CHAPTER 40 Magnetic Resonance Imaging of the Aorta and Left Ventricular Function in Inherited and Congenital Aortic Disease
Intrinsic aortic wall abnormalities have been described in inherited connective tissue disorders such as Marfan syndrome and bicuspid aortic valve disease, but recent reports indicate similar aortic involvement in patients with classic congenital heart disease (CHD) entities, such as coarctation of the aorta, tetralogy of Fallot, and transposition of the great arteries, suggesting a degenerative process that results in structural weakness of the aortic wall.1,2 The aortic media consists of smooth muscle and an extracellular matrix that is composed of ground substance, in which elastic fibers and collagen are embedded in a hydrated gel comprising glycosaminoglycans, proteoglycans, and adhesive glycoproteins.1 Smooth muscle cells govern vasodilation and vasoconstriction, collagen provides inert aortic wall strength (stiffness), and elastin—with its low elastic modulus—is readily deformable, permitting phasic distention and recoil in response to pulsatile flow.1 Current focus on the origin of aortic wall abnormalities has shifted toward these specific medial constituents and their crucial role in the structural and functional integrity of the aorta in affected groups of patients.1
In prototypical diseases such as Marfan syndrome and bicuspid aortic valve disease, loss of fibrillin 1 microfibrils has been reported to dissociate smooth muscle cells from the medial matrix components, resulting in accelerated cell death and matrix disruption (Fig. 40-1).2 Matrix metalloproteinases, endogenous enzymes that degrade matrix components, become activated in fibrillin 1–deficient tissues, degrading the structural support of the aorta.2 Similar focal abnormalities within the aortic media have recently been described in coarctation of the aorta, tetralogy of Fallot, and transposition of the great arteries.1 Whether these aortic wall changes result from an intrinsic medial abnormality or are secondary to hemodynamic states before and after surgical repair (or both) is unknown.1 Whatever the etiology of aortic wall disease given this heterogeneity, aortic dilation and reduced aortic elasticity will evolve when loss of structural support of the aortic wall progresses.1,2
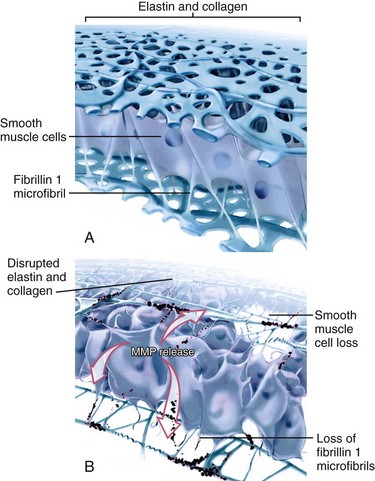
 FIGURE 40-1
FIGURE 40-1With advancing age, ascending aortic elastic fibers will fragment, collagen and ground substance will increase, and the number of smooth muscle cells will decrease.1,3 In addition, arterial hypertension, atherosclerosis, and other cardiovascular risk factors such as smoking, hypercholesterolemia, and diabetes may lead to atheroma formation, which is associated with similar degenerative processes in the extracellular matrix and the vasa vasorum.4 Therefore, the negative sequelae related to aging and numerous cardiovascular risk factors will have a superimposing impact on already existing aortic wall abnormalities.
Both reduced elasticity and dilation of the aorta have a detrimental effect on aortic valve function, especially if the aortic valve is already structurally malformed by being bicuspid or quadricuspid.5,6 As aortic valve opening occurs in concert with root expansion during the beginning of systole, aortic elasticity is crucial for aortic valve dynamics.5,6 Decreased aortic root elasticity increases leaflet stress and therefore predisposes to aortic valve dysfunction.5,6 Aortic dilation contributes to aortic valve dysfunction by loss of coaptation of the aortic valve leaflets.2,5 Moreover, with reduced aortic elasticity, the cushioning windkessel effect of the aorta is impaired. This leads to increased systolic blood pressure and pulse pressure, which in turn will increase myocardial oxygen demand by increasing LV afterload.7 Arterial stiffening is also associated with impaired coronary perfusion, the presence of coronary artery disease, and LV dysfunction.8
As life expectancy of patients with CHD has much improved during the past decades, adult cardiologists will be increasingly confronted with the challenge of concomitant aortic sequelae. Therefore, noninvasive monitoring of aortic dimensions and elasticity, in conjunction with aortic valve competence and LV function, is clinically highly desirable in patients with potential aortic wall abnormalities. Echocardiography is widely used for routine assessment of the aortic root, aortic valve, and ventricular function, but it does not permit evaluation of the entire aorta.4 Magnetic resonance imaging (MRI) provides advantages such as unlimited field of view within the entire thorax for accurate and reproducible assessment of aortic and cardiac anatomy and function.5,9,10
MARFAN SYNDROME
Prevalence and Epidemiology
Marfan syndrome is a heritable connective tissue disorder resulting in a highly variable degree of premature aortic medial degeneration with a high risk of progressive aortic dilation and subsequent aortic dissection or rupture.11,12 Marfan syndrome is caused by a mutation of the FBN1 gene on chromosome 15 that codes for fibrillin 1, in the absence of which elastin is more readily degraded by matrix metalloproteinases and smooth muscle cells will dissociate from the medial matrix components.1,4
Pathophysiology and Follow-up
Aortic dilation is the most common cause of morbidity and mortality in patients with Marfan syndrome as dilation of the sinus of Valsalva is found in 60% to 80% of adult patients (Fig. 40-2).13 The relative abundance of elastic fibers in the ascending aorta compared with other regions of the arterial tree, coupled with the repetitive stress of LV ejection, probably accounts for aortic dilation that usually occurs primarily in the aortic root.13 Therefore, the majority of patients with Marfan syndrome present with enlargement of the ascending aorta or a type A dissection and only in very rare cases with a type B dissection involving the descending aorta.11 Aortic dissection is associated with increasing aortic diameter but it may also occur in nondilated aortas.11,12 Replacement of the aortic root with a composite graft conduit has been recommended before the diameter exceeds 5.0 to 5.5 cm.11,12 Independent predictors of progressive aortic dilation that will prompt the recommendation for surgery when the aorta is smaller than 5.0 cm include rapid growth of the aortic diameter (>1 cm/yr), family history of premature aortic dissection (<5 cm), greater than mild aortic regurgitation, and pregnancy or contemplated pregnancy.11,12 Aortic regurgitation may result from distortion of the coaptation of the aortic valve cusps by the enlarged aortic root and occurs in 15% to 44% of patients.11 Especially in young children, progression of findings is more important than absolute size of the aorta as a criterion for surgery.11
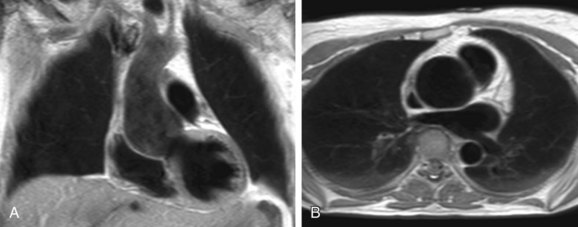
 FIGURE 40-2
FIGURE 40-2Recent MRI reports indicate that reduced aortic distensibility is an independent predictor of progressive aortic dilation in addition to aortic diameters.12 Because elastin fragmentation in the aortic media is scattered in an irregular pattern along the aorta, regional distensibility may be sensitive in the detection of regional variations in aortic stiffness.12 For optimal risk stratification, aortic stiffness may be taken into account in combination with aortic dimensions and the previously mentioned predictors of progressive aortic dilation.4,12,13 MRI has been recommended for routine assessment of aortic diameters and stiffness in patients with Marfan syndrome as well as for the follow-up of aortic complications, such as intramural hematoma and aortic aneurysms.4,11,12 Evaluation of aortic dilation should be performed every 6 months to determine the rate of progression, which can be extended to annual evaluation when the aortic size is stable over time.11 MRI can also be used to adequately monitor the beneficial effect of administration of β blockers on the progression rate of aortic dilation and reduction of aortic complications.11
BICUSPID AORTIC VALVE DISEASE
Prevalence and Epidemiology
The bicuspid aortic valve (BAV) is the most common congenital cardiac malformation, occurring in 1% to 2% of the population.14–16 BAV is the result of abnormal aortic cusp formation due to inadequate production of fibrillin 1 during valvulogenesis.2,4 Adjacent cusps fuse to form a single aberrant cusp, larger than its counterpart yet smaller than two normal cusps combined.2 BAV is probably the result of a complex developmental pathologic process rather than simply the fusion of two normal cusps.2,14,16
Pathophysiology and Follow-up
Aortic regurgitation is the most frequent (80%) complication in patients with BAV and usually occurs from cusp prolapse, fibrotic retraction, or dilation of the sinotubular junction, in many cases requiring aortic valve replacement.2,4 BAV is also present in the majority of elderly patients with significant aortic stenosis, reflecting the propensity for premature fibrosis, stiffening, and calcium deposition in these abnormally functioning valves.2,4 The vascular complications of BAV are less well understood and are associated with significant morbidity and mortality.2,16 The histology of the ascending aortic wall in patients with BAV shows strong similarities with the fibrillin 1–deficient aortas of patients with Marfan syndrome, with accelerated degeneration of the aortic media due to loss of fibrillin 1 microfibrils as well as focal abnormalities within the aortic media, such as matrix disruption and smooth muscle cell loss (see Fig. 40-1).1,14 Interestingly, BAV is present in more than 70% of patients with coarctation, and both conditions are by themselves and certainly in combination known to be associated with similar aortic wall abnormalities and concomitant aortic dilation.1,14
As a consequence of abnormal aortic wall composition in BAV, serious complications such as progressive aortic root dilation (50% to 60% of all patients with BAV; Fig. 40-3) and aneurysm formation may finally result in aortic dissection (5% of all patients with BAV).16,17 Despite this lower incidence of aortic dissection than in Marfan syndrome (40%), BAV is the more common etiology in aortic dissection because Marfan syndrome is a much rarer entity (0.01% vs. 1% to 2% of patients with BAV).2 Two different phenotypes of aortic dilation have been described. Dilation of the mid ascending aorta is most commonly present (70%) and is associated with aortic valve stenosis, suggesting a post-stenotic causative mechanism.16 Aortic root dilation is much rarer (13%), being determined by male gender and degree of aortic regurgitation.16 Aortic root replacement is generally more aggressively recommended for patients with BAV (i.e., 4 to 5 cm) than for those patients with a tricuspid aortic valve (i.e., 5 to 6 cm).2 Subdivision into the two existing phenotypes may further refine the surgical approach by suggesting aortic root sparing when only dilation of the mid ascending aorta is present.16
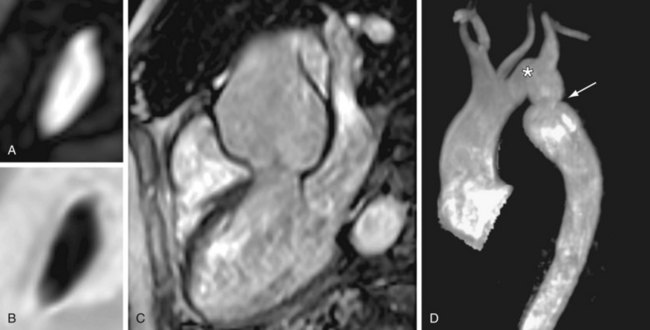
 FIGURE 40-3
FIGURE 40-3An MRI study reported frequent aortic root dilation and reduced elasticity in the entire aorta, suggesting not only that the proximal part of the aorta is affected in BAV but also that aortic wall lesions extend into the entire aorta.18 Evaluation of the elastic properties of the ascending aorta might be useful to identify patients who are at risk of progressive aortic dilation, analogous to patients with Marfan syndrome.12,18 Increased aortic stiffness was also associated with LV hypertrophy as a result of increased LV afterload.18 Sustained LV hypertrophy is associated with reduced diastolic filling; therefore, as diastolic LV dysfunction is a major contributor to congestive heart failure, LV hypertrophy might pose a future risk for LV function in patients with BAV.18 As many patients with BAV will require cardiac surgery during their lifetime, close monitoring of aortic dimensions, aortic elasticity, aortic valve competence, and LV function is mandatory during follow-up to allow timely intervention.18
COARCTATION OF THE AORTA
Prevalence and Epidemiology
Coarctation of the aorta accounts for 5% of all CHD and is defined as a congenital narrowing of the aorta, most commonly in a juxtaductal position just distal to the origin of the left subclavian artery.19 A wide spectrum of narrowing of the aorta can be observed, from a discrete narrowing to a hypoplastic aortic arch, whether or not it is associated with an intracardiac defect such as a ventricular septal defect or aortic valve disease.19 Classic symptoms are heart failure and an increased blood pressure proximal to the narrowing as well as a low perfusion status of the body distal to the coarctation.19 Coarctation of the aorta is associated with a significantly increased cardiovascular morbidity and reduced life expectancy even after successful surgical correction at a young age.7,12,20–22 Structural aortic wall abnormalities with reduced aortic elastic properties proximal and distal to the site of coarctation imply that coarctation of the aorta is a systemic vascular disease.4,20 Neonates with coarctation were found to have reduced elastic properties of the aorta before and after successful operation, suggesting a primary defect.22 Concomitant presence of BAV in 20% to 85% of patients with coarctation and the strong histologic similarity of aortic wall abnormalities between both entities are also suggestive of an inherited origin of aortic wall disease.1,14,17
Pathophysiology and Follow-up
After initially successful surgical repair, complications may occur, such as persistence of hypertension, recoarctation, aortic dilation, and aneurysm formation (Fig. 40-4).20 Persistent resting hypertension and exercise-induced hypertension have been reported in 10% to 46% and 30% to 60%, respectively, being the most important postoperative cardiovascular events.7,12 Hypertension in coarctation is accompanied by an increase in aortic medial collagen and a decrease in smooth muscle (increased stiffness) that may persist after successful repair and coincides with aortic abnormalities of a coexisting BAV.1,4 In addition, late hypertension may be caused by functional recoarctation at the site of surgical repair due to decreased distensibility.23 Impaired LV function, increased LV mass, and concomitant adverse LV remodeling even in postoperative patients with normal blood pressure can be attributed to the reduced aortic elastic properties that result in increased LV afterload.24 Furthermore, arterial hypertension and a resting pressure gradient are major contributing factors to early atherosclerotic development and should, therefore, be primary targets for therapy.20
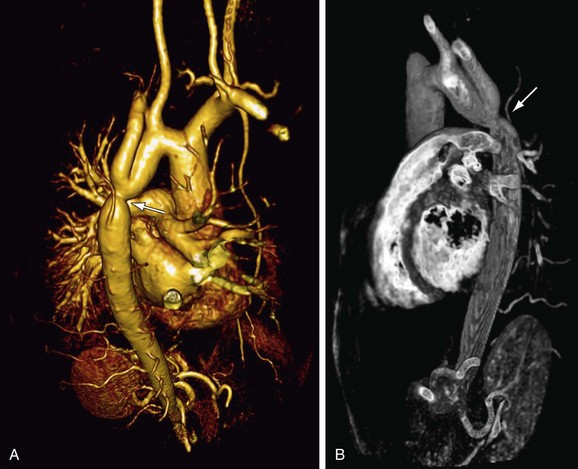
 FIGURE 40-4
FIGURE 40-4Adults after surgical repair of coarctation, especially when it is associated with BAV, should be closely monitored for detection of recoarctation as well as for progressive aortic dilation.20 Risk factors for these long-term aortic complications are the presence of BAV, advanced age, and hypertension.4 Systematic MRI screening performed at 2- to 5-year intervals has been found to be the most cost-effective approach for the follow-up of patients after coarctation repair, with early detection of aortic complications such as recoarctation.4,25 Spin-echo MRI is essential to detect abnormalities of the aortic wall and associated intracardiac abnormalities; contrast-enhanced three-dimensional MR angiography provides a highly accurate view of the entire reconstructed aorta, and this may obviate the need for invasive x-ray angiography by catheterization for planning of treatment (see Fig. 40-4).21 A combination of anatomic and flow data obtained by MRI is able to predict a catheterization peak-to-peak gradient of 20 mm Hg or more, considered to be the reference standard for hemodynamic severity of recoarctation.21 The combination of narrowest aortic cross-sectional area and heart rate–corrected mean flow deceleration in the descending aorta distinguishes between those who have transcatheter pressure gradients above and below 20 mm Hg, which can be used to determine the need for intervention.21 Velocity-encoded MRI is also useful to determine presence and hemodynamics of collateral vessels, which maintain distal aortic perfusion, depending on the severity of the aortic obstruction.26 Finally, promising utilities are MRI-guided balloon angioplasty and MRI-guided deployment of stents to repair aortic coarctation; studies have indicated that the combined use of MRI and x-ray imaging is effective for relief of stenosis with a significant reduction of radiation exposure.27
TETRALOGY OF FALLOT
Prevalence and Epidemiology
Tetralogy of Fallot (TOF) is the most commonly encountered cyanotic CHD entity with a frequency of nearly 10% of all CHD patients.28 Anterior displacement of the outflow septum is the primary defect, resulting in a ventricular septal defect, overriding of the aorta, right ventricular outflow tract obstruction, and right ventricular hypertrophy.28 Patients frequently encounter long-standing pulmonary regurgitation and associated impaired right ventricular function after primary TOF repair.28
Pathophysiology and Follow-up
Progressive dilation of the aortic root during long-term follow-up has also frequently been described after TOF repair, ranging in incidence between 15% and 88%, depending on definition of aortic root dilation (Figs. 40-5 and 40-6).28–31 Two hypotheses have been postulated to explain this observation. First, increased blood flow from both ventricles to the overriding aorta before surgical repair is thought to be an underlying pathogenic mechanism, posing increased stress on the aortic wall.1,28,30,31 This premise is supported by risk factors such as longer shunt-to-repair interval and a higher prevalence of pulmonary atresia among patients with TOF and aortic root dilation.30,31 Second, histologic changes of the aortic media, such as noninflammatory loss of smooth muscle cells and fragmentation of the elastic fibers, resembling those observed in patients with Marfan syndrome and BAV, have been reported.1,28
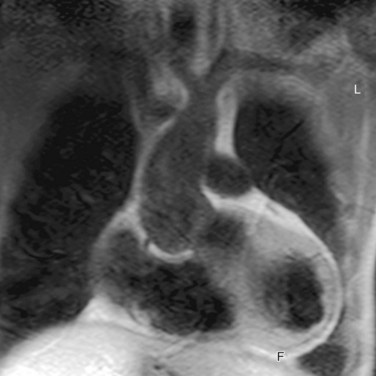
 FIGURE 40-5
FIGURE 40-5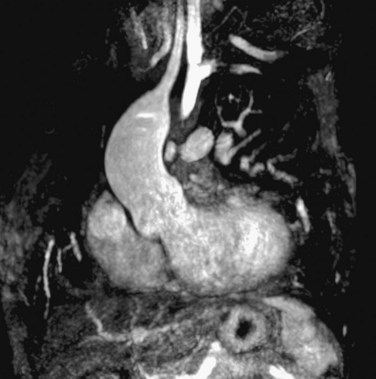
 FIGURE 40-6
FIGURE 40-6The potential for complications of aortic root dilation that may necessitate surgical intervention is increasingly recognized in patients after TOF repair.29 A study reported the progressive nature of aortic dilation in patients with TOF; aortic dilation increased at a rate of 1.7 mm/yr, in contrast to 0.03 mm/yr in healthy controls.31 Also, more marked histologic changes were observed with increasing age, suggesting that aging coupled with volume overloading on top of intrinsic aortic wall abnormalities has an additional adverse effect on the aortic histology and thus on aortic dilation.28 In addition, aortic regurgitation associated with progressive dilation of the aortic root is frequently present, and 15% to 18% of patients after TOF repair show mild degrees of aortic regurgitation (see Fig. 40-6).31 Recent case reports of aortic dissection late after TOF repair in adults whose aortic roots exceeded 6 cm in diameter indicate that close monitoring of aortic dimensions is mandatory, especially when the ascending aorta is dilated.32 Aortic root surgery may be considered for patients after TOF repair in case of progressive aortic regurgitation and aortic root dilation exceeding 5.5 cm, particularly when the primary indication for surgery is pulmonary valve replacement, and both procedures may be combined.33 At present, there is no consensus on the administration of β blockers for prevention of progressive aortic root dilation or at what stage aortic root surgery should be considered in patients after TOF repair.31
TRANSPOSITION OF THE GREAT ARTERIES
Prevalence and Epidemiology
Transposition of the great arteries is defined as atrial situs solitus, normal (concordant) connection between atria and ventricles, and abnormal (discordant) ventriculoarterial connections.34 The aorta arises from the right ventricle and the pulmonary artery arises from the left ventricle. It accounts for 4.5% of all congenital cardiac malformations.34 The arterial switch operation (ASO) has become the preferred method of surgery for transposition of the great arteries.35 Although this technique has significantly reduced the number of sequelae associated with surgical correction of transposition of the great arteries, completion of the ASO may still predispose patients to aortic root dilation and aortic regurgitation.35–37
Pathophysiology and Follow-up
Intrinsic aortic wall disease has been reported in transposition of the great arteries due to abnormal aorticopulmonary septation, damage to the vasa vasorum, and surgical manipulations during the ASO, predisposing to aortic dilation, aneurysm formation, and even aortic dissection (Fig. 40-7).35,38,39 In addition, aortic distensibility may be reduced by impaired aortic elastogenesis as well as by scar formation at the site of anastomosis.35 High-grade medial abnormalities in the ascending aorta have already been observed during the neonatal period, suggesting that they are inherited analogous to prototypical extremes, such as in patients with Marfan syndrome or BAV.1,35 Others concluded that aortic wall abnormalities develop because of structural differences in wall composition between the two great arteries; the former pulmonary arterial wall is exposed to higher systemic pressures after the ASO, posing increased stress on the neoaortic wall, which may ultimately lead to changes in structure and function of the neoaortic root.39,40 Whether medial abnormalities are inherent or acquired remains therefore difficult to distinguish.1
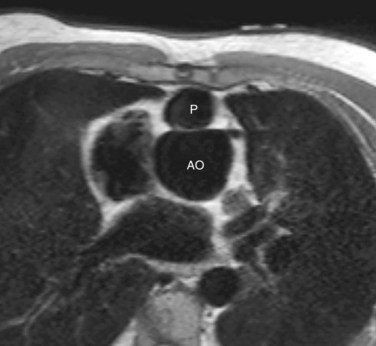
 FIGURE 40-7
FIGURE 40-7A high incidence of aortic regurgitation has been reported after ASO (30% at 6 years after ASO) and is probably the result of a multifactorial process for which aortic root geometry, surgical techniques, and preoperative size discrepancy between the two great arteries are involved.36,41 In addition, aortic regurgitation appears to be functionally correlated with aortic root dilation and reduced elasticity of the proximal aorta.37 Whether the previous existence of a ventricular septal defect plays an additional role remains controversial, although its hemodynamic effect might contribute to a size discrepancy between the aorta and pulmonary artery.35
An MRI report described not only frequent aortic root dilation and aortic regurgitation but also a cascade of events ultimately leading to LV systolic dysfunction in patients after ASO.37 Frequent aortic root dilation and reduced proximal aortic wall elasticity were associated with degree of aortic regurgitation. Aortic regurgitation subsequently led to increased LV dimensions, which consequently resulted in decreased LV ejection fraction. Therefore, LV systolic dysfunction as the endpoint in a sequence of events poses a prognostic risk for patients after ASO.37 Further elucidation of the underlying pathogenic substrate of aortic wall abnormalities and its clinical repercussions for patients after ASO is required, however, because ASO is still a relatively new surgical procedure for patients with transposition of the great arteries.37
IMAGING TECHNIQUE AND FINDINGS
Magnetic Resonance Imaging of the Aorta: Techniques and Protocol
An overview of a typical imaging protocol is listed in Table 40-1. The focus of the protocol is on depiction of aortic dimensions and the vessel wall and assessment of aortic elasticity, aortic valve competence, and systolic LV function.
| No. | Sequence Type | Scan Duration (minutes) |
|---|---|---|
| 1 | Scout images single-phase SSFP | 1 |
| 2 | Parallel imaging reference scan | 1 |
| 3 | Two-chamber LV multiphase SSFP (BH) | 1 |
| 4 | Four-chamber multiphase SSFP (BH) | 1 |
| 5 | Axial black blood TSE (BHs) | 5-10 |
| 6 | Oblique sagittal black blood along the aorta TSE (BHs) | 5-10 |
| 7 | Multislice, multiphase short-axis SSFP (BHs) | 6 |
| 8 | Oblique sagittal single-slice scout of the aorta for PWV (BH) | 1 |
| 9 | Flow mapping in ascending aorta and proximal descending aorta for PWV (FB) | 4 |
| 10 | Flow mapping in descending aorta for PWV (FB) | 4 |
| 11 | Flow mapping in descending aorta at level of diaphragm (FB), optional | 4 |
| 12 | Two sets of scout cine images of the aortic root (orthogonal) SSFP (BHs) | 2 |
| 13 | Minimal and maximal lumen area measurements at sinotubular junction SSFP (BHs) | 4 |
| 14 | Timing MRA aorta | 4 |
| 15 | MRA aorta (FB) | 7 |
| Total scan duration time | 50-60 minutes |
BH, breath-hold; FB, free breathing; MRA, magnetic resonance angiography; PWV, pulse wave velocity; SSFP, steady-state free precession sequence; TSE, turbo spin-echo.
Aortic Anatomy and Dimensions
An axial stack of black blood turbo spin-echo images is acquired to generally outline cardiac and noncardiac anatomy. Aortic root dimensions can be assessed by double oblique transverse images perpendicular to the aorta, at the levels of the annulus of the aortic valve, the sinus of Valsalva, the sinotubular junction, and the ascending aorta at the crossing of the right pulmonary artery (Fig. 40-8). Oblique black blood turbo spin-echo images along the course of the aorta (see Figs. 40-2, 40-5, and 40-7) and contrast-enhanced three-dimensional MR angiography may be used to visualize aortic dimensions and the potentially complex nature of any coarctation lesion (see Figs. 40-3, 40-4, and 40-6) as well as for diagnosis and follow-up of aortic complications, such as aortic dissection, intramural hematoma, and aortic aneurysms.42–45 Reference values of aortic dimensions are unfortunately scarce, especially for children, making individual follow-up even more valuable for depiction of aortic dilation and rate of progression.43,46
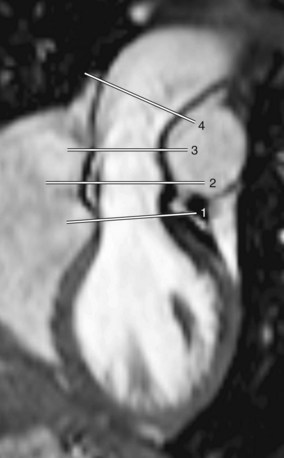
 FIGURE 40-8
FIGURE 40-8Aortic Elasticity
Intrinsic aortic wall abnormalities and progressive atherosclerotic disease are associated with increased aortic stiffness, which is represented by locally reduced aortic distensibility and increased regional pulse wave velocity.45–48 Distensibility (mm Hg−1) of the aortic root can be measured at the level of the sinotubular junction, for which two separate images are acquired for the maximal and minimal aortic lumen area, measured at the peak of aortic systolic flow and before the systolic flow curve (coinciding with the isovolumetric contraction phase), respectively (Fig. 40-9).47 Pulse wave velocity can be measured for the aortic arch and the descending aorta,45,46 for which velocity-encoded MRI is applied at the levels of the ascending aorta and proximal descending aorta and just above the bifurcation of the abdominal aorta (Fig. 40-10).45,46 The addition of velocity-encoded MRI at the level of the diaphragm can be used to quantify collateral flow often present in patients with coarctation.49
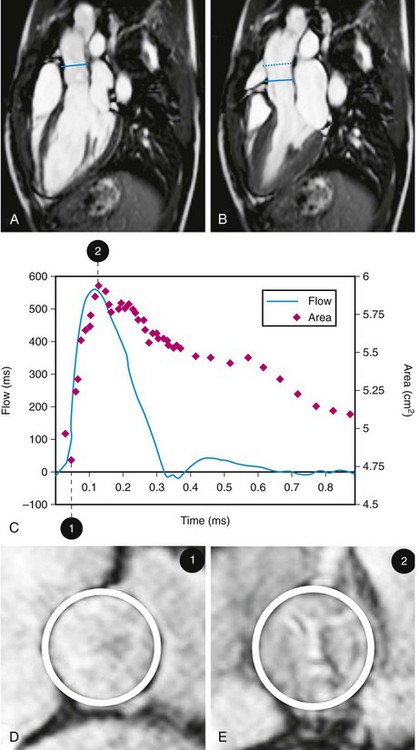
 FIGURE 40-9
FIGURE 40-9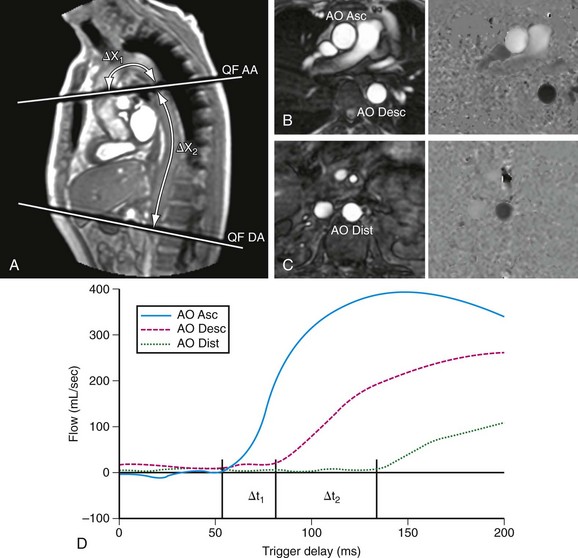
 FIGURE 40-10
FIGURE 40-10Aortic Valve and Left Ventricular Function
Quantification of aortic flow with velocity-encoded MRI allows accurate calculation of aortic valve regurgitation fraction (as percentage from aortic forward flow).47 Velocity-encoded MRI allows peak flow velocity assessment across stenotic lesions, such as aortic valve stenosis and coarctation, and thus an estimate of peak gradients by the simplified Bernoulli equation.43 Systolic LV function can be assessed by a steady-state free precession cine sequence in the short-axis or axial orientation.50 The end-diastolic volume (EDV) and end-systolic volume (ESV) are calculated after the endocardial contours are manually drawn at end-diastole and end-systole, respectively; epicardial borders may additionally be drawn for determination of LV mass.50 Ejection fraction is calculated by dividing stroke volume (EDV − ESV) by EDV.




Fedak PW, David TE, Borger M, et al. Bicuspid aortic valve disease: recent insights in pathophysiology and treatment. Expert Rev Cardiovasc Ther. 2005;3:295-308.
Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005:111-e150.
Niwa K, Perloff JK, Bhuta SM, et al. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation. 2001;103:393-400.
Nollen GJ, Mulder BJ. What is new in the Marfan syndrome? Int J Cardiol. 2004;97(Suppl 1):103-108.
Pemberton J, Sahn DJ. Imaging of the aorta. Int J Cardiol. 2004;97(Suppl 1):53-60.
Rosenthal E. Coarctation of the aorta from fetus to adult: curable condition or lifelong disease process? Heart. 2005;91:1495-1502.
Tan JL, Gatzoulis MA, Ho SY. Aortic root disease in tetralogy of Fallot. Curr Opin Cardiol. 2006;21:569-572.
Therrien J, Thorne SA, Wright A, et al. Repaired coarctation: a “cost-effective” approach to identify complications in adults. J Am Coll Cardiol. 2000;35:997-1002.
Ward C. Clinical significance of the bicuspid aortic valve. Heart. 2000;83:81-85.
Warnes CA. Transposition of the great arteries. Circulation. 2006;114:2699-2709.
1 Niwa K, Perloff JK, Bhuta SM, et al. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation. 2001;103:393-400.
2 Fedak PW, Verma S, David TE, et al. Clinical and pathophysiological implications of a bicuspid aortic valve. Circulation. 2002;106:900-904.
3 Schlatmann TJ, Becker AE. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm. Am J Cardiol. 1977;39:13-20.
4 Pemberton J, Sahn DJ. Imaging of the aorta. Int J Cardiol. 2004;97(Suppl 1):53-60.
5 Grotenhuis HB, Westenberg JJ, Doornbos J, et al. Aortic root dysfunctioning and its effect on left ventricular function in Ross procedure patients assessed with magnetic resonance imaging. Am Heart J. 2006;152:975-978.
6 Schmidtke C, Bechtel J, Hueppe M, et al. Size and distensibility of the aortic root and aortic valve function after different techniques of the Ross procedure. J Thorac Cardiovasc Surg. 2000;119:990-997.
7 Kim GB, Kang SJ, Bae EJ, et al. Elastic properties of the ascending aorta in young children after successful coarctoplasty in infancy. Int J Cardiol. 2004;97:471-477.
8 Arnett DK, Evans GW, Riley WA. Arterial stiffness: a new cardiovascular risk factor? Am J Epidemiol. 1994;140:669-682.
9 Mohrs OK, Petersen SE, Voigtlaender T, et al. Time-resolved contrast-enhanced MR angiography of the thorax in adults with congenital heart disease. AJR Am J Roentgenol. 2006;187:1107-1114.
10 van der Geest RJ, Reiber JH. Quantification in cardiac MRI. J Magn Reson Imaging. 1999;10:602-608.
11 Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005:111-e150.
12 Nollen GJ, Groenink M, Tijssen JG, et al. Aortic stiffness and diameter predict progressive aortic dilatation in patients with Marfan syndrome. Eur Heart J. 2004;25:1146-1152.
13 Nollen GJ, Mulder BJ. What is new in the Marfan syndrome? Int J Cardiol. 2004;97(Suppl 1)):103-108.
14 Fedak PW, David TE, Borger M, et al. Bicuspid aortic valve disease: recent insights in pathophysiology and treatment. Expert Rev Cardiovasc Ther. 2005;3:295-308.
15 Warren AE, Boyd ML, O’Connell C, Dodds L. Dilation of the ascending aorta in pediatric patients with bicuspid aortic valve: frequency, rate of progression and risk factors. Heart. 2006;92:1496-1500.
16 Della CA, Bancone C, Quarto C, et al. Predictors of ascending aortic dilatation with bicuspid aortic valve: a wide spectrum of disease expression. Eur J Cardiothorac Surg. 2007;31:397-404.
17 Ward C. Clinical significance of the bicuspid aortic valve. Heart. 2000;83:81-85.
18 Grotenhuis HB, Ottenkamp J, Westenberg JJ, et al. Reduced aortic elasticity and dilatation are associated with aortic regurgitation and left ventricular hypertrophy in nonstenotic bicuspid aortic valve patients. J Am Coll Cardiol. 2007;49:1660-1665.
19 Rosenthal E. Coarctation of the aorta from fetus to adult: curable condition or lifelong disease process? Heart. 2005;91:1495-1502.
20 Meyer AA, Joharchi MS, Kundt G, et al. Predicting the risk of early atherosclerotic disease development in children after repair of aortic coarctation. Eur Heart J. 2005;26:617-622.
21 Nielsen JC, Powell AJ, Gauvreau K, et al. Magnetic resonance imaging predictors of coarctation severity. Circulation. 2005;111:622-628.
22 Vogt M, Kuhn A, Baumgartner D, et al. Impaired elastic properties of the ascending aorta in newborns before and early after successful coarctation repair: proof of a systemic vascular disease of the prestenotic arteries? Circulation. 2005;111:3269-3273.
23 Ong CM, Canter CE, Gutierrez FR, et al. Increased stiffness and persistent narrowing of the aorta after successful repair of coarctation of the aorta: relationship to left ventricular mass and blood pressure at rest and with exercise. Am Heart J. 1992;123:1594-1600.
24 Krogmann ON, Rammos S, Jakob M, et al. Left ventricular diastolic dysfunction late after coarctation repair in childhood: influence of left ventricular hypertrophy. J Am Coll Cardiol. 1993;21:1454-1460.
25 Therrien J, Thorne SA, Wright A, et al. Repaired coarctation: a “cost-effective” approach to identify complications in adults. J Am Coll Cardiol. 2000;35:997-1002.
26 Araoz PA, Reddy GP, Tarnoff H, et al. MR findings of collateral circulation are more accurate measures of hemodynamic significance than arm-leg blood pressure gradient after repair of coarctation of the aorta. J Magn Reson Imaging. 2003;17:177-183.
27 Krueger JJ, Ewert P, Yilmaz S, et al. Magnetic resonance imaging–guided balloon angioplasty of coarctation of the aorta: a pilot study. Circulation. 2006;113:1093-1100.
28 Tan JL, Davlouros PA, McCarthy KP, et al. Intrinsic histological abnormalities of aortic root and ascending aorta in tetralogy of Fallot: evidence of causative mechanism for aortic dilatation and aortopathy. Circulation. 2005;112:961-968.
29 Cheung YF, Ou X, Wong SJ. Central and peripheral arterial stiffness in patients after surgical repair of tetralogy of Fallot: implications for aortic root dilatation. Heart. 2006;92:1827-1830.
30 Niwa K. Aortic root dilatation in tetralogy of Fallot long-term after repair—histology of the aorta in tetralogy of Fallot: evidence of intrinsic aortopathy. Int J Cardiol. 2005;103:117-119.
31 Niwa K, Siu SC, Webb GD, Gatzoulis MA. Progressive aortic root dilatation in adults late after repair of tetralogy of Fallot. Circulation. 2002;106:1374-1378.
32 Rathi VK, Doyle M, Williams RB, et al. Massive aortic aneurysm and dissection in repaired tetralogy of Fallot; diagnosis by cardiovascular magnetic resonance imaging. Int J Cardiol. 2005;101:169-170.
33 Therrien J, Gatzoulis M, Graham T, et al. Canadian Cardiovascular Society Consensus Conference 2001 update: Recommendations for the Management of Adults with Congenital Heart Disease—Part II. Can J Cardiol. 2001;17:1029-1050.
34 Warnes CA. Transposition of the great arteries. Circulation. 2006;114:2699-2709.
35 Hwang HY, Kim WH, Kwak JG, et al. Mid-term follow-up of neoaortic regurgitation after the arterial switch operation for transposition of the great arteries. Eur J Cardiothorac Surg. 2006;29:162-167.
36 Formigari R, Toscano A, Giardini A, et al. Prevalence and predictors of neoaortic regurgitation after arterial switch operation for transposition of the great arteries. J Thorac Cardiovasc Surg. 2003;126:1753-1759.
37 Grotenhuis HB, Ottenkamp J, Fontein D, et al. Aortic elasticity and left ventricular function after arterial switch operation: MR imaging—initial experience. Radiology. 2008;249:801-809.
38 Murakami T, Nakazawa M, Momma K, Imai Y. Impaired distensibility of neoaorta after arterial switch procedure. Ann Thorac Surg. 2000;70:1907-1910.
39 Hourihan M, Colan SD, Wernovsky G, et al. Growth of the aortic anastomosis, annulus, and root after the arterial switch procedure performed in infancy. Circulation. 1993;88:615-620.
40 Schoof PH, Gittenberger-de Groot AC, de Heer E, et al. Remodeling of the porcine pulmonary autograft wall in the aortic position. J Thorac Cardiovasc Surg. 2000;120:55-65.
41 Haas F, Wottke M, Poppert H, Meisner H. Long-term survival and functional follow-up in patients after the arterial switch operation. Ann Thorac Surg. 1999;68:1692-1697.
42 Mohrs OK, Petersen SE, Voigtlaender T, et al. Time-resolved contrast-enhanced MR angiography of the thorax in adults with congenital heart disease. AJR Am J Roentgenol. 2006;187:1107-1114.
43 Pemberton J, Sahn DJ. Imaging of the aorta. Int J Cardiol. 2004;97(Suppl 1):53-60.
44 Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005:111-e150.
45 Nollen GJ, Groenink M, Tijssen JG, et al. Aortic stiffness and diameter predict progressive aortic dilatation in patients with Marfan syndrome. Eur Heart J. 2004;25:1146-1152.
46 Grotenhuis HB, Ottenkamp J, Westenberg JJ, et al. Reduced aortic elasticity and dilatation are associated with aortic regurgitation and left ventricular hypertrophy in nonstenotic bicuspid aortic valve patients. J Am Coll Cardiol. 2007;49:1660-1665.
47 Grotenhuis HB, Westenberg JJ, Doornbos J, et al. Aortic root dysfunctioning and its effect on left ventricular function in Ross procedure patients assessed with magnetic resonance imaging. Am Heart J. 2006;152:975-978.
48 Arnett DK, Evans GW, Riley WA. Arterial stiffness: a new cardiovascular risk factor? Am J Epidemiol. 1994;140:669-682.
49 Araoz PA, Reddy GP, Tarnoff H, et al. MR findings of collateral circulation are more accurate measures of hemodynamic significance than arm-leg blood pressure gradient after repair of coarctation of the aorta. J Magn Reson Imaging. 2003;17:177-183.
50 van der Geest RJ, Reiber JH. Quantification in cardiac MRI. J Magn Reson Imaging. 1999;10:602-608.
