[level-membership-for-neurosurgery-category]
CHAPTER 121 Low-Grade Gliomas
Astrocytoma, Oligodendroglioma, and Mixed Glioma
Epidemiology
Glial tumors constitute about half of newly diagnosed primary brain tumors, with low-grade gliomas (LGGs) accounting for about 15% of all brain tumors in adults.1 The subset of tumors classified as LGG is a heterogeneous group of tumors with astrocytic, oligodendroglial, ependymal, or mixed cellular histology. In adults, the term LGG typically refers to the diffuse, infiltrating variety of tumors classified as World Health Organization (WHO) grade II lesions—specifically, low-grade astrocytomas, oligodendrogliomas, or mixed oligoastrocytomas.2 Among low-grade astrocytomas, the most common histologic subtypes are the fibrillary, protoplasmic, and gemistocytic variants. There is no indication in the literature that LGGs are more prevalent in a specific ethnic or national group.
About 1500 new cases of LGG are diagnosed in North America each year.3 Age-specific data show that low-grade astrocytomas constitute 15% of brain tumors in adults and 25% of brain tumors in children.1 Pediatric LGGs, which include cerebellar astrocytomas, optic pathway and hypothalamic gliomas, brainstem gliomas, and hemispheric LGGs, are discussed elsewhere in this book. These tumors demonstrate a slight male predominance and a biphasic age distribution, with the first peak occurring during childhood (age 6 to 12 years) and a second peak in adulthood (between the third and fifth decades). The median age of presentation in adults is 35 years.
Clinical Presentation
LGGs typically arise in the frontal lobes, followed by temporal and parietal lobe lesions, in order of decreasing incidence. Fifty percent to 80% of patients present with seizures as their initial symptom, with most remaining otherwise neurologically intact.4 Patients may present with or develop other signs and symptoms, which are largely dictated by the tumor’s size and location. These include signs and symptoms of raised intracranial pressure (headache, nausea, vomiting, lethargy, papilledema), focal neurological deficits (weakness, sensory disturbance or neglect, visual neglect, agnosia, aphasia), and impaired executive function (altered personality, disinhibition, apathy).
In some studies, seizures have accompanied presentation of LGG in up to 81% of patients.5 Of the patients who presented with seizures, about 50% have uncontrolled seizures at the time of resection despite antiepileptic treatment. Partial seizure type, temporal location, and longer seizure duration also appear to predispose patients to poorer preoperative seizure control.5 Careful consideration of a patient’s seizure status is of paramount importance for LGG patients because seizures significantly affect quality of life. Beyond antiepileptic agents, surgical resection is an effective means of reducing seizure burden on patients with LGGs. Postoperatively, the factors associated with freedom from seizures are gross total tumor resection, preoperative seizure history of less than 1 year, and non–simple partial seizure type. However, continued use of antiepileptic drugs can be necessary, and in some patients, an additional operation may be required for persistent seizure activity. In our experience, optimal control of intractable epilepsy without the use of postoperative anticonvulsants is possible when perioperative (i.e., extraoperative or intraoperative) electrocorticographic mapping of separate seizure foci accompanies the tumor resection. In most cases of epilepsy, as in patients with occasional breakthrough seizures, such mapping is not needed, but complete tumor resection is. When mapping is not used and radical tumor resection (including adjacent brain) is carried out, seizures occur less frequently, but most patients continue to take antiepileptic drugs.6
Conventional Neuroimaging Studies
The typical computed tomography (CT) appearance is one of an either discrete or diffuse hypodense to isodense mass lesion, showing minimal or no enhancement with intravenous contrast administration. In about 15% to 30% of patients, however, tumor enhancement can be discerned.7,8 Calcifications may also occur, particularly among oligodendrogliomas or mixed oligoastrocytomas. In addition, cystic changes may be seen with any histologic subtype.
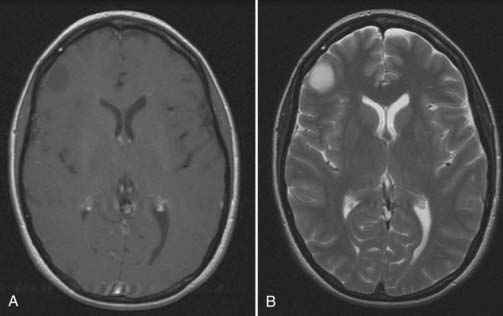
Magnetic resonance imaging (MRI) is the diagnostic procedure of choice for LGG, delineating the lesion as hypointense to isointense on T1-weighted images and as hyperintense on T2-weighted images (Fig. 121-1). Similar to their appearance on CT scans, most LGGs do not show gadolinium enhancement on MRI. LGGs are intra-axial lesions but do not typically exert significant mass effect on surrounding structures. They do, however, display a tendency to reside within and extend along white matter tracts (e.g., corpus callosum, subcortical white matter). Neuroimaging is not diagnostic but may suggest a particular pathologic subtype of LGG by virtue of the tumor’s location and imaging characteristics. Oligodendrogliomas, for example, are frequently located within the frontal lobes, involve the cortex, and display calcifications, in contrast to other LGGs. Importantly, T1-weighted MRI with gadolinium contrast may underestimate the extent of an LGG. The true extent is shown on the T2-weighted sequences, although on these sequences, tumor extent and surrounding edema are indistinguishable. More recently, diffusion tensor MRI has been used as a surrogate marker of glioma infiltration.9,10
Emerging Neuroimaging Technologies
Magnetic Resonance Imaging
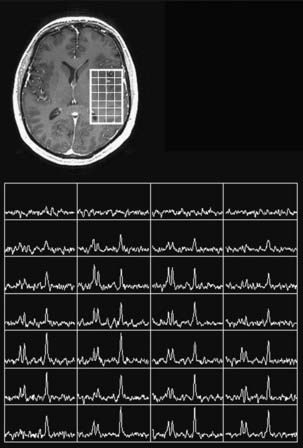
Continued improvement in the resolution of anatomic imaging and innovations in functional and physiologic imaging modalities have the potential to improve our ability to diagnose, treat, follow up, and predict outcome in LGG patients. The increasing use of 3-tesla (3-T) MRI (instead of the standard 1.5-T magnet) will provide more anatomic detail and exquisite cytoarchitectural data on intracranial lesions.11,12 Proton magnetic resonance spectroscopy (MRS) allows for the noninvasive assessment of metabolite levels within intracranial lesions (Fig. 121-2). Of particular interest are the metabolites N-acetyl aspartate (NAA), choline (Cho), creatine (Cr), and lipids. In contrast to normal brain, gliomas typically demonstrate a decrease in NAA and Cr levels and a rise in Cho levels, indicative of their proliferative potential, cellular heterogeneity, and high cell turnover. In general, higher grade lesions display higher Cho/NAA and Cho/Cr ratios than do lower-grade tumors. The utility and reliability of MRS in predicting tumor grade noninvasively is being evaluated,13–15 but it does not supplant the need for tissue diagnosis. Nevertheless, MRS may facilitate the identification of targets for surgical biopsy, focusing our attention on regions with elevated Cho peaks, suggestive of increased cellular proliferation, and thereby on regions of maximal tumor activity. In addition, MRS has proved useful in monitoring LGG patients after radiotherapy because it can often distinguish between tumor recurrence and radiation necrosis.
Magnetic resonance techniques have also been developed for assessment of cerebral blood volume (CBV). A 2- to 3-minute dynamic acquisition of T2-weighted images during intravenous injection of a bolus of gadolinium–diethylenetriamine pentaacetic acid (DTPA) allows estimations of CBV. A voxel-by-voxel CBV map can be created by integrating the area under the dynamic contrast uptake curve and provides a relative measure of CBV with a spatial resolution of about 1 × 2 × 5 mm3 or better. Magnetic resonance perfusion has already demonstrated utility in predicting histopathologic diagnosis and tumor grade noninvasively16,17 (Fig. 121-3) and will probably play a role in selecting biopsy locations, evaluating treatment response, and differentiating effects of treatment from those produced by recurrent tumor effects.
Positron Emission Tomography
Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are additional functional and metabolic imaging modalities that contribute to the diagnosis and management of LGG patients. These modalities supplement the characterization of tumor grade because LGGs are typically hypometabolic compared with high-grade lesions. Preoperatively, these techniques are also used to identify motor- or language-related regions of cortical function, but with limited specificity. In addition to predicting tumor grade and helping with preoperative planning, indications for the use of PET or SPECT in LGG include follow-up monitoring of patients for evidence of tumor recurrence or dedifferentiation.18
Magnetoencephalography
MEG has also been increasingly used for preoperative functional mapping. Compared with fMRI and PET, MEG has the advantage of higher temporal resolution by directly measuring neuronal activation rather than an indirect hemodynamic change. Previous studies have also suggested that MEG is more accurate than fMRI in identifying functional cortices that have been distorted by a nearby tumor. Overall, MEG is a robust and reliable functional imaging modality that is now used to identify the cortical location of motor and sensory pathways. Integrating MEG data with diffusion tensor imaging (DTI) information in a neuronavigational workstation directs the neurosurgeon toward potential functional brain sites that can be intraoperatively confirmed using stimulation mapping. Magnetic source imaging (MSI), which is based on the magnetoencephalographic detection of the late neuromagnetic field elicited by simple speech sounds,19 is another adjunct to mapping techniques that can be useful for mapping the somatosensory cortex and determining hemispheric dominance, and it may serve as a replacement to the Wada test.
Patient Outcome and Survival
The median overall survival time for LGG patients is about 6.5 to 8 years.20,21 Published survival estimates for patients diagnosed with LGG range from 3 to more than 20 years.7,20,22–28 Overall, 5- and 10-year survival rates of about 70% and 50%, respectively, have been reported.29 Interestingly, the clinical course of individual LGGs can demonstrate substantial heterogeneity, with certain lesions tending to behave more aggressively, whereas others follow a more indolent course. This diversity of clinical behavior is matched by the anatomic and histopathologic diversity inherent in LGGs. Not surprisingly, this contributes to the controversy among experts regarding the most appropriate strategy for treating patients with LGG. However, Smith and colleagues30 recently demonstrated that after adjusting for the effects of age, Karnofsky Performance Scale (KPS) score, tumor location, and tumor subtype, extent of resection was a significant predictor of overall survival and showed a trend toward predicting progression-free survival. This volumetric extent of resection analysis revealed that patients with greater than or equal to 90% resection had an 8-year overall survival rate of 91% and a progression-free survival rate of 43%, whereas patients with less than 90% resection had an 8-year overall survival rate of 60% and a progression-free survival rate of 21%.
Prognostic Factors
Clinical characteristics associated with improved LGG patients’ survival outcomes include age younger than 40 years at diagnosis, seizures present at diagnosis, the absence of additional neurological deficits at diagnosis, KPS score of no less than 70, and Folstein Mini-Mental Status Examination (MMSE) score greater than 26/30.20,26,27,29,31–34 Imaging factors predictive of poor survival include maximum tumor diameter greater than 5 to 6 cm and the presence of contrast enhancement.20,27 Increasingly, the extent of surgical resection has been found to be a significant predictor of outcome and progression-free survival among LGG patients (see “Surgical Resection” section under “Treatment Options”). Furthermore, histopathologic factors associated with better prognosis include an MIB-1 labeling index of less than 8% and a histologic diagnosis of either low-grade oligodendroglioma or oligoastrocytoma (especially if the tumor is harboring chromosome 1p deletions, an indication of chemosensitivity).26,27,35
Dedifferentiation or malignant transformation is a well-described phenomenon observed in LGGs. In the literature, 13% to 86% of tumors initially diagnosed as low grade were observed to recur at a higher histologic grade.4,24,33,36–40 Similar to its broad range of incidence, the time to malignant differentiation is also variable, ranging from 28 to 60 months.4,37,40–42 However, the factors resulting in the transformation to a malignant phenotype are unclear, and the effect of treatment on this malignant transformation remains controversial. In one series, 58% of patients who did not initially undergo biopsy and treatment of a tumor suspected to be an LGG after diagnostic imaging studies eventually required surgery at a median interval of 29 months, and 50% of the tumors then showed anaplastic features.41 Although these patients had a higher incidence of malignant transformation at the time of operation and shorter time to tumor progression relative to patients who were operated on initially, the study concluded that no difference was observed in overall survival. Nevertheless, it is likely that the timing of malignant transformation affects patient outcome, and this phenomenon may be detected by future studies that are more robust. Furthermore, extent of resection studies suggest that the natural history of malignant transformation can be altered by greater extent of resection.30
Genetic Expression Profile
The cause of LGG is unknown, and with the exception of patients with one of the phakomatoses, there is no defined genetic predisposition that leads to the development of these tumors. The only genetic alteration consistently observed in patients with low-grade astrocytomas is mutation of the p53 gene.43 The p53 gene is located on chromosome 17 at 17p13.1, and this site is often deleted in astrocytomas of all grades. The remaining copy of p53 is usually inactivated through a subtle mutation. Because this gene is essential in the regulation of apoptosis and cell cycle progression, loss of normal p53 function promotes the accelerated growth and malignant differentiation of astrocytes.44,45 Astrocytomas are the only brain tumors displaying significant p53 mutation rates. Fifty percent to 60% of grade 2 and grade 3 astrocytomas exhibit p53 mutations, suggesting that inactivation of this tumor suppressor gene is an early lesion among gene alterations associated with the development of malignant gliomas.46 Although some glioblastomas exhibit p53 mutations, a significant subset of them do not and instead have amplification of the epidermal growth factor receptor (EGFR) gene, suggesting that this subset arises from a different genetic pathway. Other common alterations observed in adult low-grade astrocytomas are gain of chromosome 7 and structural abnormalities, including possessing double-minute chromosomes. Losses of chromosomes 10, 13, 15, 20, and 22 and structural rearrangements involving chromosomes 4, 11, 12, 13, 16, 18, and 21 have also been reported in patients.47
Although p53 is only rarely mutated in oligodendrogliomas, more than half of these tumors show a characteristic loss of the long arm of chromosome 1 and the short arm of chromosome 19. Because loss of 1p and 19q is not seen in astrocytic tumors (with rare exception), analysis of the presence or absence of p53, 1p, and 19q can be used to distinguish an astrocytic from an oligodendroglial genotype in cases that are difficult to distinguish histologically. Similarly, most mixed oligoastrocytomas appear to segregate genetically into astrocytic or oligodendroglial genotypes, suggesting that such mixed tumors may not be a distinct biologic entity.48
Treatment Options
Observation
Little evidence exists to support this treatment strategy, although it has not been refuted, either. In one small, retrospective case-controlled study, no difference was observed in rates of malignant transformation, overall survival, or quality of life between patients initially only observed and those undergoing immediate resection.41 Similarly, in 30 patients presenting only with seizure, early versus late surgical resection did not affect overall survival.49 Importantly, the small cohort number (<50) in both studies limits our ability to extrapolate these findings to a broader patient population.
Surgical Intervention
Operative strategies for patients with LGG include open surgical resection and open or stereotactic biopsy. The choice depends in part on the patient’s clinical status, the anatomic location of the tumor, and the surgeon’s preference. Goals of surgical intervention include establishing a diagnosis, treating neurological symptoms, decompressing mass effect, and tumor cytoreduction. Currently, the only agreed-on surgical standard for adults with suspected or known supratentorial non–optic-pathway LGGs is to obtain a tissue diagnosis before active treatment commences.50
Biopsy
Stereotactic or image-guided biopsy can acquire tissue for histologic diagnosis in a minimally invasive fashion. This is particularly suitable for patients in whom open surgical resection is declined, deferred, or carries unacceptably high risks. The advantage of performing an early biopsy is that it allows for the identification of patients harboring more aggressive lesions, for which a course of observation alone may be inappropriate.51 Additionally, the tissue can be analyzed for oligodendroglial characteristics, such as chromosome 1p loss.
In general, reported surgical risks associated with stereotactic biopsy in LGG patients have been low, with morbidity and mortality rates of less than 1%.52 Mortality occurs as a result of intracranial hemorrhage, subarachnoid hemorrhage, or uncontrollable cerebral edema, although this generally has been observed only among biopsies of high-grade lesions.53
One pitfall of relying on stereotactic biopsy for tissue diagnosis is the possibility of misdiagnosis or inaccurate tumor grading owing to tumor heterogeneity and diagnosis bias resulting from limited tumor sampling. The concordance between biopsy and open resection specimens is lower in patients with larger tumors,54 suggesting that multiple biopsies, which can be collected in a single trajectory pass, may be useful in this subpopulation.
Surgical Resection
In the subset of patients with accessible LGG who suffer from symptoms of local mass effect, increased intracranial pressure, and intractable seizures, the role for open surgical resection is well established. Resection serves several purposes in these circumstances, including alleviation of mass effect, cytoreduction, and providing tissue for diagnosis. Cytoreduction can also reduce cerebral edema and potentially improve radiosensitivity and chemosensitivity. The degree of tumor removal afforded by open surgical resection also offers the advantage of providing more tissue for histologic analysis, increasing the accuracy of pathologic diagnosis. Theoretically, cytoreduction also reduces the number of tumor cells at risk for accumulating additional genetic aberrations, thereby reducing the risk for tumor progression and decreasing the potential for malignant transformation.30
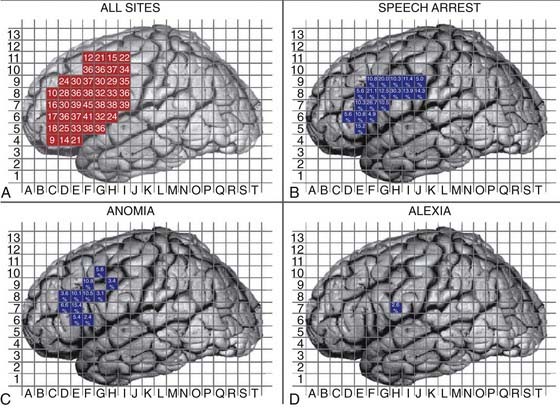
Open surgical interventions for the treatment of LGG are conducted using general neurosurgical principles of tumor surgery. Contemporary neurosurgical methods, including ultrasonography, functional mapping, frameless navigational resection devices, and intraoperative imaging techniques enable the neurosurgeon to achieve more extensive resections with less morbidity. Intraoperative ultrasonography provides real-time intraoperative data and is helpful in detecting the tumor, delineating its margins, and differentiating tumor from peritumoral edema, cyst, necrosis, and adjacent normal brain tissue. Although its use is limited by artifact from blood and surgical trauma at the margin of resection, postresection tumor volumes based on intraoperative ultrasonography significantly correlate with those determined by postoperative MRI.55 Similarly, intraoperative MRI may allow for greater extent of resection, particularly when tumor-infiltrated tissue cannot be grossly distinguished from normal tissue.56 Stimulation mapping techniques are essential to minimize morbidity and to achieve radical resections of tumors located in or around cortical and subcortical functionally eloquent sites (see Fig. 121-3).57 For lesions in and around language pathways, awake mapping remains the gold standard for minimizing morbidity and maximizing extent of resection. Intraoperative corticography can also be a useful adjunct, but it is primarily reserved for patients with intractable epilepsy.
Despite the stated benefits for surgical resection, the role for surgery among LGG patients who are minimally symptomatic or asymptomatic remains somewhat controversial. Historically, this has been due, in part, to conflicting reports regarding whether the extent of resection actually confers any survival advantage for these patients. More recently, however, there is a growing body of evidence suggesting that more extensive resection at the time of initial diagnosis is a favorable prognostic factor (Fig. 121-4). Although most reports are retrospective, it is unlikely that the necessary prospective randomized studies will be conducted to address the role of extent of resection on outcome in LGG patients owing to the relatively limited numbers of patients, the typically long survival times, and a general lack of equipoise with regard to treatment options among care providers.
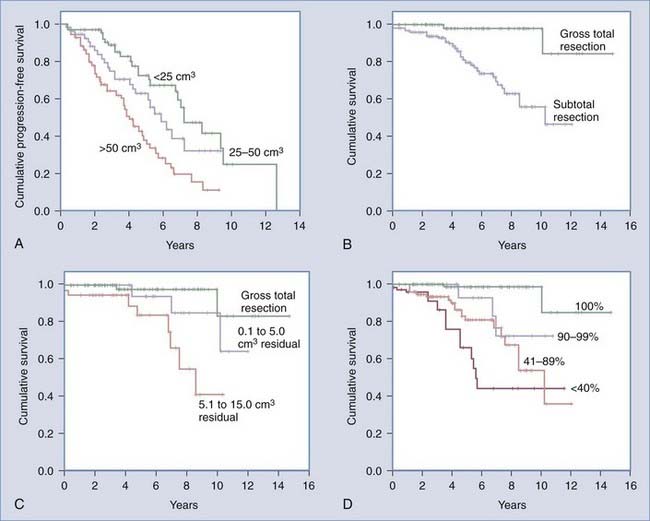
In the modern neurosurgical era, a number of studies have applied statistical analysis to examine the efficacy of extent of resection in improving survival and delaying tumor progression among LGG patients (Table 121-1).21,29,30,49,56,58–68 Five of these studies included volumetric analysis of extent of resection (Table 121-2).23,30,49,56,66 Of the studies not employing volumetric methods, 12 demonstrated evidence supporting extent of resection as a statistically significant predictor of either 5-year survival or 5-year progression-free survival. These studies were published from 1990 to 2005, and most commonly employed a combination of multivariate and univariate analyses to determine statistical significance. In most instances, extent of resection was defined in terms of gross total and subtotal resection. However, in a recent volumetric LGG extent of resection analysis, Smith and colleagues demonstrated that a more aggressive resection does predict significant improvement in overall survival compared with a simple debulking procedure.30 Interestingly, predicted overall survival was shown to be negatively affected by residual tumor volumes as small as 10 cm3 (see Fig. 121-4).
TABLE 121-1 Nonvolumetric Low-Grade Glioma Extent of Resection Studies in the Modern Neurosurgical Literature
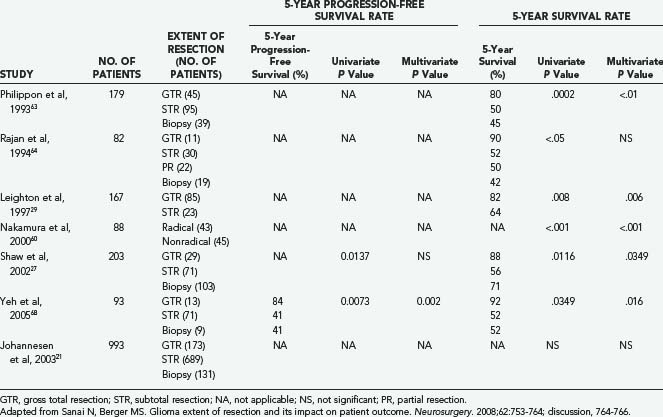
TABLE 121-2 Volumetric Low-Grade Glioma Extent of Resection Studies in the Modern Neurosurgical Literature
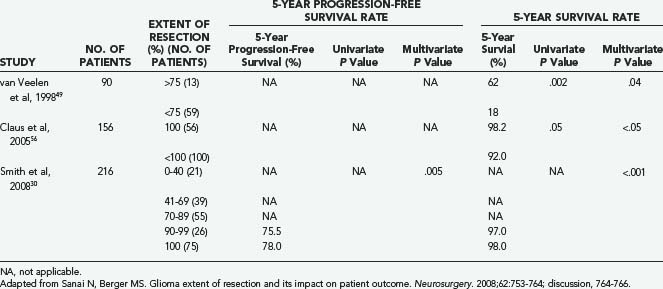
In light of this evidence, and in an effort to estimate the resectability of LGG, Chang and associates69 generated a preoperative scoring system for the long-term prognostication of outcome in patients with hemispheric LGGs. Four variables (tumor location in eloquent brain, age older than 50 years, KPS score of no more than 80, and tumor maximum diameter greater than 4 cm) were predictive of survival on multivariate analyses and were therefore used for the scoring system, with the total score being inversely proportional to predicted survival.
Thus, mounting evidence in the modern neurosurgical literature suggests that a more extensive surgical resection may be associated with a more favorable life expectancy for LGG patients.70 More aggressive resections for low-grade gliomas also affect the risk for malignant transformation,30 and they take advantage of an opportunity to treat the disease when the neoplasm is at its earliest stage of evolution.
Radiotherapy
The European Organization for Research and Treatment of Cancer (EORTC) published the first prospective, randomized clinical trial (EORTC 22844) addressing whether LGG exhibited a dose response to radiotherapy.23 In this study, 379 adult patients with LGG were randomized to receive either a low-dose regimen of 45 Gy over a 5-week period or a high-dose regimen of 59.4 Gy over 6.6 weeks, after either open surgical resection or biopsy. After a median follow-up of 74 months, patients in the low- and high-dose groups did not differ with respect to rates of overall 5-year survival (58% versus 59%, respectively; P = .73) or progression-free survival (47% versus 50%, respectively; P = .94).
A similar randomized trial, addressing the question of whether a dose response to radiotherapy existed for LGG patients, was published by Shaw and coworkers in 2002.27 This trial (NCCTG 86-72-51) was organized jointly by the North Central Cancer Treatment Group (NCCTG), Radiation Therapy Oncology Group (RTOG), and European Cooperative Oncology Group (ECOG). From 1986 to 1994, 203 patients were randomized to receive either a low-dose (50.4 Gy over 28 fractions) or a high-dose (64.8 Gy over 36 fractions) radiotherapy regimen. Similar to the EORTC 22844 study, no dose response was seen after a median follow-up interval of 6.4 years. Additionally, patients in the high-dose group were found to have a significantly increased risk for developing radionecrosis. As a result of these and other studies, the accepted dose range for LGG patients receiving radiotherapy is about 50 to 54 Gy in 1.8-Gy fractions.
To determine the benefit of early compared with delayed radiotherapy in LGG patients, another prospective trial (EORTC 22845) randomized patients to receive either early radiotherapy (54 Gy over 6 weeks) or observation alone after initial surgical resection or biopsy.28 Interim analysis and long-term follow-up both demonstrated no benefit to early irradiation in terms of overall survival. A significant increase in progression-free survival was seen in the early radiotherapy group relative to the observation alone group (4.8 years and 3.4 years, respectively; P = .02). Therefore, it was concluded that withholding radiotherapy until the time of disease progression is a safe and effective strategy because it demonstrated no adverse impact on overall survival. The absence of any difference in overall survival between the two arms of the trial was attributed in part to the effectiveness of radiation given as a salvage strategy on disease progression.
The rationale for delaying radiotherapy in patients with LGG is based in part on a desire to avoid iatrogenic radiotherapy-induced side effects, such as delayed cognitive impairment, neuroendocrine dysfunction, radionecrosis, tumor dedifferentiation, and induction of secondary malignancies. However, these concerns may need to be reassessed in view of the advances in the field of radiotherapy, the change in strategy from whole-brain irradiation to focused-dose delivery, and modern studies suggesting that the risk for adverse events is lower than reported in historical studies.71,72 Moreover, there is no current evidence to indicate that stereotactic radiosurgery is effective in treating low-grade gliomas.
Chemotherapy
The recognition of the responsiveness of oligodendroglial tumors to chemotherapy, as well as the identification of chromosomal markers predicting increased chemosensitivity, has helped to renew interest in employing chemotherapy in the management of LGG patients with other histopathologies.73 The most commonly used chemotherapeutic regimens in adult LGG patients are temozolomide, initially, and PCV (combination of procarbazine, lomustine [CCNU], and vincristine) for tumors that fail to respond to temozolomide.
An early randomized study by the Southwest Oncology Group looked at the utility of treating LGG patients with single-agent CCNU after radiotherapy.74 This study found no added benefit of including CCNU in the treatment regimen. In addition, patients in the CCNU arm of the study commonly developed hematologic side effects related to chemotherapy.
The RTOG is currently conducting a randomized phase 3 study examining the efficacy of postoperative radiotherapy with or without PCV therapy in treating LGG patients categorized as high risk. The high-risk arm will consist of patients older than 40 years or those having had only subtotal resection or biopsy as their initial surgical procedure. The results of this study are pending.75
Using temozolomide, an oral alkylating agent, for treating LGG patients is currently a mainstay of adjuvant treatment but is also under scrutiny in a variety of studies. Several small studies have explored the use of temozolomide as the initial postsurgical therapy for LGGs, using temozolomide in place of fractionated radiotherapy either immediately after surgery or when the tumor has progressed.76–79 Response rates, when minor responses are included, range from 31% to 61%. Although follow-up is short, the median time to progression ranges from 31 months to more than 36 months.77,79 Brada and associates77 conducted a phase 2 trial assessing the role of temozolomide as a primary chemotherapeutic agent in LGG patients previously treated with surgery alone and concluded that temozolomide does have single-agent activity against LGG and may help control seizures in these patients as well. Temozolomide has also shown efficacy in treatment of patients with progressive LGG.80 Importantly, temozolomide is administered orally and has a favorable side-effect profile, allowing for its use in a variety of clinical scenarios.
Berger MS, Deliganis AV, Dobbins J, et al. The effect of extent of resection on recurrence in patients with low grade cerebral hemisphere gliomas. Cancer. 1994;74:1784-1791.
Bernstein M, Parrent AG. Complications of CT-guided stereotactic biopsy of intra-axial brain lesions. J Neurosurg. 1994;81:165-168.
Brown PD, Buckner JC, O’Fallon JR, et al. Importance of baseline mini-mental state examination as a prognostic factor for patients with low-grade glioma. Int J Radiat Oncol Biol Phys. 2004;59:117-125.
Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473-1479.
Cha S, Tihan T, Crawford F, et al. Differentiation of low-grade oligodendrogliomas from low-grade astrocytomas by using quantitative blood-volume measurements derived from dynamic susceptibility contrast-enhanced MR imaging. AJNR Am J Neuroradiol. 2005;26:266-273.
Chang EF, Potts MB, Keles GE, et al. Seizure characteristics and control following resection in 332 patients with low-grade gliomas. J Neurosurg. 2008;108:227-235.
Claus EB, Horlacher A, Hsu L, et al. Survival rates in patients with low-grade glioma after intraoperative magnetic resonance image guidance. Cancer. 2005;103:1227-1233.
Ito S, Chandler KL, Prados MD, et al. Proliferative potential and prognostic evaluation of low-grade astrocytomas. J Neurooncol. 1994;19:1-9.
Jeremic B, Grujicic D, Antunovic V, et al. Hyperfractionated radiation therapy (HFX RT) followed by multiagent chemotherapy (CHT) in patients with malignant glioma: a phase II study. Int J Radiat Oncol Biol Phys. 1994;30:1179-1185.
Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36:549-556.
Lunsford LD, Somaza S, Kondziolka D, et al. Brain astrocytomas: biopsy, then irradiation. Clin Neurosurg. 1995;42:464-479.
Lunsford LD, Somaza S, Kondziolka D, et al. Survival after stereotactic biopsy and irradiation of cerebral nonanaplastic, nonpilocytic astrocytoma. J Neurosurg. 1995;82:523-529.
McKnight TR, Lamborn KR, Love TD, et al. Correlation of magnetic resonance spectroscopic and growth characteristics within Grades II and III gliomas. J Neurosurg. 2007;106:660-666.
Philippon JH, Clemenceau SH, Fauchon FH, et al. Supratentorial low-grade astrocytomas in adults. Neurosurgery. 1993;32:554-559.
Piepmeier J, Christopher S, Spencer D, et al. Variations in the natural history and survival of patients with supratentorial low-grade astrocytomas. Neurosurgery. 1996;38:872-878.
Price SJ, Jena R, Burnet NG, et al. Improved delineation of glioma margins and regions of infiltration with the use of diffusion tensor imaging: an image-guided biopsy study. AJNR Am J Neuroradiol. 2006;27:1969-1974.
Rey JA, Bello MJ. Cytogenetics. In: Berger MS, Wilsons CW, editors. The Gliomas. Philadelphia: Saunders; 1999:25-37.
Sanai N, Berger MS. Glioma extent of resection and its impact on patient outcome. Neurosurgery. 2008;62:753-764.
Sanai N, Mirzadeh Z, Berger MS. Functional outcome after language mapping for glioma resection. N Engl J Med. 2008;358:18-27.
Schiff D. Temozolomide and radiation in low-grade and anaplastic gliomas: temoradiation. Cancer Invest. 2007;25:776-784.
Shaw E, Arusell R, Scheithauer B, et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20:2267-2276.
Smith JS, Chang EF, Lamborn KR, et al. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J Clin Oncol. 2008;26:1338-1345.
van den Bent MJ, Afra D, de Witte O, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366:985-990.
van Veelen ML, Avezaat CJ, Kros JM, et al. Supratentorial low grade astrocytoma: prognostic factors, dedifferentiation, and the issue of early versus late surgery. J Neurol Neurosurg Psychiatry. 1998;64:581-587.
Watanabe K, Sato K, Biernat W, et al. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res. 1997;3:523-530.
1 Guthrie BL, Laws ERJr. Supratentorial low-grade gliomas. Neurosurg Clin N Am. 1990;1:37-48.
2 Kleihues P, Cavenee WK. Pathology and Genetics of Tumours of the Nervous System. Lyon: IARC Press; 2000.
3 Davis FG, Malinski N, Haenszel W, et al. Primary brain tumor incidence rates in four United States regions, 1985-1989: a pilot study. Neuroepidemiology. 1996;15:103-112.
4 McCormack BM, Miller DC, Budzilovich GN, et al. Treatment and survival of low-grade astrocytoma in adults—1977-1988. Neurosurgery. 1992;31:636-642.
5 Chang EF, Potts MB, Keles GE, et al. Seizure characteristics and control following resection in 332 patients with low-grade gliomas. J Neurosurg. 2008;108:227-235.
6 Bloom HJ. Intracranial tumors: response and resistance to therapeutic endeavors, 1970-1980. Int J Radiat Oncol Biol Phys. 1982;8:1083-1113.
7 Piepmeier J, Christopher S, Spencer D, et al. Variations in the natural history and survival of patients with supratentorial low-grade astrocytomas. Neurosurgery. 1996;38:872-878.
8 Magalhaes A, Godfrey W, Shen Y, et al. Proton magnetic resonance spectroscopy of brain tumors correlated with pathology. Acad Radiol. 2005;12:51-57.
9 Price SJ, Jena R, Burnet NG, et al. Improved delineation of glioma margins and regions of infiltration with the use of diffusion tensor imaging: an image-guided biopsy study. AJNR Am J Neuroradiol. 2006;27:1969-1974.
10 Price SJ, Pena A, Burnet NG, et al. Detecting glioma invasion of the corpus callosum using diffusion tensor imaging. Br J Neurosurg. 2004;18:391-395.
11 Di Costanzo A, Scarabino T, Trojsi F, et al. Multiparametric 3T MR approach to the assessment of cerebral gliomas: tumor extent and malignancy. Neuroradiology. 2006;48:622-631.
12 Di Costanzo A, Trojsi F, Giannatempo GM, et al. Spectroscopic, diffusion and perfusion magnetic resonance imaging at 3.0 Tesla in the delineation of glioblastomas: preliminary results. J Exp Clin Cancer Res. 2006;25:383-390.
13 Jeun SS, Kim MC, Kim BS, et al. Assessment of malignancy in gliomas by 3T 1H MR spectroscopy. Clin Imaging. 2005;29:10-15.
14 McKnight TR, Lamborn KR, Love TD, et al. Correlation of magnetic resonance spectroscopic and growth characteristics within Grades II and III gliomas. J Neurosurg. 2007;106:660-666.
15 Shimizu H, Kumabe T, Tominaga T, et al. Noninvasive evaluation of malignancy of brain tumors with proton MR spectroscopy. AJNR Am J Neuroradiol. 1996;17:737-747.
16 Cha S, Tihan T, Crawford F, et al. Differentiation of low-grade oligodendrogliomas from low-grade astrocytomas by using quantitative blood-volume measurements derived from dynamic susceptibility contrast-enhanced MR imaging. AJNR Am J Neuroradiol. 2005;26:266-273.
17 Hakyemez B, Erdogan C, Ercan I, et al. High-grade and low-grade gliomas: differentiation by using perfusion MR imaging. Clin Radiol. 2005;60:493-502.
18 Minn H. PET and SPECT in low-grade glioma. Eur J Radiol. 2005;56:171-178.
19 Szymanski MD, Rowley HA, Roberts TP. A hemispherically asymmetrical MEG response to vowels. Neuroreport. 1999;10:2481-2486.
20 Bauman G, Lote K, Larson D, et al. Pretreatment factors predict overall survival for patients with low-grade glioma: a recursive partitioning analysis. Int J Radiat Oncol Biol Phys. 1999;45:923-929.
21 Johannesen TB, Langmark F, Lote K. Progress in long-term survival in adult patients with supratentorial low-grade gliomas: a population-based study of 993 patients in whom tumors were diagnosed between 1970 and 1993. J Neurosurg. 2003;99:854-862.
22 Janny P, Cure H, Mohr M, et al. Low grade supratentorial astrocytomas. Management and prognostic factors. Cancer. 1994;73:1937-1945.
23 Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36:549-556.
24 Laws ERJr, Taylor WF, Bergstralh EJ, et al. The neurosurgical management of low-grade astrocytoma. Clin Neurosurg. 1986;33:575-588.
25 Lote K, Egeland T, Hager B, et al. Survival, prognostic factors, and therapeutic efficacy in low-grade glioma: a retrospective study in 379 patients. J Clin Oncol. 1997;15:3129-3140.
26 Pignatti F, van den Bent M, Curran D, et al. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol. 2002;20:2076-2084.
27 Shaw E, Arusell R, Scheithauer B, et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20:2267-2276.
28 van den Bent MJ, Afra D, de Witte O, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366:985-990.
29 Leighton C, Fisher B, Bauman G, et al. Supratentorial low-grade glioma in adults: an analysis of prognostic factors and timing of radiation. J Clin Oncol. 1997;15:1294-1301.
30 Smith JS, Chang EF, Lamborn KR, et al. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J Clin Oncol. 2008;26:1338-1345.
31 Franzini A, Leocata F, Cajola L, et al. Low-grade glial tumors in basal ganglia and thalamus: natural history and biological reappraisal. Neurosurgery. 1994;35:817-820.
32 Jeremic B, Grujicic D, Antunovic V, et al. Hyperfractionated radiation therapy (HFX RT) followed by multiagent chemotherapy (CHT) in patients with malignant glioma: a phase II study. Int J Radiat Oncol Biol Phys. 1994;30:1179-1185.
33 Soffietti R, Chio A, Giordana MT, et al. Prognostic factors in well-differentiated cerebral astrocytomas in the adult. Neurosurgery. 1989;24:686-692.
34 Brown PD, Buckner JC, O’Fallon JR, et al. Importance of baseline mini-mental state examination as a prognostic factor for patients with low-grade glioma. Int J Radiat Oncol Biol Phys. 2004;59:117-125.
35 Schiffer D, Cavalla P, Chio A, et al. Proliferative activity and prognosis of low-grade astrocytomas. J Neurooncol. 1997;34:31-35.
36 Piepmeier JM. Observations on the current treatment of low-grade astrocytic tumors of the cerebral hemispheres. J Neurosurg. 1987;67:177-181.
37 Berger MS, Deliganis AV, Dobbins J, et al. The effect of extent of resection on recurrence in patients with low grade cerebral hemisphere gliomas. Cancer. 1994;74:1784-1791.
38 Muller W, Afra D, Schroder R. Supratentorial recurrences of gliomas. Morphological studies in relation to time intervals with astrocytomas. Acta Neurochir (Wien). 1977;37:75-91.
39 North CA, North RB, Epstein JA, et al. Low-grade cerebral astrocytomas. Survival and quality of life after radiation therapy. Cancer. 1990;66:6-14.
40 Vertosick FTJr, Selker RG, Arena VC. Survival of patients with well-differentiated astrocytomas diagnosed in the era of computed tomography. Neurosurgery. 1991;28:496-501.
41 Recht LD, Lew R, Smith TW. Suspected low-grade glioma: is deferring treatment safe? Ann Neurol. 1992;31:431-436.
42 Shafqat S, Hedley-Whyte ET, Henson JW. Age-dependent rate of anaplastic transformation in low-grade astrocytoma. Neurology. 1999;52:867-869.
43 James CD, Carlbom E, Nordenskjold M, et al. Mitotic recombination of chromosome 17 in astrocytomas. Proc Natl Acad Sci U S A. 1989;86:2858-2862.
44 Bogler O, Huang HJ, Cavenee WK. Loss of wild-type p53 bestows a growth advantage on primary cortical astrocytes and facilitates their in vitro transformation. Cancer Res. 1995;55:2746-2751.
45 Yahanda AM, Bruner JM, Donehower LA, et al. Astrocytes derived from p53-deficient mice provide a multistep in vitro model for development of malignant gliomas. Mol Cell Biol. 1995;15:4249-4259.
46 Watanabe K, Sato K, Biernat W, et al. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res. 1997;3:523-530.
47 Rey JA, Bello MJ. Cytogenetics. In: Berger MS, Wilsons CW, editors. The Gliomas. Philadelphia: Saunders; 1999:25-37.
48 Reifenberger J, Reifenberger G, Liu L, et al. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994;145:1175-1190.
49 van Veelen ML, Avezaat CJ, Kros JM, et al. Supratentorial low grade astrocytoma: prognostic factors, dedifferentiation, and the issue of early versus late surgery. J Neurol Neurosurg Psychiatr. 1998;64:581-587.
50 Practice parameters in adults with suspected or known supratentorial nonoptic pathway low-grade glioma. Neurosurg Focus. 1998;4:e10.
51 Lunsford LD, Somaza S, Kondziolka D, et al. Brain astrocytomas: biopsy, then irradiation. Clin Neurosurg. 1995;42:464-479.
52 Lunsford LD, Somaza S, Kondziolka D, et al. Survival after stereotactic biopsy and irradiation of cerebral nonanaplastic, nonpilocytic astrocytoma. J Neurosurg. 1995;82:523-529.
53 Bernstein M, Parrent AG. Complications of CT-guided stereotactic biopsy of intra-axial brain lesions. J Neurosurg. 1994;81:165-168.
54 Woodworth GF, McGirt MJ, Samdani A, et al. Frameless image-guided stereotactic brain biopsy procedure: diagnostic yield, surgical morbidity, and comparison with the frame-based technique. J Neurosurg. 2006;104:233-237.
55 Hammoud MA, Ligon BL, elSouki R, et al. Use of intraoperative ultrasound for localizing tumors and determining the extent of resection: a comparative study with magnetic resonance imaging. J Neurosurg. 1996;84:737-741.
56 Claus EB, Horlacher A, Hsu L, et al. Survival rates in patients with low-grade glioma after intraoperative magnetic resonance image guidance. Cancer. 2005;103:1227-1233.
57 Sanai N, Mirzadeh Z, Berger MS. Functional outcome after language mapping for glioma resection. N Engl J Med. 2008;358:18-27.
58 Bauman G, Pahapill P, Macdonald D, et al. Low grade glioma: a measuring radiographic response to radiotherapy. Can J Neurol Sci. 1999;26:18-22.
59 Ito S, Chandler KL, Prados MD, et al. Proliferative potential and prognostic evaluation of low-grade astrocytomas. J Neurooncol. 1994;19:1-9.
60 Nakamura M, Konishi N, Tsunoda S, et al. Analysis of prognostic and survival factors related to treatment of low-grade astrocytomas in adults. Oncology. 2000;58:108-116.
61 Nicolato A, Gerosa MA, Fina P, et al. Prognostic factors in low-grade supratentorial astrocytomas: a uni-multivariate statistical analysis in 76 surgically treated adult patients. Surg Neurol. 1995;44:208-221.
62 Peraud A, Ansari H, Bise K, et al. Clinical outcome of supratentorial astrocytoma WHO grade II. Acta Neurochir (Wien). 1998;140:1213-1222.
63 Philippon JH, Clemenceau SH, Fauchon FH, et al. Supratentorial low-grade astrocytomas in adults. Neurosurgery. 1993;32:554-559.
64 Rajan B, Pickuth D, Ashley S, et al. The management of histologically unverified presumed cerebral gliomas with radiotherapy. Int J Radiat Oncol Biol Phys. 1994;28:405-413.
65 Scerrati M, Roselli R, Iacoangeli M, et al. Prognostic factors in low grade (WHO grade II) gliomas of the cerebral hemispheres: the role of surgery. J Neurol Neurosurg Psychiatry. 1996;61:291-296.
66 Shibamoto Y, Kitakabu Y, Takahashi M, et al. Supratentorial low-grade astrocytoma. Correlation of computed tomography findings with effect of radiation therapy and prognostic variables. Cancer. 1993;72:190-195.
67 Whitton AC, Bloom HJ. Low grade glioma of the cerebral hemispheres in adults: a retrospective analysis of 88 cases. Int J Radiat Oncol Biol Phys. 1990;18:783-786.
68 Yeh SA, Ho JT, Lui CC, et al. Treatment outcomes and prognostic factors in patients with supratentorial low-grade gliomas. Br J Radiol. 2005;78:230-235.
69 Chang EF, Smith JS, Chang SM, et al. Preoperative prognostic classification system for hemispheric low-grade gliomas in adults. J Neurosurgery. 2008;109:817-824.
70 Sanai N, Berger MS. Glioma extent of resection and its impact on patient outcome. Neurosurgery. 2008;62:753-764.
71 Taphoorn MJ, Schiphorst AK, Snoek FJ, et al. Cognitive functions and quality of life in patients with low-grade gliomas: the impact of radiotherapy. Ann Neurol. 1994;36:48-54.
72 Laack NN, Brown PD, Ivnik RJ, et al. Cognitive function after radiotherapy for supratentorial low-grade glioma: a North Central Cancer Treatment Group prospective study. Int J Radiat Oncol Biol Phys. 2005;63:1175-1183.
73 Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473-1479.
74 Eyre HJ, Crowley JJ, Townsend JJ, et al. A randomized trial of radiotherapy versus radiotherapy plus CCNU for incompletely resected low-grade gliomas: a Southwest Oncology Group study. J Neurosurg. 1993;78:909-914.
75 Stieber VW. Low-grade gliomas. Curr Treat Options Oncol. 2001;2:495-506.
76 Pouratian N, Gasco J, Sherman JH, et al. Toxicity and efficacy of protracted low dose temozolomide for the treatment of low grade gliomas. J Neurooncol. 2007;82:281-288.
77 Brada M, Viviers L, Abson C, et al. Phase II study of primary temozolomide chemotherapy in patients with WHO grade II gliomas. Ann Oncol. 2003;14:1715-1721.
78 Hoang-Xuan K, Capelle L, Kujas M, et al. Temozolomide as initial treatment for adults with low-grade oligodendrogliomas or oligoastrocytomas and correlation with chromosome 1p deletions. J Clin Oncol. 2004;22:3133-3138.
79 Levin N, Lavon I, Zelikovitsh B, et al. Progressive low-grade oligodendrogliomas: response to temozolomide and correlation between genetic profile and O6-methylguanine DNA methyltransferase protein expression. Cancer. 2006;106:1759-1765.
80 Schiff D. Temozolomide and radiation in low-grade and anaplastic gliomas: temoradiation. Cancer Invest. 2007;25:776-784.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 121 Low-Grade Gliomas
Astrocytoma, Oligodendroglioma, and Mixed Glioma
Epidemiology
Glial tumors constitute about half of newly diagnosed primary brain tumors, with low-grade gliomas (LGGs) accounting for about 15% of all brain tumors in adults.1 The subset of tumors classified as LGG is a heterogeneous group of tumors with astrocytic, oligodendroglial, ependymal, or mixed cellular histology. In adults, the term LGG typically refers to the diffuse, infiltrating variety of tumors classified as World Health Organization (WHO) grade II lesions—specifically, low-grade astrocytomas, oligodendrogliomas, or mixed oligoastrocytomas.2 Among low-grade astrocytomas, the most common histologic subtypes are the fibrillary, protoplasmic, and gemistocytic variants. There is no indication in the literature that LGGs are more prevalent in a specific ethnic or national group.
About 1500 new cases of LGG are diagnosed in North America each year.3 Age-specific data show that low-grade astrocytomas constitute 15% of brain tumors in adults and 25% of brain tumors in children.1 Pediatric LGGs, which include cerebellar astrocytomas, optic pathway and hypothalamic gliomas, brainstem gliomas, and hemispheric LGGs, are discussed elsewhere in this book. These tumors demonstrate a slight male predominance and a biphasic age distribution, with the first peak occurring during childhood (age 6 to 12 years) and a second peak in adulthood (between the third and fifth decades). The median age of presentation in adults is 35 years.
Clinical Presentation
LGGs typically arise in the frontal lobes, followed by temporal and parietal lobe lesions, in order of decreasing incidence. Fifty percent to 80% of patients present with seizures as their initial symptom, with most remaining otherwise neurologically intact.4 Patients may present with or develop other signs and symptoms, which are largely dictated by the tumor’s size and location. These include signs and symptoms of raised intracranial pressure (headache, nausea, vomiting, lethargy, papilledema), focal neurological deficits (weakness, sensory disturbance or neglect, visual neglect, agnosia, aphasia), and impaired executive function (altered personality, disinhibition, apathy).
In some studies, seizures have accompanied presentation of LGG in up to 81% of patients.5 Of the patients who presented with seizures, about 50% have uncontrolled seizures at the time of resection despite antiepileptic treatment. Partial seizure type, temporal location, and longer seizure duration also appear to predispose patients to poorer preoperative seizure control.5 Careful consideration of a patient’s seizure status is of paramount importance for LGG patients because seizures significantly affect quality of life. Beyond antiepileptic agents, surgical resection is an effective means of reducing seizure burden on patients with LGGs. Postoperatively, the factors associated with freedom from seizures are gross total tumor resection, preoperative seizure history of less than 1 year, and non–simple partial seizure type. However, continued use of antiepileptic drugs can be necessary, and in some patients, an additional operation may be required for persistent seizure activity. In our experience, optimal control of intractable epilepsy without the use of postoperative anticonvulsants is possible when perioperative (i.e., extraoperative or intraoperative) electrocorticographic mapping of separate seizure foci accompanies the tumor resection. In most cases of epilepsy, as in patients with occasional breakthrough seizures, such mapping is not needed, but complete tumor resection is. When mapping is not used and radical tumor resection (including adjacent brain) is carried out, seizures occur less frequently, but most patients continue to take antiepileptic drugs.6
Conventional Neuroimaging Studies
The typical computed tomography (CT) appearance is one of an either discrete or diffuse hypodense to isodense mass lesion, showing minimal or no enhancement with intravenous contrast administration. In about 15% to 30% of patients, however, tumor enhancement can be discerned.7,8 Calcifications may also occur, particularly among oligodendrogliomas or mixed oligoastrocytomas. In addition, cystic changes may be seen with any histologic subtype.
Magnetic resonance imaging (MRI) is the diagnostic procedure of choice for LGG, delineating the lesion as hypointense to isointense on T1-weighted images and as hyperintense on T2-weighted images (Fig. 121-1). Similar to their appearance on CT scans, most LGGs do not show gadolinium enhancement on MRI. LGGs are intra-axial lesions but do not typically exert significant mass effect on surrounding structures. They do, however, display a tendency to reside within and extend along white matter tracts (e.g., corpus callosum, subcortical white matter). Neuroimaging is not diagnostic but may suggest a particular pathologic subtype of LGG by virtue of the tumor’s location and imaging characteristics. Oligodendrogliomas, for example, are frequently located within the frontal lobes, involve the cortex, and display calcifications, in contrast to other LGGs. Importantly, T1-weighted MRI with gadolinium contrast may underestimate the extent of an LGG. The true extent is shown on the T2-weighted sequences, although on these sequences, tumor extent and surrounding edema are indistinguishable. More recently, diffusion tensor MRI has been used as a surrogate marker of glioma infiltration.9,10
Emerging Neuroimaging Technologies
Magnetic Resonance Imaging
Continued improvement in the resolution of anatomic imaging and innovations in functional and physiologic imaging modalities have the potential to improve our ability to diagnose, treat, follow up, and predict outcome in LGG patients. The increasing use of 3-tesla (3-T) MRI (instead of the standard 1.5-T magnet) will provide more anatomic detail and exquisite cytoarchitectural data on intracranial lesions.11,12 Proton magnetic resonance spectroscopy (MRS) allows for the noninvasive assessment of metabolite levels within intracranial lesions (Fig. 121-2). Of particular interest are the metabolites N-acetyl aspartate (NAA), choline (Cho), creatine (Cr), and lipids. In contrast to normal brain, gliomas typically demonstrate a decrease in NAA and Cr levels and a rise in Cho levels, indicative of their proliferative potential, cellular heterogeneity, and high cell turnover. In general, higher grade lesions display higher Cho/NAA and Cho/Cr ratios than do lower-grade tumors. The utility and reliability of MRS in predicting tumor grade noninvasively is being evaluated,13–15 but it does not supplant the need for tissue diagnosis. Nevertheless, MRS may facilitate the identification of targets for surgical biopsy, focusing our attention on regions with elevated Cho peaks, suggestive of increased cellular proliferation, and thereby on regions of maximal tumor activity. In addition, MRS has proved useful in monitoring LGG patients after radiotherapy because it can often distinguish between tumor recurrence and radiation necrosis.
Magnetic resonance techniques have also been developed for assessment of cerebral blood volume (CBV). A 2- to 3-minute dynamic acquisition of T2-weighted images during intravenous injection of a bolus of gadolinium–diethylenetriamine pentaacetic acid (DTPA) allows estimations of CBV. A voxel-by-voxel CBV map can be created by integrating the area under the dynamic contrast uptake curve and provides a relative measure of CBV with a spatial resolution of about 1 × 2 × 5 mm3 or better. Magnetic resonance perfusion has already demonstrated utility in predicting histopathologic diagnosis and tumor grade noninvasively16,17 (Fig. 121-3) and will probably play a role in selecting biopsy locations, evaluating treatment response, and differentiating effects of treatment from those produced by recurrent tumor effects.
Positron Emission Tomography
Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are additional functional and metabolic imaging modalities that contribute to the diagnosis and management of LGG patients. These modalities supplement the characterization of tumor grade because LGGs are typically hypometabolic compared with high-grade lesions. Preoperatively, these techniques are also used to identify motor- or language-related regions of cortical function, but with limited specificity. In addition to predicting tumor grade and helping with preoperative planning, indications for the use of PET or SPECT in LGG include follow-up monitoring of patients for evidence of tumor recurrence or dedifferentiation.18
Magnetoencephalography
MEG has also been increasingly used for preoperative functional mapping. Compared with fMRI and PET, MEG has the advantage of higher temporal resolution by directly measuring neuronal activation rather than an indirect hemodynamic change. Previous studies have also suggested that MEG is more accurate than fMRI in identifying functional cortices that have been distorted by a nearby tumor. Overall, MEG is a robust and reliable functional imaging modality that is now used to identify the cortical location of motor and sensory pathways. Integrating MEG data with diffusion tensor imaging (DTI) information in a neuronavigational workstation directs the neurosurgeon toward potential functional brain sites that can be intraoperatively confirmed using stimulation mapping. Magnetic source imaging (MSI), which is based on the magnetoencephalographic detection of the late neuromagnetic field elicited by simple speech sounds,19 is another adjunct to mapping techniques that can be useful for mapping the somatosensory cortex and determining hemispheric dominance, and it may serve as a replacement to the Wada test.
Patient Outcome and Survival
The median overall survival time for LGG patients is about 6.5 to 8 years.20,21 Published survival estimates for patients diagnosed with LGG range from 3 to more than 20 years.7,20,22–28 Overall, 5- and 10-year survival rates of about 70% and 50%, respectively, have been reported.29 Interestingly, the clinical course of individual LGGs can demonstrate substantial heterogeneity, with certain lesions tending to behave more aggressively, whereas others follow a more indolent course. This diversity of clinical behavior is matched by the anatomic and histopathologic diversity inherent in LGGs. Not surprisingly, this contributes to the controversy among experts regarding the most appropriate strategy for treating patients with LGG. However, Smith and colleagues30 recently demonstrated that after adjusting for the effects of age, Karnofsky Performance Scale (KPS) score, tumor location, and tumor subtype, extent of resection was a significant predictor of overall survival and showed a trend toward predicting progression-free survival. This volumetric extent of resection analysis revealed that patients with greater than or equal to 90% resection had an 8-year overall survival rate of 91% and a progression-free survival rate of 43%, whereas patients with less than 90% resection had an 8-year overall survival rate of 60% and a progression-free survival rate of 21%.
Prognostic Factors
Clinical characteristics associated with improved LGG patients’ survival outcomes include age younger than 40 years at diagnosis, seizures present at diagnosis, the absence of additional neurological deficits at diagnosis, KPS score of no less than 70, and Folstein Mini-Mental Status Examination (MMSE) score greater than 26/30.20,26,27,29,31–34 Imaging factors predictive of poor survival include maximum tumor diameter greater than 5 to 6 cm and the presence of contrast enhancement.20,27 Increasingly, the extent of surgical resection has been found to be a significant predictor of outcome and progression-free survival among LGG patients (see “Surgical Resection” section under “Treatment Options”). Furthermore, histopathologic factors associated with better prognosis include an MIB-1 labeling index of less than 8% and a histologic diagnosis of either low-grade oligodendroglioma or oligoastrocytoma (especially if the tumor is harboring chromosome 1p deletions, an indication of chemosensitivity).26,27,35
Dedifferentiation or malignant transformation is a well-described phenomenon observed in LGGs. In the literature, 13% to 86% of tumors initially diagnosed as low grade were observed to recur at a higher histologic grade.4,24,33,36–40 Similar to its broad range of incidence, the time to malignant differentiation is also variable, ranging from 28 to 60 months.4,37,40–42 However, the factors resulting in the transformation to a malignant phenotype are unclear, and the effect of treatment on this malignant transformation remains controversial. In one series, 58% of patients who did not initially undergo biopsy and treatment of a tumor suspected to be an LGG after diagnostic imaging studies eventually required surgery at a median interval of 29 months, and 50% of the tumors then showed anaplastic features.41 Although these patients had a higher incidence of malignant transformation at the time of operation and shorter time to tumor progression relative to patients who were operated on initially, the study concluded that no difference was observed in overall survival. Nevertheless, it is likely that the timing of malignant transformation affects patient outcome, and this phenomenon may be detected by future studies that are more robust. Furthermore, extent of resection studies suggest that the natural history of malignant transformation can be altered by greater extent of resection.30
Genetic Expression Profile
The cause of LGG is unknown, and with the exception of patients with one of the phakomatoses, there is no defined genetic predisposition that leads to the development of these tumors. The only genetic alteration consistently observed in patients with low-grade astrocytomas is mutation of the p53 gene.43 The p53 gene is located on chromosome 17 at 17p13.1, and this site is often deleted in astrocytomas of all grades. The remaining copy of p53 is usually inactivated through a subtle mutation. Because this gene is essential in the regulation of apoptosis and cell cycle progression, loss of normal p53 function promotes the accelerated growth and malignant differentiation of astrocytes.44,45 Astrocytomas are the only brain tumors displaying significant p53 mutation rates. Fifty percent to 60% of grade 2 and grade 3 astrocytomas exhibit p53 mutations, suggesting that inactivation of this tumor suppressor gene is an early lesion among gene alterations associated with the development of malignant gliomas.46 Although some glioblastomas exhibit p53 mutations, a significant subset of them do not and instead have amplification of the epidermal growth factor receptor (EGFR) gene, suggesting that this subset arises from a different genetic pathway. Other common alterations observed in adult low-grade astrocytomas are gain of chromosome 7 and structural abnormalities, including possessing double-minute chromosomes. Losses of chromosomes 10, 13, 15, 20, and 22 and structural rearrangements involving chromosomes 4, 11, 12, 13, 16, 18, and 21 have also been reported in patients.47
Although p53 is only rarely mutated in oligodendrogliomas, more than half of these tumors show a characteristic loss of the long arm of chromosome 1 and the short arm of chromosome 19. Because loss of 1p and 19q is not seen in astrocytic tumors (with rare exception), analysis of the presence or absence of p53, 1p, and 19q can be used to distinguish an astrocytic from an oligodendroglial genotype in cases that are difficult to distinguish histologically. Similarly, most mixed oligoastrocytomas appear to segregate genetically into astrocytic or oligodendroglial genotypes, suggesting that such mixed tumors may not be a distinct biologic entity.48
