:445-451.
16 Buxton AE, Lee KL, DiCarlo L, et al. Electrophysiologic testing to identify patients with coronary artery disease who are at risk of sudden death. N Engl J Med. 2000;342:1937-1945.
17 Gilman JK, Jalal S, Naccarelli GV. Predicting and preventing sudden cardiac death from cardiac causes. Circulation. 1994;90:1083-1092.
18 Kennedy HL, Whitlock JA, Spraugue MK, et al. Long-term follow up of asymptomatic healthy subjects with frequent and complex ventricular ectopy. N Engl J Med. 1985;312:193-197.
19 Jouven X, Zuriek M, Desnos M, et al. Long-term outcome in asymptomatic men with exercise-induced premature ventricular depolarisations. N Engl J Med. 2000;343:826-833.
20 Teo KK, Yusuf S, Furberg CD. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction: an overview of results from randomized controlled trials. JAMA. 1993;270:1589-1595.
21 The American Heart Association in Collaboration with the International Liasion Committee on Resuscitation (ILCOR). Guidelines 2000 for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2000;102(suppl. I):I172-203.
22 Ganz LI, Friedman PL. Supraventricular tachycardia. N Engl J Med. 1995;322:162-173.
23 Chauhan VS, Krahn AD, Klein GJ, et al. Supraventricular tachycardia. Med Clin North Am. 2001;85:193-223.
24 Camm AJ, Garratt CJ. Adenosine and supraventricular tachycardia. N Engl J Med. 1991;325:1621-1629.
25 The American Heart Association in Collaboration with the International Liasion Committee on Resuscitation (ILCOR). Guidelines 2000 for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2000;102(suppl. I):I158-165.
26 Cappato R, Calkins H, Chen SA, et al. Worldwide survey on the methods, efficacy and safety of catheter ablation for human atrial fibrillation. Circulation. 2005;111:1100-1105.
27 Garratt C, Antoniou A, Ward D, et al. Misuse of verapamil in pre-existent atrial fibrillation. Lancet. 1989;1:367-369.
28 Scher DL, Arsura EL. Multifocal atrial tachycardia: mechanisms, clinical correlates and treatment. Am Heart J. 1989;118:574-580.
29 McCord JK, Borzak S, Davis T, et al. Usefulness of intravenous magnesium for multifocal atrial tachycardia in patients with chronic obstructive pulmonary disease. Am J Cardiol. 1998;81:91-93.
30 Arsura E, Lefkin AS, Scher DL, et al. A randomized double-blind placebo-controlled study of verapamil and metoprolol in treatment of multifocal atrial tachycardia. Am J Med. 1988;85:519-524.
31 Olshansky B, Wilber DJ, Hariman RJ. Atrial flutter – update on the mechanism and treatment. Pace. 1992;15:2308-2335.
32 Falk RH. Medical progress: atrial fibrillation. N Engl J Med. 2001;344:1067-1078.
33 Ozcon C, Jahangir A, Frudman PA, et al. Significant effects of atrioventricular node ablation and pacemaker implantation on left ventricular function and long-term survival in patients with atrial fibrillation and left ventricular dysfunction. Am J Cardiol. 2003;92:33-37.
34 Prichett ELC. Management of atrial fibrillation. N Engl J Med. 1992;326:1264-1271.
35 Prystowski EN, Benson DW, Fuster V, et al. Management of patients with atrial fibrillation. A statement for healthcare professionals. From the subcommittee on electrocardiography and electrophysiology: American Heart Association. Circulation. 1996;93:1262-1277.
36 Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) Investigations. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347:1825-1833.
37 Rienstra M, Van Veldhiusen DJ, Crijns HJ, et al. Enhanced cardiovascular morbidity and mortality during rhythm control treatment in persistent atrial fibrillation in hypertensives: data of the RACE study. Eur Heart J. 2007;6:741-751.
38 Carlsson J, Miketic S, Windeler J, et al. Randomized trial of rate-control in persistent atrial fibrillation: the Strategies of Treatment of Atrial Fibrillation (STAF) study. J Am Coll Cardiol. 2003;41:1690-1696.
39 Holmloser S, Kuick K, Lilienthal J. Rhythm or rate control in atrial fibrillation: Pharmacological Intervention in Atrial Fibrillation (PIAF): a randomized trial. Lancet. 2000;356:1789-1794.
40 Predersen OD, Brendorp B, Elnung H, et al. Does conversion and prevention of atrial fibrillation enhance survival in patients with left ventricular dysfunction? Evidence from the Danish Investigations of Arrhythmia and Mortality On Dofetilide (DIAMOND) study. Card Electrophysiol Rev. 2003;7:220-224.
41 Deedwandia PC, Singh B, Ellenbogen K, et al. Spontaneous conversion and maintenance of sinus rhythm by amiodarone in patients with heart failure and atrial fibrillation: observations from the Veterans Affairs Congestive Heart Failure Survival Trial of Antiarrhythmic Therapy (CHF-STAT). Circulation. 1998;98:2574-2579.
42 Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC 2006 Guidelines for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines. Circulation. 2006;114:e257-354.
43 Hylek EM, Skates SJ, Sheehan MA, et al. An analysis of the lowest effective intensity of prophylactic anticoagulation for patients with non-rheumatic atrial fibrillation. N Engl J Med. 1996;335:540-546.
44 Klein AL, Grimm RA, Murray RD, et al. Assessment of cardioversion using transesophageal echocardiography investigators. Use of transesophageal echocardiography to guide cardioversion in patients with atrial fibrillation. N Engl J Med. 2001;344:1411-1420.
45 Oral H, Souza JJ, Michaud GF, et al. Facilitating transthoracic cardioversion of atrial fibrillation with Ibutilide pre-treatment. N Engl J Med. 1999;340:1849-1854.
46 McCellan KJ, Markham A. Dofetilide: a review of its use in atrial fibrillation and atrial flutter. Drugs. 1999;58:1043-1059.
47 Roy D, Talajie M, Dorian P, et al. The Canadian Trial of Atrial Fibrillation Investigators. Amiodarone to prevent recurrence of atrial fibrillation. N Engl J Med. 2000;342:913-920.
48 Haissaguerre M, Jais P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beat originating in the pulmonary veins. N Engl J Med. 1998;339:659-666.
49 Verma A, Natale A. Why atrial fibrillation ablation should be considered first-line therapy for some patients. Circulation. 2005;112:1214-1222.
50 Pappone C, Rosamo S, Augello G, et al. Mortality, morbidity and quality of life after circumferential pulmonary vein ablation for atrial fibrillation: outcomes from a controlled non randomized long-term study. J Am Coll Cardiol. 2003;42:185-197.
51 Gage BF, Waterman AD, Shannon W, et al. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285:2864-2870.
52 Hart RG, Benavente O, McBride R, et al. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med. 1999;131:492-501.
53 Wellens HJJ, Bar FWHM, Lie KL. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a wide QRS complex. Am J Med. 1978;64:27-33.
54 Brugada P, Brugada J, Mont L, et al. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991;83:1649-1659.
55 Antunes E, Brugada J, Steurer G, et al. The differential diagnosis of a regular tachycardia with a wide QRS complex on the 12-lead ECG: ventricular tachycardia, supraventricular tachycardia with aberrant intraventricular conduction, and supraventricular tachycardia with anterograde conduction over an accessory pathway. Pacing Clin Electrophysiol. 1994;17:1515-1524.
56 Griffith MJ, Garratt CJ, Mounsey P, et al. Ventricular tachycardia as a default diagnosis in broad complex tachycardia. Lancet. 1994;343:386-388.
57 The American Heart Association in Collaboration with the International Liasion Committee on Resuscitation (ILCOR). Guidelines 2000 for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2000;102(suppl. I):I142-157.
58 Kudenchuk PJ, Cobb LA, Copass MK, et al. Amiodarone for resuscitation after out-of-hospital cardiac arrest due to ventricular fibrillation. N Engl J Med. 1999;341:871-878.
59 Dorian P, Cass D, Schwartz B, et al. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2000;346:884-890.
60 Artucio H, Pereira M. Cardiac arrhythmias in critically ill patients: epidemiologic study. Crit Care Med. 1990;18:1383-1388.
61 Nasraway SA, Rackow EC, Astiz ME, et al. Inotropic response to digoxin and dopamine in patients with severe sepsis, cardiac failure and systemic hypoperfusion. Chest. 1989;95:612-615.
62 Holt AW. Hemodynamic responses to amiodarone in critically ill patients receiving catecholamine infusions. Crit Care Med. 1989;17:1270-1276.
63 Moran JL, Gallagher J, Peake SL, et al. Parenteral magnesium sulfate versus amiodarone in the therapy of atrial tachyarrhythmias: a prospective, randomized study. Crit Care Med. 1995;23:1816-1824.
64 Horner SM. Efficacy of intravenous magnesium in acute myocardial infarction in reducing arrhythmias and mortality: meta-analysis of magnesium in acute myocardial infarction. Circulation. 1992;86:774-779.
65 Woods KL, Fletcher S, Roffe C, et al. Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet. 1992;339:1553-1558.
66 Maggioni AP, Zuanetti G, Franzosi MG, et al. Prevalence and prognostic significance of ventricular arrhythmias after acute myocardial infarction in the thrombolytic era: GISSI-2 results. Circulation. 1993;87:312-322.
67 Solomon SD, Ridker PM, Antman EM. Ventricular arrhythmias in trials of thrombolytic therapy for acute myocardial infarction: a meta-analysis. Circulation. 1993;88:2575-2581.
68 The American Heart Association in Collaboration with the International Liasion Committee on Resuscitation (ILCOR). Guidelines 2000 for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2000;102(suppl. I):I112-128.
69 Radford MJ, Krumholz HM. Beta-blockers after myocardial infarction – for few patients, or many? N Engl J Med. 1998;339:551-553.
70 Waldo AL, Camm AJ, de Ruyter H, et al. Effect of D-sotalol on mortality in patients with left ventricular dysfunction after recent or remote myocardial infarction. The SWORD investigators. Survival with oral D-sotalol. Lancet. 1996;348:7-12.
71 Cairns JA, Connolly SJ, Roberts R, et al. Randomized trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarizations: CAMIAT. Canadian Amiodarone Myocardial Infarction Arrhythmia Trial investigators. Lancet. 1997;349:675-682.
72 Julian DG, Camm AJ, Frangin G, et al. Randomized trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. European Myocardial Infarction Amiodarone Trial investigators. Lancet. 1997;349:667-674.
73 Boutitie F, Boissel JP, Connolly SJ, et al. Amiodarone interaction with beta-blockers: analysis of merged EMIAT (European Myocardial Infarction Amiodarone Trial) and CAMIAT (Canadian Amiodarone Myocardial Infarction Arrhythmia Trial) databases. The EMIAT and CAMIAT investigators. Circulation. 1999;99:2268-2275.
74 Ommen SR, Odell JA, Stanton MS. Atrial arrhythmias after cardiothoracic surgery. N Engl J Med. 1997;336:1429-1434.
75 Andrews TC, Reimold SC, Berlin JA, et al. Prevention of supraventricular arrhythmias after coronary artery bypass surgery: a meta-analysis of randomized control trials. Circulation. 1991;84(suppl. III):III236-244.
76 Kowey PR, Taylor JE, Rials SJ, et al. Meta-analysis of the effectiveness of prophylactic drug therapy in preventing supraventricular arrhythmia early after coronary artery bypass grafting. Am J Cardiol. 1992;69:963-965.
77 Burgess DC, Kilborn MJ, Keech AC. Interventions for prevention of post-operative atrial fibrillation and its complications after cardiac surgery: a meta-analysis. Eur Heart J. 2006;27:2846-2857.
78 Patti G, Chello M, Candura D, et al. Randomized trial of atorvastatin for reduction of post operative atrial fibrillation in patients undergoing cardiac surgery: results of the ARMYDA-3 (Atorvastatin for Reduction of MYocardial Dysrhythmia After cardiac surgery) study. Circulation. 2006;114:1455-1461.
79 Ferraris VA, Ferraris SP, Gilliam HS, et al. Predictors of postoperative ventricular dysrrhythmias: a multivariate study. J Cardiovasc Surg (Torino). 1991;32:12-20.
80 Moss AJ. Prolonged QT syndromes. JAMA. 1986;256:2985-2987.
81 Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: prospective longitudinal study of 328 families. Circulation. 1991;84:1136-1144.
82 Moss AJ, Robinson J. Clinical features of the idiopathic long QT syndrome. Circulation. 1992;85(suppl. 1):140-144.
83 Tzivoni D, Bonai S, Schuger C, et al. Treatment of torsade de pointes with magnesium sulphate. Circulation. 1988;77:392-397.
84 Huikuri HV, Castellanos A, Myerburg RJ. Medical progress: sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473-1482.
85 Holmes DR, Davis KB, Mock MB, et al. The effect of medical and surgical treatment on subsequent cardiac death in patients with coronary artery disease: a report from the Coronary Artery Surgery Study. Circulation. 1986;73:1254-1263.
86 The Antiarrhythmics Versus Implantable Defibrillators (AVID) investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576-1584.
87 Moss AJ, Zareba W, Hall WJ, et al. Multicenter Automatic Implantable Defibrillator Trial II Investigators. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.
88 Maron BJ, Shen W-K, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;342:365-373.
89 Watanabe Y, Dreifus L. Electrophysiological effects of magnesium and its interactions with potassium. Cardiovasc Res. 1972;6:79-88.
90 Ghani MF, Rabah M. Effects of magnesium chloride on electrical stability of the heart. Am Heart J. 1977;94:600-602.
91 Mostow ND, Rakita L, Vrobel TR, et al. Amiodarone: intravenous loading for rapid suppression of complex ventricular arrhythmias. J Am Coll Cardiol. 1984;4:97-104.
92 Schwartz A, Shen E, Morady F, et al. Hemodynamic effects of intravenous amiodarone in patients with depressed left ventricular function and recurrent ventricular tachycardia. Am Heart J. 1983;106:848-855.
93 Cote P, Bourassa MG, Delaye J, et al. Effects of amiodarone on cardiac and coronary hemodynamics and on myocardial metabolism in patients with coronary artery disease. Circulation. 1954;67:1347-1355.
94 Torp-Pedersen C, Moller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. N Engl J Med. 1999;341:857-865.
95 Roukoz H, Saliba W. Dofetilide: a new class III antiarrhythmic agent. Exp Rev Cardiovasc Ther. 2007;5:9-19.
96 The American Heart Association in Collaboration with the International Liasion Committee on Resuscitation (ILCOR). Guidelines 200 or cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2000;102(suppl. I):I112-28.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 18 Management of cardiac arrhythmias
CARDIAC ELECTROPHYSIOLOGY
The electrophysiological properties of cardiac cells are important in understanding cardiac arrhythmias and their management. Cardiac cells undergo cyclical depolarisation and repolarisation to form an action potential. The shape and duration of each action potential are determined by the activity of ion channel protein complexes on the myocyte surface. These highly selective ion channels determine the rate of ion flux which in turn determines the magnitude and rate of change of myocyte membrane potential. Many of these ion channels are the molecular targets for antiarrhythmic drugs.
Ion channel function can be affected by:
The spectrum of cardiac action potentials varies from fast-response cells – conducting and contractile myocytes (Figure 18.1a) – to slow-response cells of pacemaker myocytes – sinoatrial (SA) and atrioventricular (AV) nodes (Figure 18.1b). Fast myocytes lose their characteristic action potential and behave more like slow myocytes when ischaemic. The action potential is divided into five phases, as follows.
PHASE 0
In fast myocytes (Figure 18.1a) rapid depolarisation occurs due to activation of voltage-dependent Na+ channels. Activation is initiated in an all-or-none response once the threshold is reached. The Na+ channels are inactivated as membrane potential rises to +30 mV and remain inactivated until repolarisation occurs. Rapidity of depolarisation determines speed of conduction. In slow-response myocytes depolarisation does not involve Na+ channels and the slower rate of depolarisation is due to a slow inward Ca2+ current via L- and T-type voltage-dependent Ca2+ channels.
PHASE 1
Early rapid incomplete repolarisation to approximately 0 mV occurs due to activation of transient outward current from ITO1 and ITO2 K+ channels. Slow myocytes do not exhibit phase 1 or 2 characteristics (Figure 18.1b).
PHASE 4
This is a stable electrical state in fast non-pacemaker myocytes. In slow pacemaker myocytes the resting membrane potential (RMP) slowly depolarises until the action potential threshold is reached (Figure 18.1b). This inward or pacemaker current is due to If K+ channels.
Fast-response and slow-response myocytes also have important differences in properties of refractoriness. In fast myocytes, Na+ channels are progressively reactivated during phase 3 repolarisation as the membrane potential becomes more negative. When an extra stimulus occurs during phase 3, the magnitude of the resulting inward Na+ current and likelihood of impulse propagation depend on the number of reactivated Na+ channels. Refractoriness is therefore determined by the voltage-dependent recovery of Na+ channels. The absolute refractory period (Figure 18.1) is that minimum time needed for recovery of sufficient Na+ channels for a stimulus to result in impulse propagation. However, once propagation in fast myocytes occurs, conduction velocity is normal. In contrast, slow-response or Ca2+ channel-dependent myocytes exhibit time-dependent refractoriness. Even after full repolarisation further time is needed before all Ca2+ channels are reactivated. Stimuli during this period produce reduced Ca2+ current and the propagation velocity of any resulting impulse is reduced. The conduction velocity independence of premature action potentials with fast-response myocytes is lost in the setting of Na+ channel-blocking drugs or ischaemia because they behave increasingly like slow-response myocytes with resulting slowed impulse conduction.
GENETIC BASIS TO ARRHYTHMIA1
MOLECULAR BASIS TO ARRHYTHMIA1
Tachycardic remodelling of the atrium is associated with:
Heart failure is associated with:
ARRHYTHMOGENIC MECHANISMS2–4
Many factors in isolation or combination give rise to the substrate of arrhythmogenesis (Figure 18.2). Arrhythmia may arise from abnormalities of impulse generation or conduction. Table 18.1 demonstrates the relationship between mechanism and type of arrhythmia, and desired antiarrhythmic effect.
ABNORMAL IMPULSE GENERATION (Table 18.2)
ENHANCED NORMAL AUTOMATICITY
| Enhanced normal automaticity | Adrenergic stimulation |
| Abnormal automaticity | Ischaemia |
| Early afterdepolarisations | Hypoxia |
| Hypercapnia | |
| Catecholamines | |
| Class IA antiarrhythmic drugs | |
| Class III antiarrhythmic drugs | |
| Other drugs that prolong repolarisation | |
| Delayed afterdepolarisations | Digoxin toxicity |
| Increased intracellular Na+ | |
| Decreased extracellular K+ | |
| Increased intracellular Ca2+ | |
| Intracellular Ca2+ overload | |
| Myocardial infarction | |
| Reperfusion after ischaemia |
TRIGGERED ACTIVITY
ABNORMAL IMPULSE CONDUCTION5
Abnormal impulse conduction may cause an arrhythmia by the phenomena of re-entry. Re-entry describes the re-excitation of an area or entire heart by a circulating impulse. Although the classic ‘bifurcating Purkinje fibre’ model of Schmitt and Erlanger has given way to a much more complex picture, the essential electrophysiological requirements for re-entrant excitation remain. Requirements for re-entry are (Figure 18.3):
When these properties are present, the chance of a circulating impulse producing re-entrant excitation depends on pathway geometry, the electrical properties and length of the depressed area and conduction velocity within each component. The segment of the re-entry pathway that is initially refractory and therefore blocks conduction down one limb and recovers in time to conduct the return impulse is termed the ‘excitable gap’. Therefore, the generation and subsequent maintenance of a circuit depend on this excitable gap of non-refractory tissue circulating between the advancing depolarising wave front and the repolarising tail. The resulting re-entrant impulse can be self-terminating, causing ectopic beats, or lead to atrial or ventricular tachyarrhythmias.
Re-entry may be terminated by:
The cellular properties that lead to impaired conduction include:
ELECTROLYTE ABNORMALITIES AND ARRHYTHMIA6
POTASSIUM
Hyper- and hypokalaemia both cause arrhythmia mediated by the resultant changes in RMP (Table 18.3). In ischaemia, hyperkalaemia at the local tissue level caused by a pathological extracellular shift of K+ is the major factor contributing to ventricular arrhythmia in this setting. In hypokalaemia, the dispersion of pacemaker activity and the effect on repolarisation are similar to the electrophysiological effects of cardiac glycosides and β-adrenergic agonists, and it is not surprising that a combination of these factors is associated with an increased incidence of arrhythmia. The increased risk of death in hypertensive patients treated with thiazide diuretics has been attributed to hypokalaemic (and possibly hypomagnesaemic)-induced arrhythmia (Multiple Risk Factor Intervention Trial). Thiazide-induced hypokalaemic ventricular ectopy is worsened by exercise.7 Hypokalaemia is associated with VF and VT following acute myocardial infarction (AMI). The increased incidence of VF/VT with a serum K+ less than 3.5 mmol/l is clearly established and the probability of VT increases as the serum K+ decreases. During AMI the incidence of VF/VT was 15% at 4.5 mmol/l, 38% at 3.5 mmol/l, 55% at 3.0 mmol/l and 67% at 2.5 mmol/l.8
MAGNESIUM
The antiarrhythmic properties of Mg2+ are clearly established but a causal relationship between hypomagnesaemia and arrhythmia is largely circumstantial. Decreased extracellular Mg2+ by itself has little effect on the electrophysiological properties of myocytes or the ECG. Hypomagnesaemia has been implicated in the genesis of VT/VF in patients with hypertension and heart failure receiving thiazide or loop diuretics, acute alcohol intoxication or withdrawal and possibly with AMI. The product of K+ and Mg2+ is the best predictor of arrhythmia in hypertensive patients taking thiazide diuretics.9
AUTONOMIC NERVOUS SYSTEM AND VENTRICULAR ARRHYTHMIA10
PROARRHYTHMIC EFFECTS OF ANTIARRHYTHMIC DRUGS11,12
Concomitant proarrhythmia with the use of antiarrhythmic drugs is increasingly recognised. The ‘quinidine syncope’ due to VF and polymorphic VT at therapeutic concentrations was also seen with disopyramide. The Cardiac Arrhythmia Suppression Trial (CAST) clearly defined the magnitude of this deleterious side-effect in drugs that were previously perceived to be of benefit.13 This study, which involved flecainide, encainide and morizicine (a class IA drug), was terminated early because of adverse outcome in the flecainide and encainide groups (relative risk of arrhythmic death or non-fatal cardiac arrest of 3.6, 95% confidence interval (CI) 1.7–8.5). Proarrhythmia is reported between 5.9% and 15.8% depending on agent, clinical setting and definition of proarrhythmia, and now considered ubiquitous with all antiarrhythmic drugs.
Proarrhythmia has been defined as an increase in frequency of ventricular ectopic beat (VEB) or aggravation of the target arrhythmia on Holter monitor or exercise test. Manifestations of proarrhythmia not only include VEB, monomorphic and polymorphic VT and VF, but also bradyarrhythmias and Afl with 1:1 AV conduction. Most proarrhythmic events occur soon after starting the drug, but late arrhythmias are also a significant problem.
The mechanism of drug proarrhythmia is probably via both slowing of conduction and abnormal automaticity. Paradoxically slowing conduction, which may block a re-entry circuit, may also create the very substrate needed for re-entry, unidirectional block and an excitable gap. The existence of a re-entrant circuit requires the circulating wave front of the impulse not to catch up with the refractory tissue behind the tail. Re-entry is more likely to occur with a shorter refractory period and reduced conduction velocity (Figure 18.4).11
Antiarrhythmic drugs are effective at suppressing abnormal automaticity, with the exception of triggered automaticity due to EAD. Class IA, class III and many non-antiarrhythmic drugs can produce proarrhythmia via EAD. These drugs increase not only the frequency of EAD, but also the likelihood of them leading to triggered tachyarrhythmias. Slowing repolarisation, which leads to QT prolongation and slower heart rate, is central to this increased frequency and sensitivity to EAD. EAD manifests as prominent and bizarre T-U waves on the ECG and, if triggered activity results, VEB and ventricular tachyarrhythmias may occur. Torsade de pointes is the classical resulting arrhythmia, although less classical polymorphic VT and VF result. Risk of proarrhythmia via this mechanism correlates with the degree of QT prolongation.
Antiarrhythmic drug proarrhythmia is facilitated by several factors, which are frequently found in patients on antiarrhythmic drugs or with heart disease (Table 18.4).
Table 18.4 Factors facilitating antiarrhythmic drug proarrhythmia
| Toxic blood levels due to excessive dose or reduced clearance from old age, heart failure, renal disease or hepatic disease |
| Severe left ventricular dysfunction. Ejection fraction less than 35% |
| Pre-existing arrhythmia or arrhythmia substrate |
| Digoxin therapy |
| Hypokalaemia or hypomagnesaemia |
| Bradycardia |
| Combinations of antiarrhythmic drugs and concomitant drugs with similar toxicity |
(Adapted from Campbell TJ. Proarrhythmic actions of antiarrhythmic drugs: a review. Aust NZ J Med 1990; 20: 275–82, with permission.)
MANAGEMENT OF THE PATIENT WITH A CARDIAC ARRHYTHMIA
INVESTIGATIONS
A 12-lead ECG should be recorded with a longer rhythm strip (usually lead II or V1). If P-waves are not visible, atrial activity may be recorded using an oesophageal electrode or pacing lead, or via a central venous catheter or the right atrial injectate port of a pulmonary artery catheter, using 20% saline and a bedside monitor.14
EPS, which involves invasive electrophysiological testing with programmed electrical stimulation, attempts to reproduce the spontaneously occurring arrhythmia.15,16 EPS is not clearly superior to Holter monitoring in evaluating drug treatment for ventricular arrhythmias.
Other investigative techniques being studied include signal-averaged ECG, heart rate variability and electrical alternans measurement.10,17
MANAGEMENT OF SPECIFIC ARRHYTHMIAS
Treatment has two aspects: acute termination of the arrhythmia and long-term prophylaxis. The decision whether to treat depends on the rhythm diagnosis, haemodynamic consequences, aetiology of the arrhythmia and the prognosis (e.g. risks of sudden death or long-term complications).
PREMATURE VENTRICULAR ECTOPIC BEATS
ECG
Occasionally VEB may not produce any pause, and are said to be interpolated (see Figure 18.6). Interpolated VEB occur when the background sinus rhythm is slow. The retrograde conduction into the AV node renders it partially refractory to the next impulse and its conduction through the AV node is slowed and the PR interval is prolonged. A VEB following each sinus beat is ventricular bigeminy (Figure 18.7). Ventricular trigeminy refers to recurring sequences of a VEB followed by two sinus beats. Two VEB in succession are a couplet (Figure 18.8), and three, a triplet.
CLINICAL
Even when frequent, complex, or in short runs of non-sustained VT, VEB are not associated with risk of sudden death in asymptomatic healthy adults.18 However, there is increased risk of cardiovascular death with:
SUPRAVENTRICULAR TACHYCARDIAS22,23 (Table 18.5)
| Atrioventricular (AV) node-dependent |
| AV nodal re-entry tachycardia: re-entry within the AV node |
| AV re-entry tachycardia: re-entry includes accessory pathway between atria and ventricles |
| Accelerated idionodal rhythm: increased automaticity of AV nodea |
| AV node-independent |
| Atrial flutter: re-entry confined to atria |
| Atrial fibrillation: multiple re-entry circuits confined to atria |
| Unifocal atrial tachycardia: usually due to increased automaticity |
| Multifocal atrial tachycardia: increased automaticity or triggered activity |
| Others: sinus node re-entry tachycardia |
A clinically useful classification divides SVT into AV node-dependent and AV node-independent.
AV NODAL RE-ENTRY TACHYCARDIA (AVNRT)(Figure 18.9)
ECG
There is regular narrow-complex tachycardia (140–220 beats/min) with abrupt onset and termination. P-waves are not usually observed as they are buried in the QRS complexes (Figure 18.10).
TREATMENT
Vagal manoeuvres slow conduction through the AV node and may ‘break’ the tachycardia. If carotid sinus massage fails, adenosine is the drug of choice and nearly all AVNRT will revert with adenosine.24,25 Verapamil has been used in the past, but causes hypotension, which may be prolonged if cardiac function is depressed or patients are receiving β-adrenergic blockers. Sotalol, amiodarone and flecainide may also be effective but are rarely used. Rapid atrial pacing will usually terminate AVNRT but is rarely needed.
AV RE-ENTRY TACHYCARDIA (SEE Figure 18.9)
ECG
The ECG is similar to AVNRT. The length of the re-entry circuit is however greater, and the accessory AV pathway is some distance from the AV node. It therefore takes longer for the impulse to be conducted backwards to the atria, and so the retrograde P-wave usually occurs after the QRS, sometimes at some distance, and is inverted in leads II, III and aVF (Figure 18.11 and 18.12).
ACCELERATED IDIONODAL RHYTHM
ECG
There are narrow complexes on the ECG at a regular rate (60–130 beats/min) (Figure 18.13), often with independent atrial activity. With isorhythmic dissociation, P-wave is either fixed relative to the QRS complex (usually just after) or oscillates to and fro across the QRS in a rhythmical manner.
UNIFOCAL ATRIAL TACHYCARDIA
ECG
P-wave morphology is abnormal but monomorphic. Atrial rate is often 130–160 beats/min, and may occasionally exceed 200 beats/min. Atrial rate distinguishes unifocal atrial tachycardia from atrial flutter (Afl), with Afl greater than 250 beats/min. The QRS complexes will usually be narrow (Figure 18.14). AV block is common (Figure 18.15).
CLINICAL
Digitalis intoxication is the most common cause, especially when AV block is present. Other causes include myocardial infarction, chronic lung disease and metabolic disturbances.
MULTIFOCAL ATRIAL TACHYCARDIA28
ECG
There are irregular atrial rates, usually 100–130 beats/min, with varying P-wave morphology (at least three different P-wave morphologies and varying PR interval) and some degree of AV block (Figure 18.16). Most P-waves are conducted to the ventricles, usually with narrow QRS complexes.
TREATMENT
Treatment should correct the underlying cause (e.g. treatment of cardiorespiratory failure, electrolyte and acid–base abnormalities and theophylline toxicity). Spontaneous reversion is common, and few patients require antiarrhythmic therapy. Magnesium is the drug of choice for acute control.29 β-Blockers are probably more effective than diltiazem, but because of the common association of MAT with obstructive lung disease have limited utility.30 Digoxin and cardioversion are ineffective, which highlights the need to differentiate MAT from AF. Longer-term control is best achieved with diltiazem in patients with good left ventricular (LV) function and amiodarone in those without.
ATRIAL FLUTTER31
ECG
Afl waves (characteristic sawtooth appearance with no isoelectric baseline) are best seen in V1 (Figure 18.17) or aVF, but leads II and III may also be useful. The flutter waves are usually negative in aVF. Rapid QRS waves may obscure typical flutter waves, and vagal manoeuvres may unmask them (see Figure 18.5). AV conduction block (usually 2:1) is usually present, so that alternate flutter waves are conducted to the ventricles, with a ventricular rate close to 150 beats/min. Frequently flutter waves are not obvious and a ventricular rate of 150 beats/min leads to the presumption of Afl (Figure 18.18). Type II Afl results in greater atrial and ventricular rates (Figure 18.19). Treatment with drugs that affect AV node conduction may lead to higher degrees of AV block (Figure 18.20) and/or variable AV block with irregular QRS duration. Rarely, Afl with 1:1 conduction occurs. This is usually associated with sympathetic overactivity or class I antiarrhythmic drugs (which slow atrial discharge rate to 200 beats/min, thereby allowing each atrial impulse to be conducted) (Figure 18.21). QRS complexes are usually narrow, as conduction through the bundle branches is normal.
TREATMENT25
No drug will reliably terminate Afl, although ibutillide and dofetilide have been shown to be most likely to result in pharmacological reversion. Attempts at slowing ventricular rate by drugs that will increase the degree of AV block are worthwhile in the first instance. Drugs such as digoxin, diltiazem, β-adrenergic blockers, sotalol and amiodarone may be tried; the choice depends on LV function. Flecainide and procainamide may occasionally be effective at terminating Afl. However, class IA and IC drugs may lead to 1:1 AV conduction. Class I drugs should probably be avoided unless ventricular response has been slowed with calcium channel or β-adrenergic blocking drugs.
Anticoagulation guidelines are the same as that for AF, although there are less supporting data.
PREVENTION
Prevention is difficult. Drugs used include sotalol and amiodarone at low doses. Class IC agents (e.g. flecainide) may be used in patients without significant structural heart disease. Increasingly recurrent or refractory Afl may be cured by radiofrequency ablation to create a linear lesion between the inferior tricuspid annulus and the eustachian ridge at the anterior margin of the inferior vena cava to interrupt the re-entry circuit.26
ATRIAL FIBRILLATION32
ECG
Atrial activity is chaotic with rapid (350–600 beats/min) and irregular depolarisations varying in amplitude and morphology (fibrillation waves). Ventricular response is irregularly irregular (Figure 18.22). Most atrial impulses are not conducted to the ventricles, resulting in an untreated ventricular rate of 100–180 beats/min. QRS complexes will usually be narrow. When the ventricular rate is very rapid or very slow, ventricular irregularity may be missed (Figure 18.23).
CLINICAL
TREATMENT25,34,35
The goals of treatment include ventricular rate control, anticoagulation where appropriate and conversion to sinus rhythm. There is increasing evidence available on the ‘rate versus rhythm’ control debate. Results from several recent major studies have challenged the previous belief that achievement of sinus rhythm is important in the long term (Table 18.6). When comparing control of ventricular rate versus reversion to sinus rhythm no clear survival benefit is apparent. However composite end-points of death, stroke and recurrent hospilisation favour rate control only.36–39
The possible reasons why rhythm control has not been shown to be superior include:
However rhythm control (if possible) appears superior in patients with LV dysfunction, with both amiodarone and dofetilide reducing mortality when sinus rhythm is achieved.40,41 The paucity of data in younger patients (less than 60 years) favours initial attempts at rhythm control, particularly in those with structurally normal hearts, in the hope that progressive atrial electrical and anatomical remodelling is prevented.
RECENT ONSET OR PAROXYSMAL AF
VENTRICULAR RATE CONTROL
CONVERSION TO SINUS RHYTHM
Antiarrhythmic drugs or DC shock cardioversion can be used. The likelihood of short- and long-term success depends on the clinical situation. Conversion to sinus rhythm is more important in young patients and those with heart failure. Maintenance of sinus rhythm is problematic: sinus rhythm at 1 year is 60% with amiodarone and 40% with sotalol, and associated with significant drug cardiac and extracardiac toxicities. The risk of stroke and need for antithrombotic therapy due to frequent AF recurrences, which may be asymptomatic, remain. Achieving sinus rhythm (especially greater than 60 years) is less important than previously thought.36
DC SHOCK CARDIOVERSION
Critically ill patients who are septic, postoperative or on drugs such as catecholamines are likely to relapse.
ANTIARRHYTHMIC DRUGS
Quinidine is more effective than placebo, but increases mortality through proarrhythmia (generally class IA and IC antiarrhythmic drugs are contraindicated). Ibutilide and dofetilide are newer antiarrhythmic drugs with particular success at pharmacological cardioversion. Pretreatment with ibutilide increased DC shock cardioversion from 72% for placebo to 100%. In placebo failures, cross-over to ibutilide resulted in a 100% success rate with subsequent cardioversion. Ibutilide also resulted in reduction in DC shock energy required from 228 ± 93 J to 166 ± 80 J. However, ibutilide was associated with a 3% incidence of sustained polymorphous VT.45 Dofetilide appears to cardiovert AF and Afl pharmacologically in about a third of patients (intravenous (IV) better than oral, recent onset better than prolonged and Afl may be more responsive than AF). Dofetilide is far superior to placebo and sotalol, with similar recurrence rates to amiodarone.46
Other drugs currently used to promote onset of sinus rhythm and prevent AF relapse include amiodarone, sotalol, procainamide, flecainide and propafenone. Amiodarone was found to be superior in preventing AF recurrence with a recurrence rate of 35% compared to a recurrence rate of 63% for sotalol and propafenone.47
The factors dictating choice are:
When using amiodarone for prevention of AF recurrence there was an 18% incidence of adverse effects versus 11% for sotalol and propafenone.47
ATRIAL FIBRILLATION ABLATION THERAPY
Ablation techniques for AF have been continuously refined since the original Maze III surgical procedure which involved numerous atrial incisions to form a maze-like pattern of scarring, blocking propagation of arrhythmia. The ultility of this procedure was limited because it was surgical, with longer bypass times, postoperative bleeding and impaired atrial contractility. The magnitude of this original procedure was based on the belief that the entire atrium was involved in the initiation and maintenance of the fibrillatory conduction. This may be true for long-standing AF but paroxysmal AF appears to originate primarily at the junction of the left atrium and pulmonary veins. AF in 94% of patients is initiated by rapid discharges from one or more foci at or near the pulmonary vein orifices.48 Atrial tissue in this area has heterogeneous electrophysiological properties and there is also clustering of vagal inputs, which creates substrate for rapid discharges that initiate microre-entrant circuits or ‘rotors’. These high-frquency periodic rotors send spiral wave fronts of activation into surrounding atria. Localised ablation of a single dominant foci and rotor is inadequate as there are usually multiple foci.
Left atrial catheter (transatrial septum) AF ablation isolating all four pulmonary veins using radiofrquency is being heralded as the possible AF cure. Results are improving as all pulmonary veins are now isolated and the encircling lesion is clear of the pulmonary vein antrum (reducing pulmonary vein stenosis). Success rates of 81% (75–88%) free of AF and off drugs are reported. Success appears long-term as recurrence occurs early. A further 10–20% may become responsive to antiarrhythmic drugs which were previously ineffective. Repeating the procedure can increase success to > 90% with failure only in patients found to have extensive atrial scarring (predicting and excluding patients with this extensive atrial scarring is a major future challenge). Although not yet the universal cure the results are two- to threefold better than antiarrhythmic drugs alone.
Complication rates are also falling associated with:
Transient ischaemic attacks, strokes, tamponade/perforation and symptomatic pulmonary vein stenosis are all well below 1% respectively. Proarrhythmia resulting from re-entrant tachycardias from incomplete ablative lesions is more common. Some are advocating ablation as first-line treatment whereas most are selecting younger patients (less than 70 years) with paroxysmal AF for whom antiarrhythmic therapy has failed, left atrial diameter is less than 5 cm and ejection fraction is greater than 40%.26 Head-to-head studies comparing ablation and antiarrhythmic drugs are appearing with suggested survival benefit, improved quality of life, reduced adverse effects and cost-effectiveness after approximately 3 years with catheter AF ablation therapy.49,50
ANTICOAGULATION FOR CHRONIC ATRIAL FIBRILLATION
Consider for all patients, especially those with risk factors (Table 18.7).
Table 18.7 Prognostic factors for ischaemic stroke and systemic embolism in patients with atrial fibrillation
| High | Previous stroke, transient ischaemic attack, systemic embolism |
| Mitral stenosis | |
| Prosthetic heart valve | |
| Moderate | Age > 75 years |
| Left atrial size > 45 mm | |
| Hypertension | |
| Congestive cardiac failure | |
| Diabetes mellitus | |
| Left ventricular ejection fraction < 35% | |
| Low | Female |
| Age 65–74 years | |
| Coronary artery disease | |
| Thyrotoxicosis |
NON-VALVULAR ATRIAL FIBRILLATION
The risk of stroke is determined by CHADS2 score (assign 1 point for congestive heart failure, hypertension, age = 75 years and diabetes mellitus, and 2 points for stroke/TIA)51 (Tables 18.8 and 18.9).
Table 18.8 CHADS2 score stroke risk stratification in non-valvular atrial fibrillation
| Prognostic factor | Relative risk* | CHADS2 score |
|---|---|---|
| Congestive heart failure (ejection fraction < 35%) | 1.4 | 1 |
