Local Anaesthetic Agents
Local anaesthetics are analgesic drugs that suppress action potentials by blocking voltage-activated sodium ion (Na+) channels in excitable tissues. Examples include the anaesthetic amides (e.g. lidocaine, bupivacaine, ropivacaine) and esters (e.g. cocaine and procaine) (Table 4.1). Other drugs which can inhibit voltage-activated Na+ channels, such as diphenhydramine (a first-generation histamine H1 receptor antagonist) and amitriptyline (a tricyclic antidepressant) also have local anaesthetic properties. The blockade of voltage-activated Na+ channels accounts for both their analgesic effects, mediated through inhibition of action potentials in nociceptive neurones, and their systemic effects. The inhibition of action potentials in the heart contributes to local anaesthetic toxicity and also accounts for the antiarrhythmic actions of intravenous lidocaine (a class 1b antiarrhythmic). Unlike general anaesthetics (the pharmacology of which is described in Chs 1 and 2), local anaesthetics do not diminish consciousness when administered correctly.
TABLE 4.1
The Features of Individual Local Anaesthetic Drugs
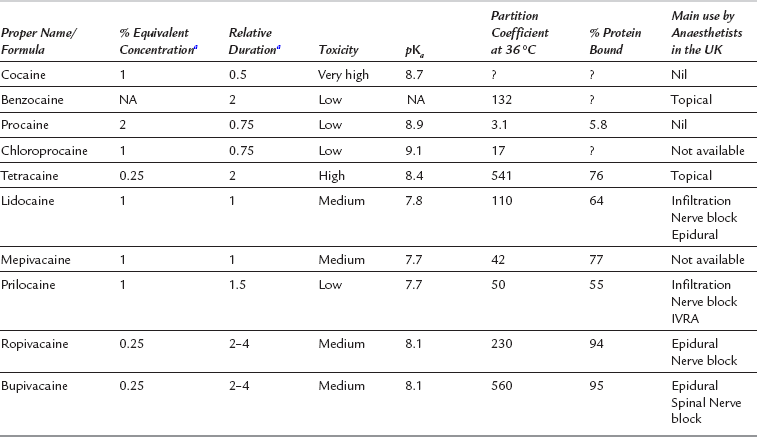
aLidocaine = 1, NA = not applicable (not used in solution), ? = information not available.
Local anaesthetics block sensation at the site of administration by inhibiting action potentials in all nociceptive fibres and therefore do not discriminate between pain modalities, unlike other analgesic drugs, such as the anti-inflammatory agents and opioids. Opioid analgesics (morphine, fentanyl, hydrocodone, etc.) and other central analgesic drugs such as the α2-adrenergic agents (clonidine, dexmedetomidine) activate metabotropic (G protein-coupled) receptors within the membranes of specific neurones located within the pain pathway. A component of the actions of these drugs is centrally mediated (as described in Ch 5).
MECHANISM OF ACTION OF LOCAL ANAESTHETICS
The primary target of local anaesthetics, the voltage- activated Na+ channel (VASC) is one of numerous membrane proteins which reside in the phospholipid bilayers encapsulating neurones (Fig. 4.1). VASCs provide selective permeability to Na+ when the cell becomes depolarized from the resting potential (approximately − 70 mV), which is maintained in quiescent neurones by the tonic activity of potassium ion (K+) channels. Local anaesthetics applied either topically to the skin or by infiltration inhibit action potentials in primary afferent nociceptive neurones, the pain-sensing neurones which transmit to the dorsal horn of the spinal cord (Fig. 4.2).
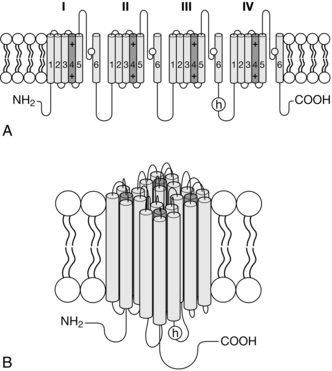
FIGURE 4.1 The topology of the VASC α-subunit. (A) The subunit has 24 membrane-spanning segments arranged in four domains with positively charged amino acid residues in the fourth segment of each domain providing voltage-sensitivity. Pore loops between segments five and six line the channel and have negatively charged amino acids which attract Na+ into the channel’s outer vestibule. The intracellular loop between domains three and four contains the inactivation gate (or h gate). (B) The four domains come together to form a channel. The ancillary β-subunits, which modulate channel function, are not shown.
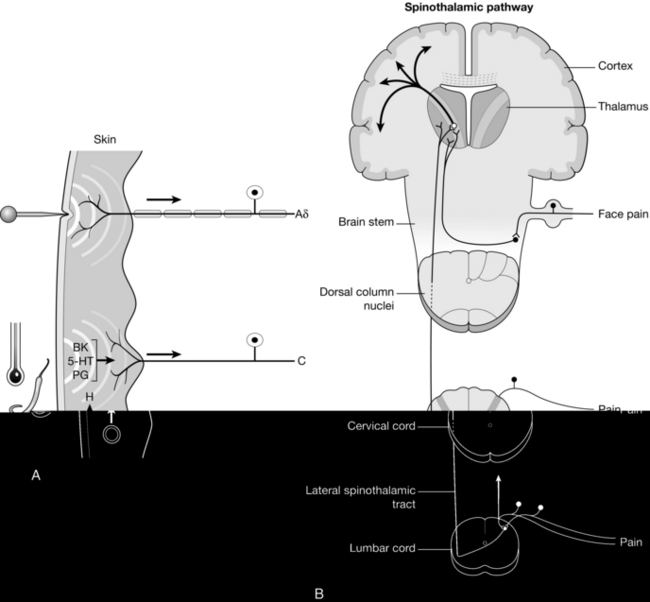
FIGURE 4.2 The ascending pain pathway. (A) Peripheral nociceptors in the skin are activated by painful stimuli. Rapid stabbing pain is transmitted by myelinated Aδ fibres. Unmyelinated C fibres, activated by inflammatory mediators (e.g. bradykinin (BK), serotonin (5-HT), prostoglandins (PG) and histamine (H)) transmit aching pain more slowly. These fibres express TRPV1 channels activated by both noxious heat and capsaicin. (B) Primary afferent fibres synapse on neurones in the dorsal horn of the spinal cord, which transmit pain stimuli to the thalamus. Thalamic neurones project to the cortex.
Pain transmission begins as a depolarization in the nerve ending of the primary afferent neurone initiated by the activation of cation channels. When the depolarization reaches the threshold for activation of VASCs (approximately − 45 mV), action potentials are generated, resulting in rapid depolarization to approximately + 20 mV (Fig. 4.3). Each action potential is brief (approximately 2 ms) because VASCs rapidly inactivate, leading to closure of their inactivation gates, and at the same time voltage-activated K+ channels activate, leading to an increase in the permeability of the cell membrane to K+. As a result, the membrane potential travels rapidly back towards the K+ equilibrium potential and this period is known as the after-hyperpolarization, a phenomenon which contributes to the refractory period during which it is unlikely that another action potential will be generated (Fig. 4.3).
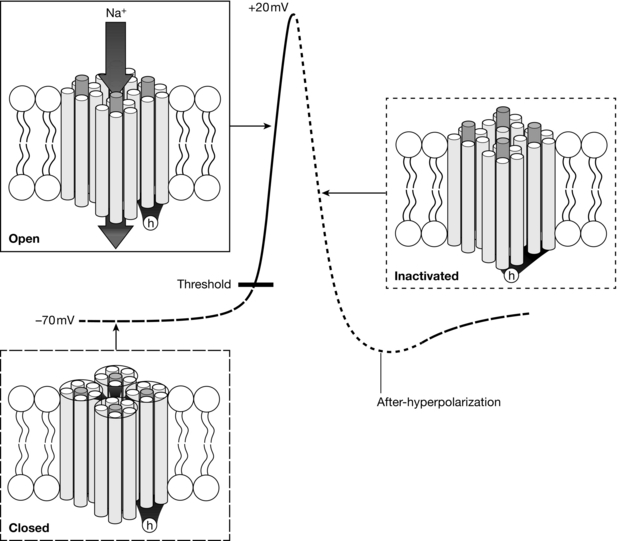
FIGURE 4.3 Most VASCs are closed at resting membrane potential (− 70 mV). Depolarization activates VASCs once the threshold potential is reached. Open VASCs enable greater depolarization until channels become inactivated and no longer support Na+ influx due to closure of the h-gate. Voltage-activated K+ channels (not shown) enable K+ efflux leading to hyperpolarization.
Mechanism of Local Anaesthetic Inhibition of the Voltage-Activated Na+ Channel
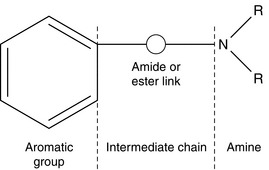
Local anaesthetics inhibit VASC activity by gaining access to the open channel from the inside of the cell and binding to specific amino acids lining the channel lumen (Fig. 4.1). They bind preferentially to the open channel and are therefore said to be use-dependent (or open channel) blockers. First, the local anaesthetic must cross the cell membrane, a passage which requires lipid solubility. The molecule must then diffuse into the aqueous environment within the ion channel. Amide and ester local anaesthetics posses both lipophilic and hydrophilic properties and are described as amphipathic (Fig. 4.4). They exist in basic (uncharged) and cationic (charged) forms and the relative proportion of each (determined using the Henderson–Hasselbalch equation) is dependent upon the pH of the solution and the pKa of the local anaesthetic:
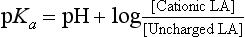
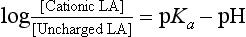
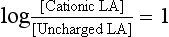
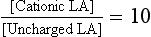
THE VOLTAGE-ACTIVATED Na+ CHANNEL
Local anaesthetics gain access to their binding site within the inner lumen of the VASC when the activation gate opens in response to depolarization. The VASC is formed by a large protein (the α-subunit) consisting of 24 membrane-spanning segments arranged in four repetitive motifs (Fig. 4.1). The fourth segment of each motif is a voltage sensor, a series of positively charged amino acids (arginine and lysine residues) lying within the membrane. Depolarization causes electrostatic repulsion of the voltage sensors, providing the energy required to open the activation gate (Fig. 4.3). Na+ ions, selected by the filter formed by the four pore loops (between the 5th and 6th segments) lining the outer vestibule of the channel, are then free to pass down their concentration gradient into the cell, generating a depolarizing electrical current. However, Na+ current is inhibited by local anaesthetic bound within the inner vestibule of the channel. The inactivation gate, formed by intracellular components of the channel, closes rapidly following depolarization (Fig. 4.3) and local anaesthetics stabilize the inactivated state.
PAIN FIBRES
Different peripheral nerve fibres have differing sensitivities to block by local anaesthetics and are classified as A, B and C according to their conduction velocities, A being the fastest conductors and C the slowest. Aδ and C fibres both conduct pain (Fig. 4.2). Other subtypes of A fibre supply skeletal muscles (α and γ) and conduct tactile sensation (β), while type B are preganglionic autonomic fibres. Aδ fibres are heavily myelinated and rapidly conduct acute stabbing pain. Myelination enables a remarkably high velocity of transmission (approximately 20 m s− 1) through a mechanism known as saltatory conduction. VASCs are segregated within the neuronal membrane of Aδ fibres at gaps in the myelin sheaths (nodes of Ranvier), enabling action potentials effectively to ‘jump’ from one node to the next. Aδ fibres are of small diameter and therefore have little ability to conduct changes in membrane potential once VASC activity has been inhibited. This makes them particularly sensitive to local anaesthetic block. Unlike Aδ fibres, C fibres are unmyelinated and their velocity of conduction from the skin to the spinal cord is relatively slow (approximately 1 m s− 1). Local anaesthetics effectively block the transmission of dull, aching pain mediated by C fibres. The fibre diameter is very small (approximately 1 μm) and therefore there is little passive conduction, making transmission reliant on the activity of VASCs. C fibres are activated by inflammatory mediators and therefore the pain resulting from their stimulation can also be treated by anti-inflammatory agents.
LOCAL ANAESTHETIC STRUCTURE
Local anaesthetics of the amide and ester classes share three structural moieties: an aromatic portion, an intermediate chain and an amine group (Fig. 4.4). The aromatic portion is lipophilic, and lipid solubility is further enhanced in local anaesthetics with lengthy intermediate chains. The amine group is a proton acceptor, providing the potential for both charged and uncharged isoforms (i.e. the source of the amphipathic nature of local anaesthetics). Amide and ester anaesthetics are so named because of their distinctive bonds within the intermediate chain. Examples of esters are cocaine, procaine, chloroprocaine and amethocaine; examples of amides are lidocaine, prilocaine, mepivacaine, etidocaine, bupivacaine, ropivacaine and levobupivacaine. A convenient mnemonic is that the names of esters contain one letter i while those of amides contain two letters i’s. The presence of either an amide or an ester bond dictates the pathway through which the local anaesthetic is metabolized and this has important implications regarding allergy potential and pharmacokinetic profile (described below). Since the introduction of cocaine into clinical practice by Koller in 1884, numerous local anaesthetics have been synthesized, beginning with procaine in 1905. Structural modifications influence pharmacokinetics. For example, replacement of the tertiary amine by a piperidine ring increased local anaesthetic lipid solubility and duration of action; addition of an ethyl group to lidocaine on the α-carbon of the amide link created etidocaine; and addition of a propyl group or butyl group to the amine end of mepivacaine resulted in [p]ropivacaine and bupivacaine respectively. Halogenation of the aromatic ring of procaine created chloroprocaine, an ester with faster hydrolysis and shorter duration of action.
PHARMACOLOGICAL PROPERTIES OF LOCAL ANAESTHETICS
A number of important factors influence the pharmacological profile of local anaesthetic drugs (Table 4.1). The pKa, molecular weight, lipid solubility, protein binding and vasoactivity influence speed of onset, potency and duration of action.
 pKa is the pH at which the ionized and non-ionized form of a compound is present in equal amounts. For basic drugs such as local anaesthetics, the greater the pKa, the greater the ionized fraction. As diffusion across the nerve sheath and nerve membrane requires non-ionized drug, a local anaesthetic with a low pKa has a fast onset of action while a high pKa causes a slow onset of action. For example, lidocaine (pKa 7.6) has a fast onset in comparison with bupivacaine (pKa 8.2), because, at pH 7.4, 35% of lidocaine exists in the non-ionized base form compared to only 20% of bupivacaine.
pKa is the pH at which the ionized and non-ionized form of a compound is present in equal amounts. For basic drugs such as local anaesthetics, the greater the pKa, the greater the ionized fraction. As diffusion across the nerve sheath and nerve membrane requires non-ionized drug, a local anaesthetic with a low pKa has a fast onset of action while a high pKa causes a slow onset of action. For example, lidocaine (pKa 7.6) has a fast onset in comparison with bupivacaine (pKa 8.2), because, at pH 7.4, 35% of lidocaine exists in the non-ionized base form compared to only 20% of bupivacaine.




PHARMACOKINETICS
The site, dose and rate of injection, and pharmacological properties, with or without addition of adrenaline, determine the absorption of a local anaesthetic drug from its site of injection. The maximum recommended clinical doses are shown in Table 4.2. The rank order of plasma concentration after injection at various sites is: intrapleural > intercostal > lumbar epidural > brachial plexus > sciatic > femoral, which reflects the vascular supply to these tissues. First-pass pulmonary metabolism limits the concentration of local anaesthetic which reaches the systemic circulation.
TABLE 4.2
Maximum Doses of Local Anaesthetics Administered as a Bolus
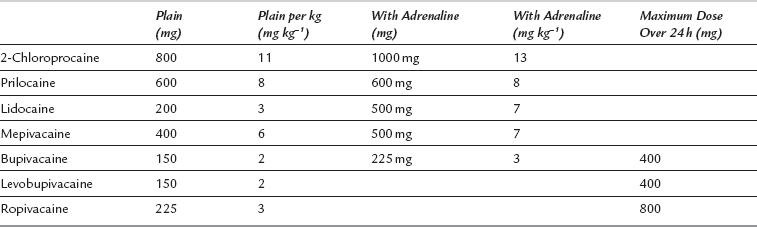
Adapted from McLeod GA, Butterworth JF, Wildsmith JAW (2008) Local anesthetic systemic toxicity. In: Cousins, Bridenbaugh, Horlecker, Carr (eds) Neural blockade. Lippincott, Williams & Wilkins, Ch 5 pp 114–132.
ENANTIOMER PHARMACOLOGY
Bupivacaine is a chiral molecule consisting of two structurally similar, non-superimposable, mirror images called enantiomers (Table 4.3). The nomenclature of enantiomers is based on the Cahn-Ingold-Prelog priority rules whereby the smallest atom is placed to the rear of the central atom about which the molecule rotates, and the sequence of the remaining three atoms is determined. For example, an increase in atomic mass in a clockwise direction is indicative of an S (sinistra) or laevo enantiomer whereas an increase in atomic mass in an anticlockwise direction is indicative of an R (rectus) or dextro enantiomer. Levobupivacaine is the S (–) or laevo enantiomer of bupivacaine whereas ropivacaine is a single enantiomer from the same series as bupivacaine, but with a propyl rather than a butyl group.
TABLE 4.3
| Chirality | Spatial arrangement of atoms, non-superimposable on each other |
| Isomer | Molecule with the same atomic composition but different stereochemical formulae and hence different physical or chemical properties |
| Stereoisomers | Identical isomers which differ in the arrangement of their atoms in space |
| Enantiomer | One of a pair of molecules which are mirror images of each other and non-superimposable |
| Racemate | An equimolar mixture of a pair of enantiomers |
LOCAL ANAESTHETIC TOXICITY
MANAGEMENT OF SEVERE LOCAL ANAESTHETIC TOXICITY
Treatment of systemic toxicity is outlined in the guidelines produced by the Association of Anaesthetists of Great Britain and Ireland (AAGBI) (Table 4.4).
TABLE 4.4
Management of Severe Local Anaesthetic Toxicity: AAGBI Safety Guideline
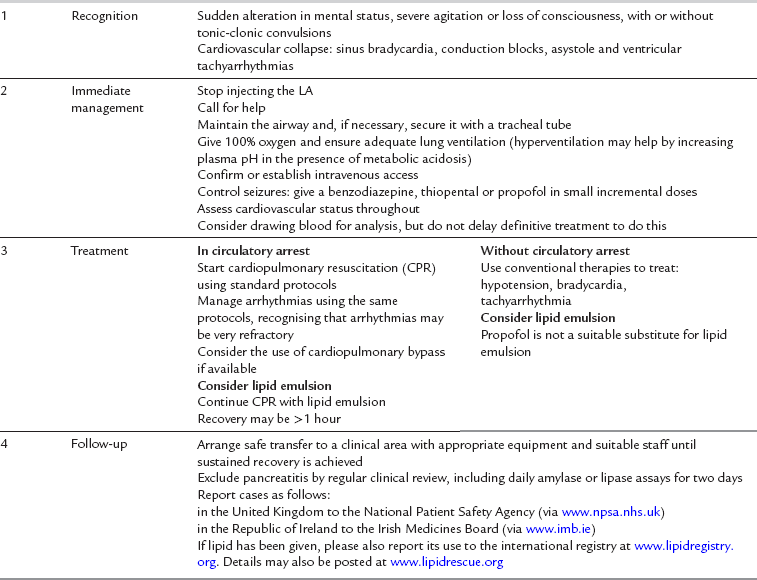
Reproduced with permission from AAGBI Safety Guideline Management of severe local anaesthetic toxicity http://www.aagbi.org/publications/guidelines/docs/la_toxicity_ 2010.pdf.
Recognition of the prodromal symptoms, such as agitation and disruption of sensory perception, is essential in order to instigate full life support measures. Immediate management should follow the Airway, Breathing, Circulation (ABC) mnemonic of resuscitation as outlined by the UK Resuscitation Council. Seizures should be controlled with small doses of thiopental, propofol or midazolam, depending on what is at hand. Hypoxaemia, hypercapnia and acidosis should be avoided because they all suppress myocardial function. If cardiac arrest occurs, cardiopulmonary resuscitation (CPR) should be started using standard protocols, but it should be recognized that arrhythmias may be very refractory. If so, consideration should be given to using an intravenous bolus dose of 20% Intralipid 1.5 mL kg− 1 over 1 min followed by an infusion of 15 mL kg h− 1. If still unresponsive, a further two boluses may be given and the infusion rate doubled (Table 4.5). The role of adrenaline in lipid-based resuscitation from local anaesthetic-induced cardiac arrest is the subject of debate and is still to be resolved. Several case reports now allude to the success of Intralipid in both paediatric and adult resuscitation.
TABLE 4.5
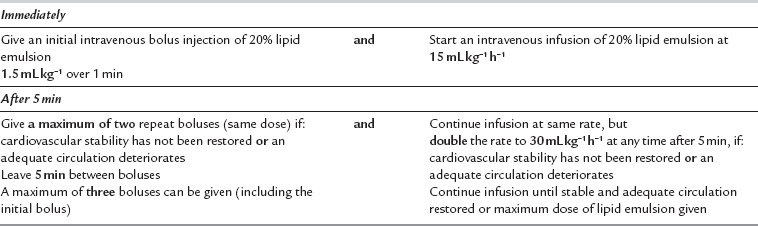
Do not exceed a maximum cumulative dose of 12 mL kg− 1.
Reproduced with permission from AAGBI Safety Guideline Management of severe local anaesthetic toxicity http://www.aagbi.org/publications/guidelines/docs/la_toxicity_ 2010.pdf.
Prevention of Severe Local Anaesthetic Toxicity








EMERGING LOCAL ANAESTHETIC APPROACHES
TRP Channels and Pain
There has been a great deal of recent interest in targeting ion channels of the transient receptor potential (TRP) family to produce analgesia. Noxious heat activates TRP channels of the TRPV1 subtype in primary afferent nociceptive neurones. TRPV1 channel activation triggers the influx of Na+ and Ca2 + ions, leading to depolarization and activation of VASCs. The resulting action potentials in pain fibres trigger the burning pain stimulus (Fig. 4.2). TRPV1 channels are also activated by the vanilloid capsaicin, a pungent substance extracted from chilli peppers. Other subtypes of TRP channel (TRPM8 and TRPA1) appear to be stimulated by noxious cold. There is interest in antagonists of TRP channels as potential analgesic drugs. Recent studies also demonstrate that TRPV1 channels can flux the positively charged quaternary lidocaine molecule QX-314. The drug is permanently charged and therefore cannot pass across the membrane of nociceptive neurones without an aqueous passage. Stimulation of TRPV1 channels by capsaicin provides a conduit for entry of QX-314 into pain fibres. Once inside neurones, the charged molecule binds to the local anaesthetic binding site within VASCs and inhibits action potentials. There is interest in this approach for targeting local anaesthesia to nociceptive neurones, thereby increasing the selectivity of block. Animal studies demonstrate that this method provides prolonged analgesia with much less motor block than is caused by equivalent analgesia using lidocaine.
AAGBI Safety Guideline. Management of severe local anaesthetic toxicity. http://www.aagbi.org/publications/guidelines/docs/la_toxicity_ 2010.pdf
Binshtok, A.M., Bean, B.P., Woolf, C.J. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449:607–610.
Catterall, W.A., Dib-Hajj, S., Meisler, M.H., Pietrobon, D. Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J. Neurosci. 2008;28:11768–11777.
Chang, D.H., Ladd, L.A., Wilson, K.A., Gelgor, L., Mather, L.E. Tolerability of large-dose intravenous laevo-bupivacaine in sheep. Anesth. Analg. 2000;91:671–679.
McLeod, G.A., Burke, D. Levobupivacaine. Anaesthesia. 2001;56:331–341.
Rao, R.B., Ely, S.F., Hoffman, R.S. Deaths related to liposuction. N. Engl. J. Med. 1999;340:1471–1475.
Rosenblatt, M.A., Abel, M., Fischer, G.W., Itzkovich, C.J., Eisenkraft, J.B. Successful use of a 20% lipid emulsion to resuscitate a patient after a presumed bupivacaine-related cardiac arrest. Anesthesiology. 2006;105:217–218.
Strichartz, G.R., Sanchez, V., Arthur, G.R., Chafetz, R., Martin, D. Fundamental properties of local anesthetics. II. Measured octanol: buffer partition coefficients and pKa values of clinically used drugs. Anesth. Analg. 1990;71:158–170.
Weinberg, G., Ripper, R., Feinstein, D.L., Hoffman, W. Lipid emulsion infusion rescues dogs from bupivacaine-induced cardiac toxicity. Reg. Anesth. Pain Med. 2003;28:198–202.
Yanagidate, F., Strichartz, G.R. Local anesthetics. Handb. Exp. Pharmacol. 2007;177:95–127.